

Abstract
A stereocontrolled and scalable synthesis of an advanced intermediate of the dysiherbaine tetrahydropyran core has been achieved in 11 steps and 27% overall yield. The key feature of this synthetic approach is the application of the Donohoe tethered aminohydroxylation reaction to install the amino diol and establish the four contiguous syn stereocenters on the tetrahydropyran ring.
Since its isolation in 1997, (–)-dysiherbaine (1) has attracted attention in the chemical and biological communities due to its interesting structure and unique biological activity.1 The molecular architecture of dysiherbaine (DH), characterized by a cis fused hexahydrofuro[3,2-b]pyran ring system displaying a glutamic acid appendage, has generated a great deal of interest as a synthetic target.2,3 Our own synthetic efforts were motivated by dysiherbaine’s impressive biological profile.4 Radioligand binding assays of DH activity at ionotropic glutamate receptors indicate that DH is a potent agonist of kainate receptors. Furthermore, DH induces epilepsy-like seizures in mice and has been shown to be the most potent epileptogenic excitatory amino acid yet identified.
Both the kainate receptor selectivity and potency of DH make it a valuable pharmacological tool for investigating the physiological roles of the various glutamate receptors in the CNS.4e Assays of dysiherbaine activity at ionotropic glutamate receptors have demonstrated that DH is a potent agonist of non-NMDA type glutamate receptors, with a particularly high affinity for the KA receptor: IC50 = 33 and 230 nM for KA and AMPA, respectively.1 When injected into mice, DH exhibited the most potent epileptogenic activity and neurotoxic symptoms among the known excitatory amino acids.4a Both the receptor selectivity and potency of DH make it an excellent probe to elucidate the roles of KA receptor subtypes in the CNS. Furthermore, as a neurotoxin and potent convulsant, DH may also provide insight as to targets for therapeutic intervention in the treatment of seizures. The low natural abundance of DH, however, has limited its availability, thereby impeding further studies on its mode of action. As a consequence, total synthesis of DH is required for further physiological studies.
Since the initial elucidation of the dysiherbaine structure, several research groups, including our own, have independently accomplished the total synthesis of DH. The synthesis of DH previously developed in our laboratory featured a Fleet-inspired ring contraction of the δ-lactone 3 to construct the bicyclic ring system and establish the stereochemistry at the tetrasubstituted γ carbon of the glutamate side chain (Scheme 1).2d
Scheme 1.
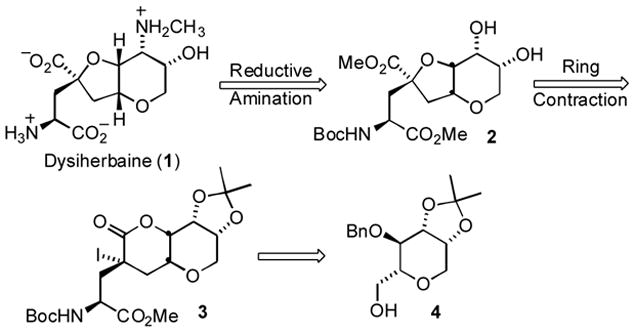
Retrosynthetic strategy for previous dysiherbaine synthesis
Overall, the synthetic route was highly convergent and it provided sufficient quantities of DH for biological evaluation; however, the final stages of the synthesis were plagued by difficulties encountered with an oxidation and reductive amination sequence that ultimately produced the N-Me amino alcohol of 1 in poor yield. To circumvent this low yielding amination step, we have explored an alternative approach to DH in which an analogous but more highly functionalized tetrahydropyran derivative, 6, would be carried through a similar sequence in place of the intermediate 4 (Scheme 2), with the critical installation of the N-Me amino alcohol moiety occurring prior to the lactone ring contraction. Moreover, we believe that the route we have developed to the tetrahydropyran 6 has considerable merit in its own right -- independent of its potential as a DH intermediate -- as one of the few examples of a tethered amino hydroxylation reaction applied to natural product structures, and more importantly, as a caveat in the application of this exceptionally selective and high-yielding reaction to construct vicinal amino alcohols. We thus describe herein an efficient and scalable synthesis of the highly functionalized intermediate 6 that embodies the tetrahydropyran (THP) core of dysiherbaine.
Scheme 2.
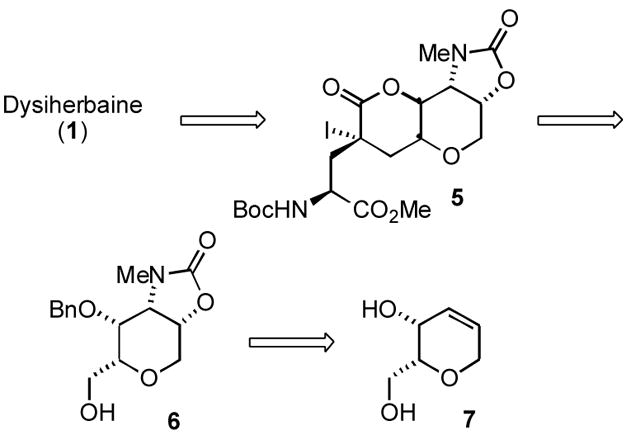
Revised retrosynthetic strategy for dysiherbaine featuring early installation of amino alcohol
Previous syntheses of advanced DH intermediates identified the four contiguous syn stereocenters and aminodiol functionality of the tetrahydropyran core as a significant synthetic challenge.2,3 The strategy reported herein was motivated by the view that a new synthetic approach to the DH pyran core would need to provide greater efficiency and implement high yielding processes in strategic roles. With this in mind, we recognized that the hydroxyamination of an unsaturated pyran derivative (7) might be ideally suited for this application. While the Sharpless asymmetric aminohydroxylation reaction of olefins has emerged as a powerful method to construct vicinal amino alcohols in the context of natural product synthesis5, it has not been employed to introduce the amino alcohol of dysiherbaine—possibly because of concerns regarding the regioselectivity of this transformation, which may detract from its potential synthetic utility. In 2002, Donohoe and co-workers introduced an innovative method of controlling aminohydroxylation regioselectivity, in which the nitrogen source is tethered to the olefin substrate. This tethered aminohydroxylation of olefins was reported to proceed with complete control of regioselectivity and was highly diastereoselective in cyclic substrates.6,7 These characteristics, in conjunction with a substitution pattern in dysiherbaine ideally suited for this process, suggested that the Donohoe aminohydroxylation reaction might be an ideal means of achieving the desired levels of regio- and stereoselectivity in the synthesis of 6. Thus, the tethered aminohydroxylation reaction figured prominently in our synthetic planning.
Our approach to 6 thus began with the preparation of the tethered aminohydroxylation substrate, allylic carbamate 11 (Scheme 3). The microwave accelerated indium catalyzed Ferrier rearrangement8 of tri-O-acetyl-D-galactal (8) in the presence of triethylsilane consistently provided good yields (85%) of olefin 99 on scales ranging from 100 mg to 20 g. Deacetylation of 9 was accomplished using NaOMe in MeOH to afford diol 7, which was selectively protected at the primary alcohol as the TBS silyl ether 10. Sequential treatment of allylic alcohol 10 with trichloroacetylisocyanate and K2CO3/MeOH afforded multi-gram quantities of carbamate 11, the substrate for the key tethered aminohydroxylation reaction, in 96% yield after purification.
Scheme 3.
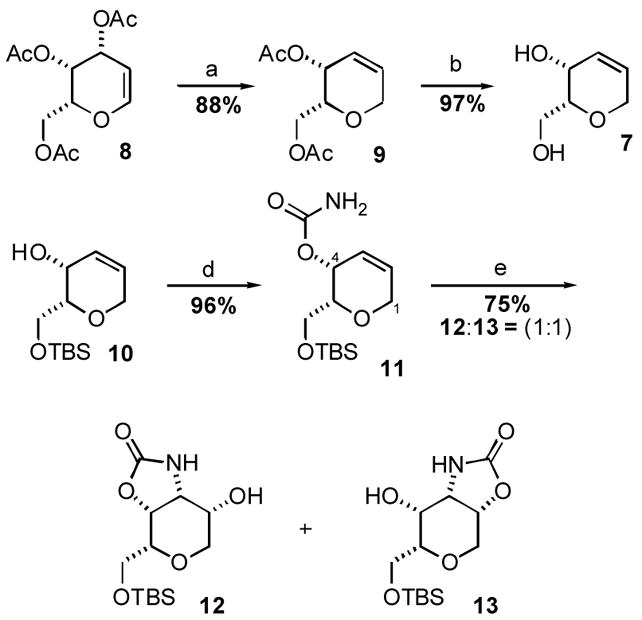
Reagents and Conditions: (a) Et3SiH, InCl3 , CH3CN, μ wave; (b) NaOMe, MeOH; (c) TBSCl, imidazole, DMF; (d) i. Cl3CCONCO, CH2Cl2 ii. K2CO3, MeOH/H2O; (e) t-BuOCl, NaOH, K2OsO2(OH)4, (DHQ)2PHAL, nPrOH/H2O
Following the successful synthesis of carbamate 11, the key tethered aminohydroxylation reaction was investigated. We first examined the tethered aminohydroxylation conditions reported by Donohoe, which were modeled after the traditional Sharpless asymmetric aminohydroxylation procedures. Satisfyingly, exposure of carbamate 11 to the standard tethered aminohydroxylation conditions (t-BuOCl, NaOH, K2OsO2(OH)4, (DHQ)2PHAL, nPrOH/H2O) cleanly installed the syn amino diol motif on the DH tetrahydropyran core in 75% yield. The remainder of the mass balance for this transformation consisted of recovered starting material that could be easily recycled through the hydroxyamination sequence. To our surprise, however, the aminohydroxylation product was not isolated as the single anticipated hydroxyoxazolid in one isomer 12. On the contrary, a 1:1 mixture of hydroxy oxazolidinone isomers 12 and 13 was observed in the crude reaction mixture. Following careful chromatography to separate the two isomers, 12 and 13 were independently resubjected to the tethered aminohydroxylation reaction conditions. Both reactions afforded identical 1:1 product mixtures regardless of the starting isomer, confirming that equilibration is quite facile and has reached thermodynamic equilibrium under the reaction conditions. The hydroxy oxazolidinone isomers 12 and 13 also undergo equilib ration upon exposure to NaOMe/MeOH or NaH/THF.
The few reports in the literature of tethered aminohydroxylation reactions of allylic carbamates do not include any examples of related equilibrations, which clearly might diminish the synthetic utility of the reaction to some extent. We believe, however, that this side-reaction is substrate dependent, and in this specific instance occurs because of the overwhelmingly axial disposition of the secondary alcohols in both the initially formed product (12) and in the migration product (13). Both alcohols are ideally positioned for direct acyl transfer; thus a plausible mechanism for equilibration in the specific case of 11 → 12 + 13 is direct attack of the axial alkoxide on the urethane carbonyl group followed by a regioselectively arbitrary collapse of the tetrahedral intermediate and facile equilibration of the two urethanes. Significantly, carbamate migrations were not observed during the tethered aminohydroxylation reaction on an analogous cyclic substrate (C4-epi-11 → 14, Figure 1) in which the allylic alcohol occupies an equatorial position.6
Figure 1.
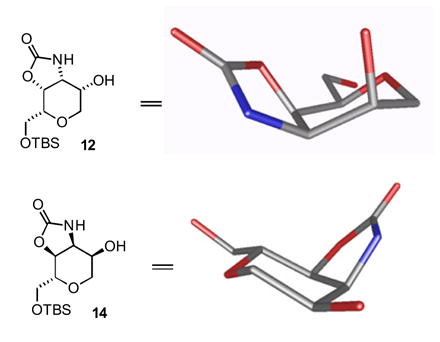
Comparison of oxazolidinone equilibration substrates (TBS group omitted for clarity)
Indeed, molecular modeling10 indicates that the equatorial alkoxide of 14 is poorly aligned for addition to π*C=O, while the axial alkoxide of 12 is well positioned for attack at the Burgi-Dunitz angle. An alternative equilibration pathway might involve deprotonation of the carbamate N-H to form an isocyanate that is attacked indiscriminately by the flanking hydroxyl groups. This pathway, however, does not account for the difference in the migratory propensity of 12 and 14 under the reaction conditions, because one would not expect a significant difference in the rates of isocyanate formation from the diastereomers 12 and 14. This analysis suggests more generally that carbamate equilibration in the tethered aminohydroxylation reaction is a consequence of relative stereochemistry, and may be facile if the participating groups are favorably positioned for acyl transfer in energetically preferred conformers of the initially formed product. A related example describing oxazolidinone migration during the tethered aminohydroxylation reaction of a homoallylic carbamate11 has recently appeared, supporting this contention.
Despite a considerable effort, conditions for preventing the equilibration of 12 could not be found; however, the problem was circumvented by processing both oxazolidinone isomers into a single advanced intermediate (17) by sequential treatment with Boc2O, TBAF, and Cs2CO3 in MeOH.12 Employing this optimized three step protcol, the conversion of 12 and 13 into THP 17 was realized on an 11 g scale in 81% overall yield (Scheme 4). In order to strategically differentiate between the two secondary axial alcohols on the pseudosymmetric 17, the DH tetrahydropyran core was selectively functionalized using benzaldehyde dimethylacetal and CSA (cat.) at 0 °C to exclusively provide the six-membered ring benzylidene acetal 18 in 78% yield. Sequential treatment of 18 with KOtBu and MeI then afforded N-Me oxazolidinone 19 as a crystalline solid in 95 % yield. X-ray crystallographic analysis of 19 verified the structure and unambiguously established the all syn stereochemistry of the substituents on the THP ring (Figure 2).13 Finally, regioselective reductive cleavage of the benzylidene acetal14 was best accomplished using PhBCl2 and Et3SiH at –78 °C to furnish multi-gram quantities of tetrahydropyran (6).
Scheme 4.
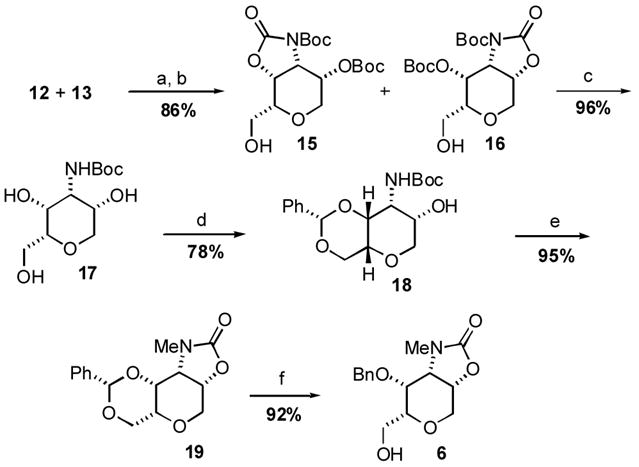
Reagents and C onditions: (a) Boc2O, Et3N, DMAP, THF; (b) TBAF, AcOH, DMF; (c) Cs2CO3, MeOH; (d) PhCH(OMe)2, CSA, CH2Cl2; (e) KOtBu, MeI, THF; (f) PhBCl2, Et3SiH, 4Å, CH2Cl2
Figure 2.
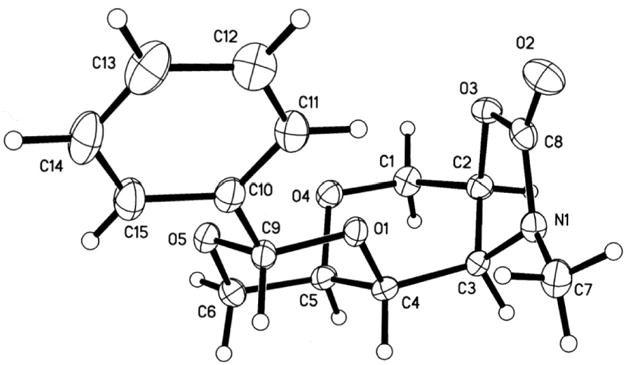
Single crystal x-ray structure of 19
In summary, we have developed an efficient, stereocontrolled, and scalable synthesis of a highly functionalized amino hydroxy tetrahydropyran derivative in 11 steps and 27 % overall yield. The key feature of this approach is the application of the Donohoe tethered aminohydroxylation reaction to install the syn amino diol motif found in dysiherbaine. We also observed, consistent with a recent literature report, that the initially formed product of this reaction can undergo facile acyl transfer to give a thermodynamic mixture of cyclic urethanes, although this process appears to be highly substrate dependent and in the case reported here, easily circumvented.
Supplementary Material
Acknowledgments
We acknowledge Professor Robert Doedens and Erin Tappan for solving the x-ray crystal structure of compound 19. We are grateful for support from the National Institute of Neurological Disorders and Stroke (NS-27600 to A.R.C.) and the National Institute of General Medical Sciences (GM07311, training grant support for J.L.C.)
Footnotes
Supplementary Material
Complete experimental procedures, product characterization, and spectral data. Supplementary data associated with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakai R, Kamiya H, Murata M, Shimamoto K. J Am Chem Soc. 1997;119:4112–4116. [Google Scholar]
- 2.Total Syntheses: Snider B, Hawryluk N. Org Lett. 2000;2:635–638. doi: 10.1021/ol991393d.Sasaki M, Koike T, Sakai R, Tachibana K. Tetrahedron Lett. 2000;41:3923–3926.Masaki H, Maeyama J, Kamada K, Esumi T, Iwabuchi Y, Hatakeyama S. J Am Chem Soc. 2000;122:5616–5217.Phillips D, Chamberlin AR. J Org Chem. 2002;67:3194–3201. doi: 10.1021/jo0107610.
- 3.Partial and Formal Syntheses: Miyata O, Iba R, Hashimoto J, Naito T. Org Biomol Chem. 2003;1:772–774. doi: 10.1039/b212556k.Kang S, Lee Y. Synlett. 2003;7:993–994.Huang J, Xu K, Loh T. Synthesis. 2003;5:755–764.Naito T, Nair S, Nishiki A, Yamashita K, Kiguchi T. Heterocycles. 2000;53:2611–2615.
- 4.Biological Activity: Sasaki R, Swanson G, Shimamoto K, Green T, Contractor A, Ghetti A, Tamura-Horikawa Y, Oiwa C, Kamiya H. J Pharm Exp Ther. 2001;296:650–658.Swanson G, Green T, Sakai R, Contractor A, Che W, Kamiya H, Heinemann S. Neuron. 2002;34:589–598. doi: 10.1016/s0896-6273(02)00676-1.Minei R. Jpn J Ophthalmol. 2002;46:153–159. doi: 10.1016/s0021-5155(01)00488-9.Sanders J, Ito K, Settimo L, Pentikainen O, Shoji M, Sasaki M, Johnson M, Sakai R, Swanson G. J Pharmacol Exp Ther. 2005;314:1068–1078. doi: 10.1124/jpet.105.086389.Sakai R, Swanson GT, Sasaki M, Shimamoto K, Kamiya H. Central Nervous System Agents in Med Chem. 2006;6:83–108.
- 5.Recent reviews on Sharpless Asymmetric Aminohydroxylation: O'Brien P. Angew Chem Int Ed. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T.Nilov D, Reiser O. Adv Syn Catal. 2002;344:1169–1173.Bodkin J, McLeod M. J Chem Soc, Perkin Trans. 2002;1:2733–2746.Bolm C, Hildebrand J, Muniz K. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. John Wiley & Sons, Inc; New York, N.Y: 2000. pp. 399–429. Chapter 6E.Muniz K. Chem Soc Rev. 2004;33:166–174. doi: 10.1039/b307102m.
- 6.Donohoe TJ, Johnson PD, Cowley A, Keenan M. J Am Chem Soc. 2002;124:12934–12935. doi: 10.1021/ja0276117. [DOI] [PubMed] [Google Scholar]
- 7.Examples of tethered aminohydroxyalations of allylic carbamates: Donohoe TJ, Johnson PD, Helliwell M, Keenan M. Chem Commun. 2001:2078–2079. doi: 10.1039/b107253f.Donohoe TJ, Johnson PD, Pye RJ, Keenan M. Org Lett. 2004;6:2583–2585. doi: 10.1021/ol049136i.Donohoe TJ, Johnson PD, Pye RJ. Org Biomol Chem. 2003;1:2025–2028. doi: 10.1039/b305189g.Kenworthy MN, McAllister GD, Taylor RJ. Tetrahedron Lett. 2004;45:6661–6664.Donohoe TJ, Johnson PD, Pye RJ, Keenan M. Org Lett. 2005;7:1275–1277. doi: 10.1021/ol0473750.
- 8.Das SK, Reddy KA, Abbineni C, Das SK, Reddy KA, Abbineni C, Roy J, Narasimha Rao KVL, Sachwani R, Iqbal J. Tetrahedron Lett. 2003;44:4507–4509. [Google Scholar]
- 9.Crimmins MT, King BW, Zuercher WJ, Choy AL. J Org Chem. 2000;65:8499–8509. doi: 10.1021/jo005535p. [DOI] [PubMed] [Google Scholar]
- 10.Molecular modeling was performed using Spartan ’02 (WaveFunction, Inc., Irvine, CA) and conformations 12 and 14 were optimized using Molecular Mechanics MMFF method.
- 11.Curtis KL, Fawcett J, Handa S. Tetrahedron Lett. 2005;46:5297–5300. [Google Scholar]
- 12.Cesium carbonate cleavage of N-Bocoxazolidinone: Ishizuki T, Kunieda T. Tetrahedron Lett. 1987;28:4185–4188.
- 13.Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 280311
- 14.Sakagami M, Hamana H. Tetrahedron Lett. 2000;41:5547–5551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.