

Abstract
Five routes to stable chlorins bearing 0 or 1 meso substituents have been investigated, among which reaction of a 9-bromo-1-formyldipyrromethane and 2,3,4,5-tetrahydro-1,3,3-trimethyldipyrrin proved most effective. Application of this route afforded metallochlorins [Cu(II), Zn(II), Pd(II)] including the chlorin lacking any β-pyrrole and meso substituents.
Keywords: Chlorin, Hydroporphyrin, Hydrodipyrrin, Dipyrromethane
1. Introduction
The dihydroporphyrins known as chlorins constitute the chromophore of plant chlorophylls. In comparison with porphyrins, chlorins absorb more strongly in the red region of the spectrum.1 The prototypical chlorins are chlorophyll a and chlorophyll b, whose structure and absorption spectra are shown in Figure 1. The spectra differ owing to the presence of the methyl group or the formyl group at the 7-position. Thus, chlorin spectra can be significantly altered upon modification of even a single substituent. To gain a deep understanding of the effects of substituents on the spectral properties of chlorins requires the ability to prepare chlorins bearing diverse patterns of substituents. To tailor chlorins for use in diverse applications also requires a fundamental understanding of how substituents alter reactivity. A comprehensive treatment of the effects of substituents requires access to chlorins bearing a systematic progression of substituents, beginning with no substituents and proceeding to one, two, or more groups at designated locations. An ultimate objective of this work is to be able to design and synthesize chlorins that exhibit desired spectral and photophysical properties for diverse applications ranging from artificial photosynthesis to photomedicine.
Figure 1.

Absorption spectra of chlorophyll a (solid line) and chlorophyll b (dashed line) in diethyl ether at room temperature.2 The chlorin numbering system is shown at right.
Surprisingly few systematic studies are available concerning the effects of substituents on chlorin properties. The dearth stems largely from synthetic limitations. Two simple routes to chlorins entail (1) derivatization of naturally occurring chlorins,3 and (2) reduction/derivatization of synthetic porphyrins.4 The former route is constrained by the numerous substituents present in the naturally occurring macrocycles. The lack of regioselectivity of the latter route typically limits the scope to the reduction of porphyrins having substitution patterns (giving 4-fold or 2-fold symmetry) wherein regioisomers cannot form. Benchmark chlorins of the latter type include meso-tetraphenylchlorin (H2TPC)5 and octaethylchlorin (H2OEC),6 which are commercially available, and 5,15-diphenylchlorin.7 Analogues such as meso-tetrakis(3-hydroxyphenyl)chlorin,8 2,3-dihydroxy-meso-tetraphenylchlorin,9 and diverse 5,15-diarylchlorins10 have been prepared for studies of photodynamic therapy.
The chlorin lacking any substituents, 2,3-dihydroporphine (known as “chlorin” itself, here termed H2Chlorin; Chart 1), has been the subject of theoretical calculations11 and numerous spectroscopic studies.12-69 The latter include fundamental studies to probe chlorin features, as well as spectral hole-burning experiments to probe the utility of chlorins in optical information storage applications. However, such a potentially valuable benchmark has not been employed for studies of reactivity (other than dehydrogenation70), presumably owing to the limited quantities of available material. The only reported synthesis of H2Chlorin entails Grignard-mediated cyclization of 2-(N,N-dimethylaminomethyl)pyrrole.71,72 Photoreduction of zinc porphine affords the corresponding ZnChlorin73 but no preparative procedure has been reported. Regardless, all of the hydroporphyrins shown in Chart 1 are susceptible in an aerobic environment to adventitious dehydrogenation to give the porphyrin, and thus have limited stability toward routine handling in the laboratory.
Chart 1.
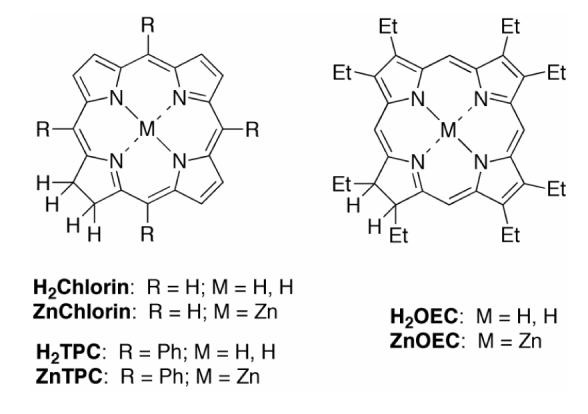
The naturally occurring chlorins bonellin (a non-photosynthetic pigment) and Faktor I (a biosynthetic intermediate) each contain a geminal dialkyl group in the pyrroline ring, which stabilizes the chlorin to dehydrogenation (Chart 2). Total syntheses of these “C-methylated chlorins” and other naturally occurring chlorins have been developed; however, such syntheses are necessarily elaborate owing to the challenges of installing the multiple β-pyrrolic and pyrrolinic substituents.74
Chart 2.
The methodology developed for preparing bonellin and Faktor I has been extended to gain access to synthetic chlorins bearing more simple substituent patterns (while retaining the geminal dimethyl group).75-78 Two routes that we developed in this regard include (I) reaction of a 9-bromodipyrromethane-1-carbinol (Eastern half) and a 1,3,3-trimethyltetrahydrodipyrrin (Western half),76 and (II) reaction of a 9-bromodipyrromethane-1-carboxaldehyde (Eastern half) and the same 1,3,3-trimethyltetrahydrodipyrrin (Western half). The two routes are shown in Scheme 1. The Western half incorporates a geminal dimethyl group that ensures the presence of the reduced, pyrroline ring in the resulting chlorin, thereby precluding adventitious dehydrogenation leading to the porphyrin. In each case, the reaction proceeds in a two-step process of condensation of the Eastern and Western halves to give a 1-methyl-19-bromo-bilane derivative, and metal-mediated oxidative cyclization of the latter to give the corresponding chlorin.
Scheme 1.

The two routes are similar in a number of respects but also differ in the conditions for condensation, nature of the acyclic intermediate, and substituent patterns in the resulting chlorins. Route I has provided access to chlorins bearing two substituents (5,10-,75,76 5,12-,79 5,8-79) or three substituents (2,5,12-,79 5,10,15-,80 5,10,20-80) at sites other than the pyrroline ring, and each chlorin has contained a 5-substituent (see Figure 1 for chlorin numbering system). Reasonable yields of chlorin macrocycle formation (12-45%) have facilitated preparation of 5,10-disubstituted chlorins in >100-mg quantities, as required for a variety of applications. Route II, developed very recently and described herein, has been applied to access zinc chlorins bearing two substituents (3,13-), three substituents (3,10,13-), and no meso- or β-pyrrole substituents.77 By contrast with route I where each chlorin contains a 5-substituent, the development of route II for the synthesis of sparsely substituted, 5-unsubstituted chlorins has presented a number of unexpected challenges.
In this paper, we describe the investigation of the synthesis of sterically uncongested, stable chlorins possessing no β-pyrrole substituents and no or only one meso substituent (at the 5- or 10-position). A 5-substituted chlorin was prepared via route I. Five routes were investigated to prepare 5-unsubstituted chlorins. The investigation entailed use of two types of Western halves and five types of Eastern halves. The routes found suitable for preparing 5-unsubstituted chlorins include route II (described above), two modified versions of route II designed to facilitate implementation and increase the scope, and a new route that entails reaction of a 9-formyltetrahydrodipyrrin and a dipyrromethane. The synthetic routes described herein have enabled examination of the fundamental reactivity, structure, and spectral properties of stable chlorins bearing few or no substituents, as are described in the companion papers.81,82
2. Results and discussion
The shorthand nomenclature for the chlorins described herein employs the following abbreviations with superscripts to denote substituents and their positions: B (3,5-di-tert-butylphenyl), P (phenyl), M (mesityl), and T (p-tolyl).
2.1. Synthesis of chlorins bearing one substituent (5-position)
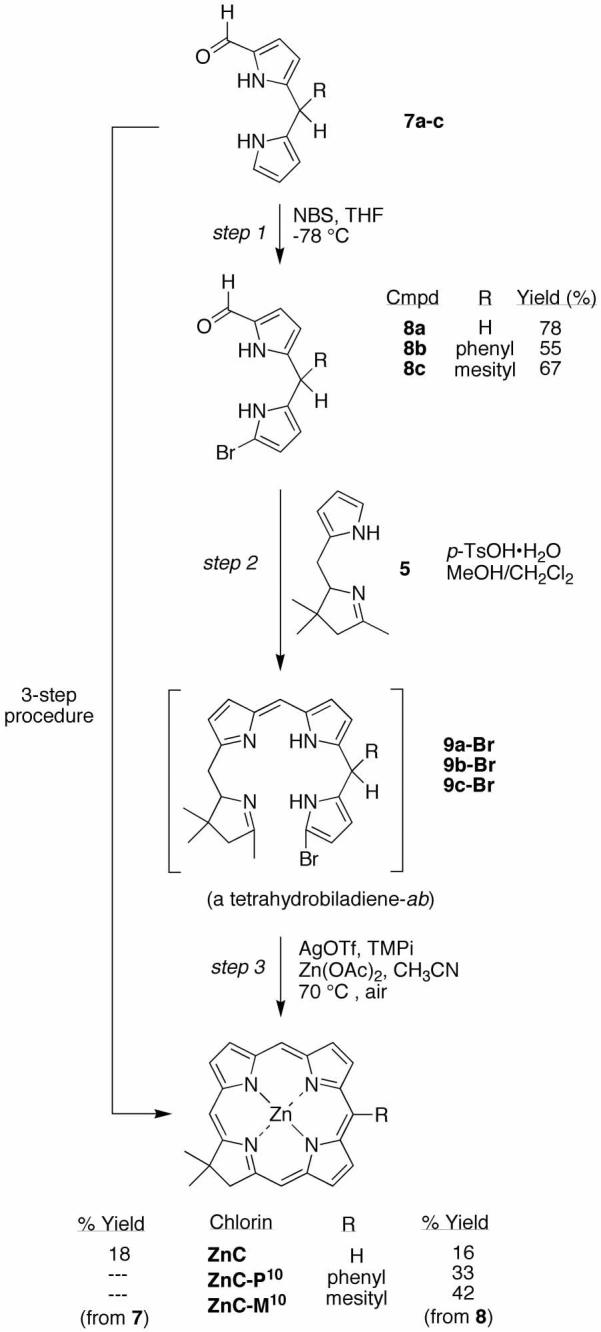
The synthesis of a chlorin bearing a lone substituent located at the 5-position was carried out following route I76 (Scheme 1). Thus, acylation83 of dipyrromethane (1a)84 using benzothioate 2-B76 or 2-T85 gave the 1-acyldipyrromethane 3-B or 3-T in 71% or 39% yield, respectively (Scheme 2). Treatment of 3-B with NBS afforded 4-B in 53% yield. Reduction of 4-B with NaBH4 gave the corresponding carbinol 4-B-OH (Eastern half), which upon condensation with 2,3,4,5-tetrahydro-1,3,3-trimethyldipyrrin (5, Western half)76 using TFA gave the tetrahydrobilene-a (6-B) in 53% yield. Treatment of the latter to metal-mediated oxidative cyclization conditions [AgOTf, 2,2,6,6-tetramethylpiperidine (TMPi) and Zn(OAc)2 in refluxing CH3CN exposed to air] afforded chlorin ZnC-B5 in 17% yield. The same approach was used with 1-acyldipyrromethane 3-T, but the limited stability of the corresponding 9-bromo species 4-T prompted in situ conversion to the chlorin. Thus, bromination of 3-T and subsequent condensation with 5 yielded putative tetrahydrobilene-a 6-T, which was converted to chlorin ZnC-T5 in 7% overall yield (from 3-T).
Scheme 2.

2.2. Synthesis of chlorins lacking a 5-substituent
(1) Retrosynthetic analysis
We investigated five routes (I–V) to the target chlorin bearing no substituents (R = H) or one substituent at the 10-position (R = aryl), as shown in Scheme 3. The routes are based on the ready availability of dipyrromethanes (1) and the tetrahydrodipyrrin 5. The 2 + 2 synthesis of a chlorin entails condensation of an Eastern half (composed of two pyrrole units) and a Western half (composed of one pyrrole and one pyrroline unit). The two halves must have complementary sets of reactive nucleophilic and electrophilic positions. The routes differ from each other in the placement of reactive groups on the two halves: the α-pyrrolic position and the pyrrolinic methyl group react as nucleophiles, whereas the α-bromo-, formyl, and hydroxymethylpyrrolic groups serve as electrophiles.
Scheme 3.

Routes to 5-unsubstituted chlorins
The routes also differ in the nature of the intermediates expected upon condensation of the Eastern half and Western half. The intermediates are shown in the retrosynthetic scheme. Intermediate A is a 2,3,4,5-tetrahydrobilene-a, B is a 2,3,4,5-tetrahydrobiladiene-ab, and C is a 2,3,4,5-tetrahydrobilatriene-abc. While the intermediates obtained upon condensation are typically not characterized but instead are treated to the conditions for metal-mediated oxidative cyclization, the differing oxidation state of A, B, and C are expected to give rise to different properties and ease of oxidation.
A key issue in the condensation concerns the lability of the Eastern half and Western half under the reaction conditions employed, particularly the level of acid present in the condensation. A requirement for stronger acidic conditions is expected for the condensation in each of routes I–V (pyrrolic-carboxaldehyde or 1° pyrrole-carbinol) to give a 5-unsubstituted chlorin versus that employed to give a 5-substituted chlorin (2° pyrrole-carbinol in route I). However, strong acid causes the Western half 5 to undergo irreversible rearrangement to a bicyclic product,76 and Eastern halves can undergo cleavage. The liberation of free pyrrolic species upon acidolysis of an Eastern half opens the door to the formation of chlorin byproducts as well as porphyrin species. Dipyrromethanes bearing meso-aryl substituents are more prone to acidolysis than those lacking a meso substituent.86 Even in the absence of acid, bromo-dipyrromethanes (e.g., many of the Eastern halves) are inherently reactive. Thus, conditions to force the reaction of Eastern and Western halves, such as strong acid or elevated temperature, are of limited utility.
(2) Route I
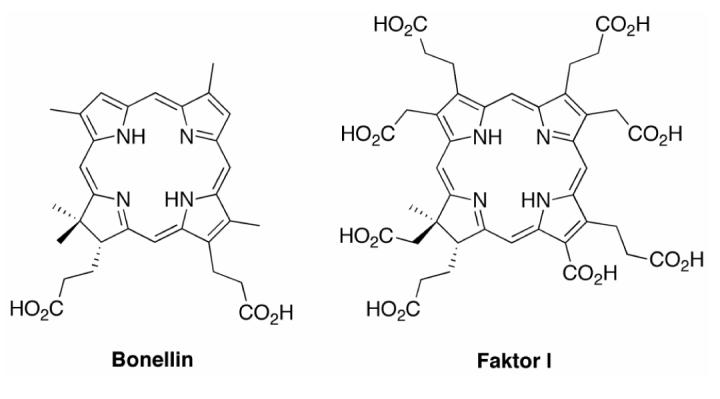
Access to a 5-unsubstituted chlorin via route I (Scheme 1) requires use of a 1° carbinol rather than a 2° carbinol. The synthesis of such chlorins required preparation of 1-formyldipyrromethanes. Thus, Grignard-mediated formylation (with phenyl formate) of dipyrromethane (1a), 5-phenyldipyrromethane (1b) and 5-mesityldipyrromethane (1c) gave the corresponding 1-formyldipyrromethanes 7a-c.87 Treatment of 7c with NBS gave the 1-bromo-9-formyldipyrromethane 8c in 67% yield. Reduction of 8c with NaBH4 afforded the 1-bromo-9-hydroxymethyldipyrromethane 8c-OH. Reaction of 8c-OH with 5 under the standard conditions75 of TFA catalysis followed by metal-mediated oxidative cyclization afforded the corresponding chlorin ZnC-M10 in <1% yield (Scheme 4). We attributed the low yield to the poor reactivity of the 1° carbinol. Related studies of the reaction of similar dipyrromethane-carbinols in porphyrin-forming conditions have also given quite low yields.88
Scheme 4.
Route I
(3) Route II
(a) Standard procedure
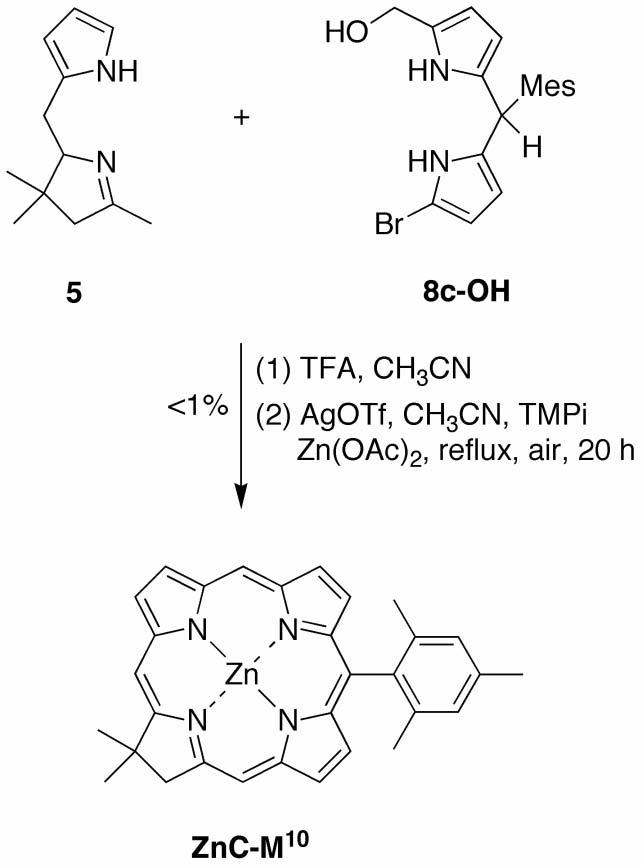
Route II utilizes a 1-bromo-9-formyldipyrromethane (Eastern half) and the tetrahydrodipyrrin (Western half). Treatment of a 1-formyldipyrromethane (7a-c) with NBS gave the corresponding 9-bromo-1-formyldipyrromethane 8a-c in yields of 55-78%. The condensation of 9-bromo-1-formyldipyrromethane (8a) and Western half 5 was carried out with 1 equiv of BF3·O(Et)2 in CH3CN, which gave no reaction after 15 minutes, while excess BF3·O(Et)2 resulted in decomposition of 5. Battersby reported the condensation of a 1-formyldipyrromethane and a tetrahydrodipyrrin using p-toluenesulfonic acid (p-TsOH·H2O) in MeOH, affording an intermediate 2,3,4,5-tetrahydrobiladiene-ab on the path to a copper chlorin.89 Note that the Eastern half and Western half reactants employed by Battersby contained several β-substituents and no meso substituents. We performed the condensation of 8a and 5 using 13.5 mM reactants and 5 molar equiv (68 mM) of p-TsOH·H2O in methanol (Scheme 5). The putative protonated 2,3,4,5-tetrahydrobiladiene-ab was observed with a maximum absorption centered at 480 nm (to be compared with Battersby's tetrahydrobiladiene-ab at 496 nm89), an absorption characteristic of dipyrrin chromophores.
Scheme 5.
Route II
The crude mixture was then subjected to the standard conditions76 for metal-mediated oxidative cyclization, affording the desired zinc chlorin ZnC in 9% yield. The similar reaction of 9-bromo-1-formyldipyrromethane 8b or 8c afforded ZnC-P10 or ZnC-M10 in 10% or 12% yield, respectively. A microscale study of the concentration dependence of the condensation of 8a and 5 (and 5 mol equiv of p-TsOH·H2O in CH2Cl2/MeOH) identified improved yields upon doubling the concentration of the condensation reactants. Thus, reaction using 26 mM Eastern and Western halves afforded workable quantities (66–104 mg) of ZnC, ZnC-P10 ZnC-M10 in 16%, 33% or 42% yield, respectively. The syntheses of ZnC and ZnC-M10 in this manner were reported in a separate publication.77
(3b) Streamlined route II
The clean bromination of 1-formyldipyrromethane 7a suggested a streamlined three-step procedure for chlorin synthesis. Thus, 7a was brominated with NBS and the crude reaction mixture was carried through the two-step procedure for chlorin formation as described above. The yield of chlorin ZnC in the three-step procedure was 18% (145 mg) versus ∼12% (104 mg)77 in the standard procedure (Scheme 5). The streamlined three-step procedure is advantageous in avoiding chromatography following step one, and employing a simple workup in step two.
(3c) AgOTf-free route II
In several cases we attempted to isolate the tetrahydrobiladiene-ab intermediate. In the reaction of 8a + 5, the resulting unsubstituted tetrahydrobiladiene-ab (9a-Br) was not isolated due to decomposition upon flash chromatography. However, the phenyl-substituted tetrahydrobiladiene-ab (9b-Br, derived from 8b + 5) was more stable and was readily isolated by flash chromatography (silica, CH2Cl2). The 1H NMR spectrum revealed a mixture of diastereomers, which made individual proton assignments difficult. However, two pyrrole NH protons were clearly observed at 7.83 and 8.18 ppm, consistent with a tetrahydrobiladiene structure.
We then turned to investigate alternative conditions for the metal-mediated, oxidative cyclization of the tetrahydrobiladiene-ab. The traditional method employs Zn(OAc)2, AgOTf and 2,2,6,6-tetramethylpiperidine in refluxing acetonitrile exposed to air. Prior omission/reconstitution experiments concerning route I revealed that omission of Zn(OAc)2 and 2,2,6,6-tetramethylpiperidine resulted in failure to form any detectable chlorin, whereas omission of AgOTf gave chlorin albeit in diminished yield.76 Here we explored the cyclization in the presence of various metal salts and bases in refluxing solvents exposed to air, but without any AgOTf (i.e., a AgOTf-free step 3 in route II). In each case, the p-TsOH-catalyzed condensation of 5 and 8b gave the crude reaction mixture containing 9b-Br. The latter was neutralized with an appropriate base, dissolved in a given solvent, and treated with a base and metal salt (Scheme 6). The resulting reaction mixture was refluxed until the absorption spectrum remained relatively unchanged. The results are summarized in Table 1.
Scheme 6.
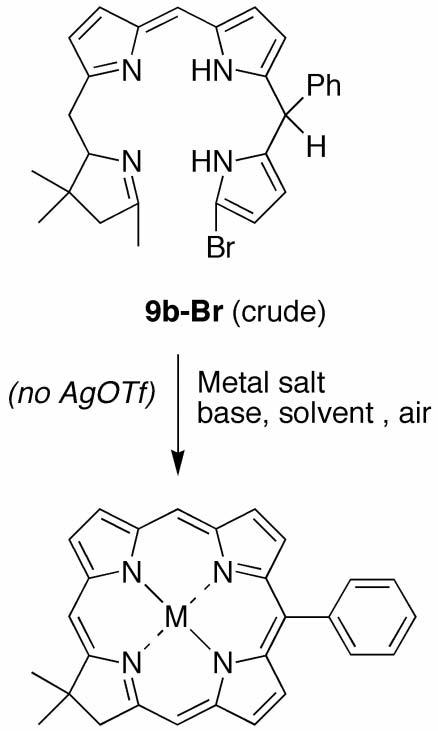
AgOTf-free step 3 of route II
Table 1.
Exploration of the metal-mediated oxidative cyclization of a tetrahydrobiladiene-aba
| Entry | Metal salt | Baseb | Solvent | Product | Yield |
|---|---|---|---|---|---|
| 1 | Zn(OAc)2 | TMPi | MeCN | ZnC-P10 | 37%c |
| 2 | Zn(OAc)2 | DBU | MeCN | ZnC-P10 | 9%c |
| 3 | Zn(OAc)2 | DIEA | MeCN | ZnC-P10 | 10%c |
| 4 | Pd(OAc)2 | TMPi | MeCN | PdC-P10 | 12%d |
| 5 | Pd(OAc)2 | KOHe | EtOH | PdC-P10 | 5.6%d |
| 6 | Pd(MeCN)2Cl2 | KOHe | EtOH | PdC-P10 | 6.2%d |
| 7 | Cu(OAc)2 | TMPi | MeCN | CuC-P10 | 4%c |
| 8 | Cu(OAc)2 | KOHe | EtOH | CuC-P10 | 14%d |
| 9 | SnCl2 | TMPi | MeCN | SnC-P10 | 5% |
| 10 | MgBr2 | TMPi | MeCN | -- | 0% |
| 11 | Co(OAc)2 | TMPi | MeCN | -- | 0% |
| 12 | NiCl2 | TMPi | MeCN | -- | 0% |
| 13 | CdCl2 | TMPi | MeCN | -- | 0% |
| 14 | No metal | TMPi | MeCN | -- | 0% |
| 15 | No metal | DBU | MeCN | -- | 0% |
| 16 | No metal | DIEA | MeCN | -- | 0% |
The crude biladiene 9b-Br was prepared by p-TsOH-catalyzed condensation of 5 and 8b under standard conditions. The reaction mixture was then neutralized by the base to be used in the metal-mediated oxidative cyclization (when KOH was used as a base in metal-mediated cyclization, crude 9b-Br was neutralized by triethylamine). Unless noted otherwise each reaction was performed at 10 mM concentration with 15 equiv of metal salt and 30 equiv of base.
Bases include 2,2,6,6-tetramethylpiperidine (TMPi), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and N,N-diisopropylethylamine (DIEA).
Spectroscopic yield (from 8b) unless noted otherwise, assuming a fixed molar absorption coefficient in all cases (see Experimental Section).
Isolated yield.
5 Equiv was employed.
With a number of metal salts, the (AgOTf-free) metal-mediated oxidative cyclization of 9b-Br gave the corresponding metallochlorin. Each metal reagent examined gave an absorption spectrum consistent with the metal complex of 9b-Br, regardless of whether the chlorin ultimately was obtained. The metal reagents that successfully gave chlorin include Zn(II) (entries 1-3), Pd(II) (entries 4-6), Cu(II) (entries 7 and 8), In(III) (entries 9 and 10), and Sn(II) (entry 11) but not Mg(II), Co(II), Ni(II), Cd(II) or no metal (entries 12-17) under the conditions examined. The best yield typically was observed when 2,2,6,6-tetramethylpiperidine was used in acetonitrile. In the case of Cu(II), a better yield was observed for KOH/EtOH, conditions (entry 8) developed previously for the synthesis of palladium porphyrins.90 The procedure of acid-catalyzed condensation followed by AgOTf-free metal-mediated oxidation was applied to give milligram quantities of CuC, PdC, CuC-P10, ZnC-P10, and PdC-P10.
A few indium(III) reagents were examined in place of zinc acetate in the AgOTf-free chlorin-forming reaction [InCl3, InBr3, In(OAc)3] to prepare X-InC-P10 where X is the counteranion. Each indium reagent afforded the corresponding indium(III)chlorin in ∼30% yield (observed spectroscopically), with the cleanest reaction observed for In(OAc)3. Contrary to other chlorin chelates examined herein, the indium(III)chlorin was highly polar and proved difficult to purify by chromatography. Moreover, we observed partial conversion of the indium(III)chlorin on silica (ethyl acetate) into another less polar chlorin. In the case of reaction with InCl3, the two indium(III)chlorin species were separated (although not isolated in pure form), and exhibited spectroscopic properties (LD-MS, UV-vis, only slightly different 1H NMR spectra) that were similar to each other. In the case of reaction with In(OAc)3, chromatography [alumina, ethyl acetate → ethyl acetate/MeOH (10:1)] followed by preparative SEC (THF) afforded the expected indium(III)chlorin in nearly pure form as determined by TLC and 1H NMR spectroscopy. However, attempts at further purification on alumina [ethyl acetate/MeOH (10:1)] caused formation of a second indium(III)chlorin in almost equal amount. The two chlorins were observed by TLC and 1H NMR spectroscopy but could not be separated. In this regard, it is noteworthy that Pandey's group observed the formation of two indium-chelated species upon metalation of a free base chlorin related to methyl pyropheophorbide.91 Thus, an indium-chlorin of unambiguous identity was not obtained.
Attempts to form chlorin directly from 8b and 5 by refluxing a mixture of both halves in EtOH in the presence of KOH and an appropriate metal salt (thereby avoiding p-TsOH-catalyzed condensation) resulted in a mixture of chlorin and porphyrin products (the latter formed by self-condensation of the Eastern half), or no macrocycle formation. We previously employed imine derivatives of 1,9-formyldipyrromethanes for condensation with zinc acetate (and no other acid) to give the trans-AB-porphyrin.92 Condensation using the N-propyliminomethyl analogue of 8b (PrN-8b) and Western half 5 (100 mM each) in ethanol afforded chlorin ZnC-P10, albeit in only 3.3% yield. Thus, the synthesis of chlorins requires the acid-catalyzed condensation of Eastern and Western halves.
In summary, following the acid-catalyzed condensation, various metallochlorins [Cu(II), Zn(II), Pd(II), and putative ClIn(III)] can be prepared via a AgOTf-free metal-mediated oxidative cyclization process. The indium chelate proved difficult to purify to homogeneity. Tetrapyrrolic complexes of Pd(II) and In(III) are potentially useful in the life sciences for photodynamic therapy, and 111In chelates may be used for photosensitization, imaging, and/or radiotherapy applications. Although palladium and indium chelates of porphyrins are well known, there are only a few reports about Pd(II)93 and In(III)91,94 complexes of chlorins, hence this straightforward route to metallochlorins warrants further investigation.
(4) Route III
Route III employs the same Western half as in routes I and II, but uses a 1-bromo-9-formyldipyrrin as an Eastern half rather than a 1-bromo-9-formyldipyrromethane. This route was considered to be attractive owing to the greater stability of dipyrrins versus dipyrromethanes. The condensation should afford a 2,3,4,5-tetrahydrobilatriene intermediate, which is more oxidized than the 2,3,4,5-tetrahydrobiladiene formed in route II, and thereby require a lesser quantity of oxidant to yield the chlorin.
Dipyrromethane 8b was treated with DDQ at room temperature in CHCl3,95 affording 1-bromo-9-formyldipyrrin 10b in 50% yield (Scheme 7). Dipyrrin 10b was subjected to p-TsOH-catalyzed condensation followed by metal-mediated oxidative cyclization. Absorption spectroscopy of the crude reaction mixture showed only a small amount of chlorin (<1%). The origin of the low yield appears to stem from the low reactivity of the formyl group in the dipyrrin moiety, because 10b remained unreacted following attempted acid-catalyzed condensation.
Scheme 7.
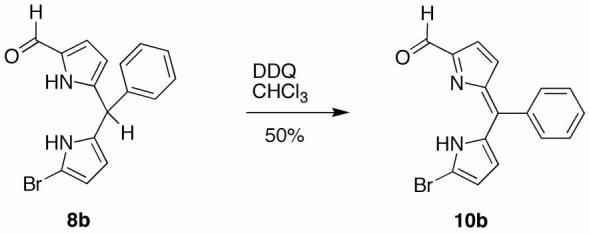
Route III
(5) Route IV
Route IV employs the 9-formyltetrahydrodipyrrin (Western half) and a 1-bromodipyrromethane (Eastern half). 9-Formyltetrahydrodipyrrin 11 was obtained by standard Vilsmeier formylation of 5.96 The reaction of 1c with NBS in THF at −78 °C afforded a mixture of the 1-bromodipyrromethane and 1,9-dibromodipyrromethane together with unreacted starting material. Column chromatography of the crude mixture afforded the known 1-bromodipyrromethane 12c75 in 35% yield. 1-Bromodipyrromethanes such as 12c (as well as 12a and 12b, derived from 1a and 1b) are prone to decomposition during purification and routine handling.
The condensation of 11 and 1-bromodipyrromethane 12c was carried out in CH2Cl2 in the presence of 5 equiv of p-TsOH·H2O. Absorption spectroscopy of the crude reaction mixture showed a strong band at λ = 478 nm, attributed to the protonated tetrahydrobiladiene. After quenching with NaHCO3, the crude biladiene was subjected to metal-mediated oxidative cyclization under standard conditions, affording the chlorin ZnC-M10 in ∼8% yield (Scheme 8). Attempts to use 1-cyano-5-phenyldipyrromethane87 or 1-carboethoxy-5-phenyldipyrromethane87 in place of 12c did not give chlorin. The statistical bromination of the dipyrromethane and the apparent instability of the resulting 1-bromodipyrromethane limited the utility of this route. Accordingly, no further studies of this route were performed.
Scheme 8.
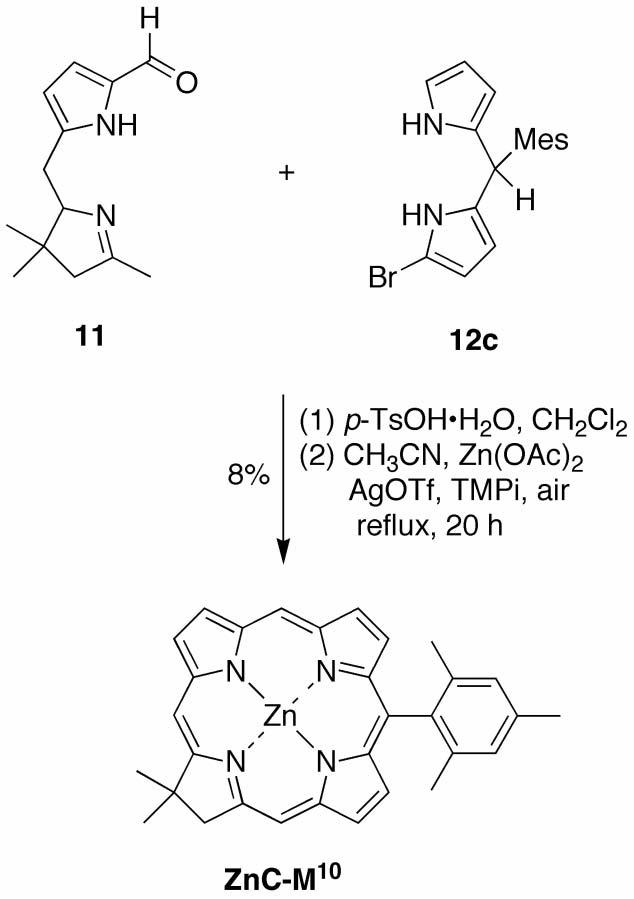
Route IV
(6) Route V
Route V avoids any functionalization of the starting dipyrromethane (Eastern half) by condensation of the dipyrromethane with a 9-formyltetrahydrodipyrrin (Western half). Subsequent 19-bromination of the resulting biladiene sets the stage for standard metal-mediated oxidative cyclization.
Treatment of a mixture of 11 and 1a with 5 equiv of p-TsOH·H2O in CH2Cl2/MeOH afforded the corresponding tetrahydrobiladiene 9b (Scheme 9). (The tetrahydrobiladiene was formed in 21% yield if the peak molar absorption coefficient is 100,000 M−1cm−1.) Treatment of crude 9b with NBS in THF at −78 °C afforded the putative 19-bromotetrahydrobiladiene 9b-Br. The bromination proved difficult to verify: LD-MS did not show a peak corresponding to the expected 9b-Br, and absorption spectroscopy did not show any changes compared with that of 9b. Regardless, the crude reaction mixture after bromination was subjected to metal-mediated oxidative cyclization for 24 h, affording chlorin in ∼8% yield. The 1H NMR and LD-MS analyses revealed the presence of various monobromochlorins in the purified sample. Separation of those species proved difficult, which makes this route completely unattractive.
Scheme 9.
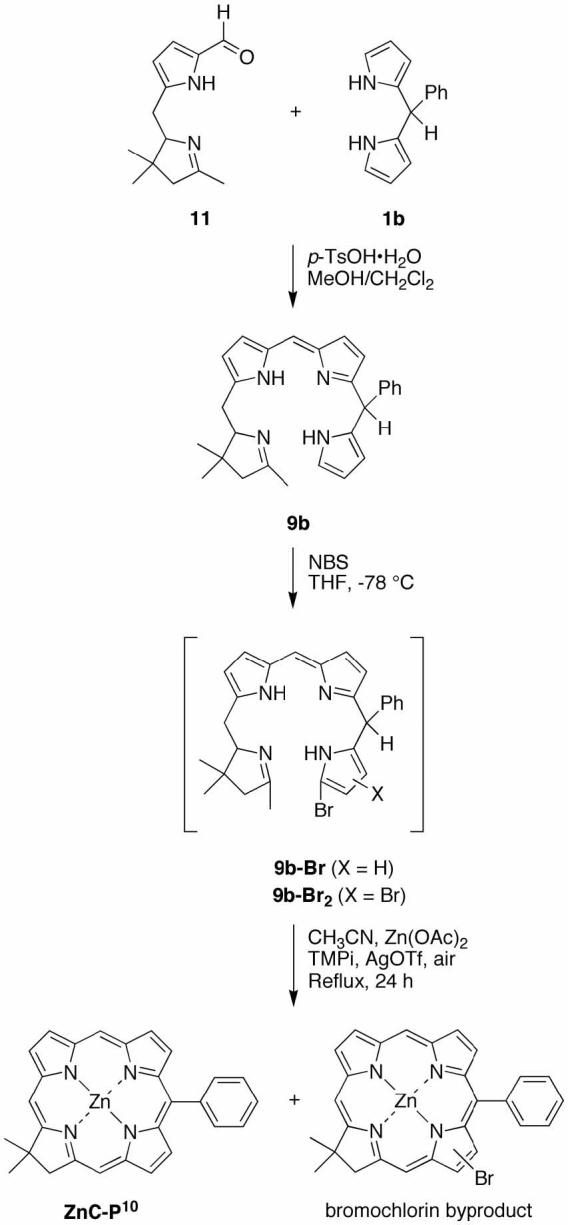
Route V
(7) Comparison of routes
Of the routes investigated, route II and its variants appear most useful. Indeed, the streamlined route II was employed to prepare 145 mg of the benchmark chlorin ZnC lacking any meso- or β-pyrrole substituents. In addition, the AgOTf-free version of route II provides direct access to metallochlorins containing Cu(II), Zn(II), Pd(II), and ClIn(III).
3. Outlook
Three distinct methods of chlorin synthesis include modification of naturally occurring chlorins, reduction/derivatization of porphyrins, and de novo synthesis. Of these, de novo synthesis affords the most versatility. The reaction of 1-formyl-9-bromo-dipyrromethane and 2,3,4,5-tetrahydro-1,3,3-trimethyldipyrrin affords the corresponding chlorin bearing only a geminal dimethyl group at the 18-position. The reaction can be carried out with several variations: (i) in a three-step process (9-bromination of the 1-formyldipyrromethane, acid-catalyzed condensation to form the tetrahydrobiladiene-ab, and metal-mediated oxidative cyclization), (ii) in a streamlined manner where the 1-formyl-9-bromo-dipyrromethane is used in crude form, and/or (iii) with a AgOTf-free metal-mediated oxidative cyclization process. The AgOTf-free route provides direct access to a selection of metallochlorins. Similar reaction enables synthesis of a chlorin bearing one additional substituent at the 10-position. In conjunction with other synthetic methods, simple routes are now available for preparing stable chlorins that bear no substituents, a single substituent at the 5- or 10-position, or two or three distinct meso substituents. All of the synthetic chlorins prepared herein were stable upon routine handling in aerobic environments, including use of procedures such as chromatography, recrystallization, and standing in solution on the open benchtop. The ready synthesis and stability of the chlorins enabled the studies of derivatization chemistry and spectroscopic properties described in the companion papers.81,82
4. Experimental section
4.1. General methods
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were collected at room temperature in CDCl3 unless noted otherwise. NMR spectroscopy was not performed on Cu(II) chelates of the chlorins. Chlorins were analyzed by laser desorption mass spectrometry without a matrix (LD-MS).76,97 Fast atom bombardment mass spectrometry (FAB-MS) data are reported for the molecule ion or protonated molecule ion. Melting points are uncorrected. NBS was recrystallized (H2O). Chromatography was performed with flash silica (80-200 mesh). Absorption and fluorescence spectra were obtained in toluene at room temperature.
4.2. Solvents
THF was distilled over sodium metal and benzophenone as required. Methanol, CH3CN, and CH2Cl2 were used as anhydrous grade. Toluene used in absorption and fluorescence studies was spectroscopic grade. All other solvents were used as received.
4.3. Noncommercial compounds
Compounds 1a,842-B,76 2-T,85 5,76,98 7a–c,87 and 1196 were prepared as described in the literature. Compound 12c was prepared as described in the Supporting Information of reference 75.
4.4. Synthesis of 5-substituted chlorins
4.4.1. 1-(3,5-Di-tert-butylbenzoyl)dipyrromethane (3-B)
Following a general procedure,83 EtMgBr (16.4 mL, 16 mmol, 1.0 M in THF) was added to a solution of dipyrromethane 1a (1.00 g, 6.48 mmol) in dry THF (10 mL) at room temperature under argon. The mixture was stirred at room temperature for 10 min and then cooled to −78 °C. A solution of S-2-pyridyl 3,5-di-tert-butylbenzothioate (2-B) (2.24 g, 6.84 mmol) in dry THF (10 mL) was added. The reaction mixture was maintained at −78 °C for 30 min, whereupon the cooling bath was removed. After 1 h, the reaction was quenched by addition of 100 mL of saturated aqueous NH4Cl. The reaction mixture was extracted with CH2Cl2. The organic extract was washed with water, dried (Na2SO4), and concentrated under reduced pressure. Column chromatography [silica; packed and eluted with hexanes/CH2Cl2 (1:2), then eluted with hexanes/ethyl acetate (5:1)] afforded a white powder (1.76 g, 71%): mp 178–180 °C; 1H NMR δ 1.34 (s, 18H), 4.09 (s, 2H), 6.04–6.08 (m, 1H), 6.09–6.13 (m, 1H), 6.14–6.17 (m, 1H), 6.55–6.59 (m, 1H), 6.78–6.82 (m, 1H), 7.61 (t, J = 1.8 Hz, 1H), 7.70 (d, J = 1.8 Hz, 2H), 8.76–8.96 (br s, 1H), 10.32–10.56 (br s, 1H); 13C NMR δ 27.0, 31.6, 35.2, 106.6, 108.4, 110.3, 117.9, 122.5, 123.7, 126.3, 127.9, 131.0, 138.0, 140.5, 151.2, 186.4; FAB-MS obsd 363.2435, calcd 363.2436 [(M + H)+, M = C24H30N2O].
4.4.2. 1-(4-Methylbenzoyl)dipyrromethane (3-T)
Following the procedure described for 3-B, EtMgBr (24.0 mL, 24 mmol, 1.0 M in THF) was added to a solution of dipyrromethane 1a (1.46 g, 10.0 mmol) in dry THF (10 mL) at room temperature under argon. The mixture was stirred at room temperature for 10 min and then cooled to −78 °C. A solution of S-2-pyridyl 4-methylbenzothioate (2-T) (2.29 g, 10.0 mmol) in dry THF (10 mL) was added. The reaction mixture was maintained at −78 °C for 30 min, whereupon the cooling bath was removed. After 1 h, the standard workup afforded a white powder (1.03 g, 39%): mp 167–169 °C; 1H NMR δ 2.43 (s, 3H), 4.08 (s, 2H), 6.03–6.06 (m, 1H), 6.08–6.12 (m, 1H), 6.14–6.17 (m, 1H), 6.53–6.56 (m, 1H), 6.80–6.83 (m, 1H), 9.04–9.20 (br, 1H), 10.72–10.88 (br, 1H); 13C NMR δ 21.8, 27.0, 106.5, 108.4, 110.3, 117.8, 122.9, 128.2, 129.37, 129.39, 130.8, 136.0, 140.9, 142.8, 185.5; FAB-MS obsd 265.1338, calcd 265.1341 [(M + H)+, M = C17H16N2O].
4.4.3. 1-Bromo-9-(3,5-di-tert-butylbenzoyl)dipyrromethane (4-B)
Following a general procedure,75 a solution of 3-B (768 mg, 2.12 mmol) in dry THF (15 mL) was cooled to −78 °C under argon. NBS (377 mg, 2.12 mmol) was added, and the reaction mixture was stirred for 1 h at −78 °C. Hexanes (100 mL) and water (100 mL) were added. The mixture was allowed to warm to room temperature. The organic layer was separated. The aqueous phase was extracted with CH2Cl2. The hexanes and CH2Cl2 extracts were combined, dried (NaHCO3) and concentrated in vacuo without heating. Column chromatography [silica packed and eluted with hexanes/CH2Cl2 (1:1), then eluted with hexanes/ethyl acetate (7:1)] afforded a light brown powder (493 mg, 53%): mp 120 °C (dec.); 1H NMR δ 1.31 (s, 18H), 4.08 (s, 2H), 5.92–5.94 (m, 1H), 5.95–5.97 (m, 1H), 6.18–6.20 (m, 1H), 6.86–6.88 (m, 1H), 7.61–7.64 (m, 1H), 7.70–7.73 (m, 2H), 9.66–9.76 (br s, 1H), 11.37–11.46 (br s, 1H); 13C NMR (dec. in CDCl3); FAB-MS obsd 441.1541, calcd 441.1541 [(M + H)+, M = C24H29BrN2O].
4.4.4. 19-Bromo-10-(3,5-di-tert-butylphenyl)-2,3,4,5-tetrahydro-1,3,3-trimethylbilene-a (6-B)
Following the procedure for preparing a tetrahydrobilene-a,76 a sample of 4-B (441 mg, 1.00 mmol) was reduced with NaBH4 (378 mg, 10.0 mmol) in anhydrous THF/methanol (20 mL, 4:1). The resulting solid was dissolved in 10 mL of anhydrous CH3CN, to which 5 (190 mg, 1.00 mmol) and TFA (77 μL, 1.0 mmol) were added. The reaction mixture was stirred at room temperature for 30 min. Then 10% aqueous NaHCO3 (50 mL) was added, and the mixture was extracted with distilled CH2Cl2. The organic extract was washed with water, dried (NaHCO3) and concentrated in vacuo without heating. The resulting brown solid was purified by chromatography [silica, hexanes/ethyl acetate (5:1) → ethyl acetate] to give a brown solid (325 mg, 53%; mixture of stereoisomers): mp 107–110 °C; 1H NMR δ 0.91 (s, 3H), 1.06 (s, 3H), 1.26 (s, 18H), 1.86–1.90 (m, 3H), 2.18–2.34 (m, 2H), 2.56 (ABX, 3J = 10.8 Hz, 2J = 14.8 Hz, 1H), 2.71 (ABX, 3J = 3.2 Hz, 2J = 14.8 Hz, 1H), 3.60–3.64 (m, 1H), 3.85 (s, 2H), 5.27–5.31 (m, 1H), 5.70–5.99 (m, 6H), 7.05 (s, 2H), 7.26 (s, 1H), 7.81–7.90 (m, 1H), 8.21–8.39 (m, 1H), 8.97–9.10 (m, 1H); FAB-MS obsd 615.3062, calcd 615.3062 (C36H47BrN4).
4.4.5. Zn(II)-5-(3,5-Di-tert-butylphenyl)-17,18-dihydro-18,18-dimethylporphyrin (ZnC-B5)
Following the procedure for preparing chlorins,76 a solution of 6-B (283 mg, 0.460 mmol) in CH3CN (46 mL) was treated with Zn(OAc)2 (1.27 g, 6.90 mmol), AgOTf (355 mg, 1.38 mmol) and 2,2,6,6-tetramethylpiperidine (1.16 mL, 6.90 mmol). The reaction mixture was refluxed for 24 h. Standard workup and chromatography [silica, hexanes/CH2Cl2 (3:1)] gave a purple solid (45 mg, 17%): 1H NMR δ 1.51 (s, 18H), 2.03 (s, 6H), 4.55 (s, 2H), 7.73–7.75 (m, 1H), 7.93–7.96 (m, 2H), 8.58–8.60 (m, 1H), 8.62 (s, 1H), 8.68–8.71 (m, 2H), 8.73–8.75 (m, 1H), 8.76–8.78 (m, 1H), 8.86–8.88 (m, 1H), 9.08–9.10 (m, 1H), 9.62 (s, 1H); LD-MS obsd 589.25; FAB-MS obsd 590.2380, calcd 590.2388 (C36H38N4Zn); λabs 405 (log ε = 5.50), 605 (4.89) nm; λem 609 nm.
4.4.6. Zn(II)-17,18-Dihydro-18,18-dimethyl-5-(4-methylphenyl)porphyrin (ZnC-T5)
Following the general procedure for preparing chlorins,75 a solution of 3-T (264 mg, 1.00 mmol) in 10 mL of dry THF was cooled to −78 °C under argon. NBS (178 mg, 1.00 mmol) was added, and the reaction mixture was stirred for 1 h at −78 °C. Hexanes (50 mL) and water (50 mL) were added, and the mixture was allowed to warm to room temperature. The organic layer was separated. The aqueous phase was extracted with CH2Cl2. The hexanes and CH2Cl2 extracts were combined, dried (NaHCO3) and concentrated in vacuo without heating, affording crude 4-T (367 mg, quantitative): 1H NMR δ 2.43 (s, 3H), 4.08 (s, 2H), 6.03–6.06 (m, 1H), 6.08–6.12 (m, 1H), 6.14–6.17 (m, 1H), 6.53–6.56 (m, 1H), 6.80–6.83 (m, 1H), 9.04–9.20 (br, 1H), 10.72–10.88 (br, 1H). Following the procedure for preparing a tetrahydrobilene-a,76 a sample of crude 4-T (343 mg, 1.00 mmol) was reduced with NaBH4 (378 mg, 10.0 mmol) in anhydrous THF/methanol (10 mL, 4:1). The resulting solid was dissolved in anhydrous CH3CN (10 mL), to which 5 (190 mg, 1.00 mmol) and TFA (77 μL, 1.0 mmol) were added. The reaction mixture was stirred at room temperature for 30 min. Then 10% aqueous NaHCO3 (50 mL) was added. The mixture was extracted with distilled CH2Cl2. The organic extract was washed with water, dried (NaHCO3) and concentrated in vacuo without heating. The resulting brown solid was purified by chromatography [silica, hexanes/ethyl acetate (5:1) → ethyl acetate] to give 6-T as a brown solid (110 mg, 21%). Following the procedure for preparing chlorins,76 a solution of 6-T (59.3 mg, 0.115 mmol) in CH3CN (11 mL) was treated with Zn(OAc)2 (317 mg, 1.73 mmol), AgOTf (88.6 mg, 0.345 mmol) and 2,2,6,6-tetramethylpiperidine (0.29 mL, 1.7 mmol). The reaction mixture was refluxed exposed to air for 24 h. Standard workup and chromatography [silica, hexanes/CH2Cl2 (3:1)] gave a purple solid (19.5 mg, 34%): 1H NMR δ 2.02 (s, 6H), 2.68 (s, 3H), 4.50 (s, 2H), 7.49–7.52 (m, 2H), 7.95–8.00 (m, 2H), 8.61 (s, 1H), 8.62 (s, 1H), 8.64–8.66 (m, 1H), 8.67–8.70 (m, 1H), 8.72–8.75 (m, 1H), 8.82–8.85 (m, 1H), 8.99–9.02 (m, 1H), 9.54 (s, 1H); 13C NMR δ 21.8, 31.2, 45.3, 50.5, 94.9, 96.3, 109.1, 124.4, 126.8, 127.1, 127.6, 128.1, 129.4, 132.8, 133.4, 133.9, 137.2, 139.7, 146.00, 146.01, 146.7, 147.4, 153.4, 153.7, 159.7, 170.6; LD-MS obsd 491.1; FAB-MS obsd 492.1295, calcd 492.1292 (C29H24N4Zn); λabs 405, 605 nm; λem 608 nm.
4.5. Precursors for examination of routes I-V
4.5.1. 1-Bromo-9-formyl-5-phenyldipyrromethane (8b)
Following the procedure for the preparation of 8c,77 bromination of 7b (150 mg, 0.600 mmol) with NBS gave a crude solid that upon recrystallization (EtOH/H2O, 4:1) afforded a light brown solid (108 mg, 55%): mp 133 °C (dec.); 1H NMR (THF-d8) δ 5.42 (s, 1H), 5.59–5.61 (m, 1H), 5.86–5.88 (m, 1H), 5.92–5.94 (m, 1H), 6.79–6.81 (m, 1H), 7.16–7.30 (m, 5H), 9.40 (s, 1H), 10.51 (br s, 1H) 11.21 (br s, 1H); 13C NMR δ 44.3, 97.2, 109.3, 109.5, 110.3, 120.4, 126.9, 128.3, 128.5, 133.5, 133.8, 141.7, 142.2, 177.9. Anal Calcd for C16H13BrN2O: C, 58.38; H, 3.98; N, 8.51. Found: C, 58.31; H, 3.92; N, 8.45.
4.5.2. 1-Bromo-9-formyl-5-phenyldipyrrin (10b)
A solution of 8b (0.033 g, 1.0 mmol) in CHCl3 (2 mL) was treated with a suspension of DDQ (0.027 g, 1.0 mmol) in CHCl3 (1 mL). The mixture was stirred at room temperature for 1 h. The mixture was concentrated. The residue was chromatographed (silica, CH2Cl2) to afford an orange solid (0.016 g, 50%): mp 121–123 °C; 1H NMR δ 6.35 (d, J = 4.4 Hz, 1H), 6.20 (d, J = 4.4 Hz, 1H), 6.75 (d, J = 4.4 Hz, 1H), 6.88 (d, J = 4.4 Hz, 1H), 7.42–7.52 (m, 5H), 9.72 (s, 1H); 13C NMR δ 119.7, 121.7, 128.3, 129.7, 129.8, 130.9, 137.6, 138.9, 150.6, 152.2, 180.3; FAB-MS obsd 327.0129, calcd 327.0133 [(M + H)+, M = C16H11N2OBr]; λabs 437 nm.
4.5.3. 2,3,4,5-Tetrahydro-9-(N-propyliminomethyl)-1,3,3-trimethyldipyrrin (PrN-8b)
Following a general procedure,92 a sample of 11 (0.044 g, 0.20 mmol) was dissolved in 1-aminopropane (1 mL) at room temperature. After stirring for 1 h, the 1-aminopropane was removed by distillation under reduced pressure. The crude product was used in the next step without further purification: 1H NMR δ 0.92 (s, 3H), 0.94 (t, J = 7.4 Hz, 3H), 1.09 (s, 3H), 2.04 (m, 3H), 1.61–1.70 (m, 2H), 2.27, 2.36 (AB, J = 16.6 Hz, 2H), 2.66 (ABX, 2J = 14.8 Hz, 3J = 10.6 Hz, 1H), 2.75 (ABX, 2J = 14.8 Hz, 3J = 4.0 Hz, 1H), 3.39–3.51 (m, 2H), 3.64–3.69 (m, 1H), 5.99 (d, J = 3.8 Hz, 1H), 6.35 (d, J = 3.8 Hz, 1H), 7.96 (s, 1H).
4.6. Route I
4.6.1. Attempted synthesis of ZnC-P10 using TFA
A solution of 7b (32 mg, 0.10 mmol) in THF/MeOH (40 mL, 3:1) was treated with NaBH4 (0.19 g, 5.0 mmol) and stirred at room temperature for 30 min. TLC [silica, CH2Cl2/ethyl acetate (5:1)] showed complete consumption of starting material. The reaction was quenched by addition of saturated aqueous NH4Cl (∼50 mL). The resulting mixture was extracted with ether. The organic extract was washed (water and brine) and dried (MgSO4). Acetonitrile (1 mL) was added, and the volatile solvent (predominantly ether) was evaporated under low vacuum. (Note that complete evaporation of solvent caused complete decomposition of the carbinol.) A sample of 5 (19 mg, 0.10 mmol) was added, and the resulting solution was treated with TFA (8 μL, 0.1 mmol). The reaction mixture was stirred at room temperature for 30 min and then diluted with CH3CN (9 mL). Samples of 2,2,6,6-tetramethylpiperidine (0.506 mL, 3.00 mmol), anhydrous Zn(OAc)2 (0.276 g, 1.5 mmol) and AgOTf (0.077 g, 0.3 mmol) were added. The resulting mixture was refluxed exposed to air for 20 h. The yield observed spectroscopically was <1%. The mixture was concentrated and filtered through silica (CH2Cl2) to afford a green solid. Characterization data (LD-MS, UV-vis) were identical as described for ZnC-P10.
4.6.2. Attempted synthesis of ZnC using BF3·O(Et)2
A solution of 5 (38.0 mg, 0.198 mmol) and 8a (50.0 mg, 0.198 mmol) in dry CH3CN (1.98 mL) was treated with BF3·O(Et)2 (25.0 μL, 0.198 mmol) and stirred for 15 min at room temperature. No reaction occurred upon examination by TLC, whereupon additional BF3·O(Et)2 (50.0 μL) was added. The mixture was stirred for 20 min, quenched with water and extracted with CH2Cl2. The solvent was removed under reduced pressure to give a brown oil. TLC analysis and 1H NMR spectroscopy of the crude mixture revealed only starting material 8a.
4.7. Route II
4.7.1. Condensation study
A solution of 5 (10.0 mg, 39.5 μmol) and 8a (7.50 mg, 39.5 μmol) in CH2Cl2 under argon was treated with p-TsOH·H2O (37.6 mg, 0.198 mmol) in MeOH. The quantity of each of 5, 8a, and p-TsOH·H2O was identical in each experiment, while the quantity of solvent was varied from 5.7 mL (6.92 mM) to 0.77 mL (51.3 mM). In each case, the ratio of CH2Cl2/methanol was held constant at 4:1. For example, samples of 5 and 8a were dissolved in CH2Cl2 (2.34 mL) and subsequently treated with p-TsOH·H2O in MeOH (0.59 mL) to give the desired concentration of 13.5 mM. The mixture was stirred for 30 min under argon and quenched by addition of 10% aqueous NaHCO3 (10 mL). After extracting the mixture with CH2Cl2, the extract was dried (Na2SO4) and filtered. The filtrate was concentrated to give a brown solid. The crude solid was dissolved in CH3CN (4 mL, 10 mM assuming quantitative formation) and treated with Zn(OAc)2 (109 mg, 0.593 mmol), AgOTf (30.4 mg, 0.119 mmol), and 2,2,6,6-tetramethylpiperidine (0.101 mL, 0.593 mmol). The reaction mixture was heated at 70 °C for 19 h exposed to air. The mixture was concentrated. The resulting black residue was chromatographed [silica, CH2Cl2], affording a bluish-green solid. The isolated chlorin yield was then recorded after drying under vacuum, affording 1.8 mg (11%) of ZnC.
A study of the concentration dependence of the condensation conditions where 5 mol equiv of p-TsOH·H2O was employed led to the following yields of ZnC: 10% (7 mM), 11% (14 mM), 22% (26 mM), and 22% (51 mM).
4.7.2. Route II: Zn(II)-17,18-Dihydro-18,18-dimethyl-10-phenylporphyrin (ZnC-P10)
A mixture of 8b (0.165 g, 0.500 mmol) and 5 (0.095 g, 0.50 mmol) in dry CH2Cl2 (10.8 mL) was treated with a solution of p-TsOH·H2O (0.475 g, 2.50 mmol) in anhydrous MeOH (8.25 mL). The resulting red solution was stirred at room temperature for 30 min. A sample of 2,2,6,6-tetramethylpiperidine (0.630 mL, 3.71 mmol) was added. The mixture was concentrated. The resulting yellow solid was dissolved in CH3CN (50 mL). Samples of 2,2,6,6-tetramethylpiperidine (2.10 mL, 12.4 mmol), anhydrous Zn(OAc)2 (1.37 g, 7.50 mmol) and AgOTf (0.385 g, 1.50 mmol) were added. The mixture was refluxed in the presence of air for 20 h. The mixture was concentrated. The resulting black residue was chromatographed [silica, CH2Cl2/hexanes (1:1)] to afford a violet-green solid (79 mg, 33%): 1H NMR δ 2.04 (s, 6H), 4.53 (s, 2H), 7.67–7.72 (m, 3H), 8.08–8.10 (m, 2H), 8.52 (d, J = 4.5 Hz, 1H), 8.62–8.64 (m, 2H), 8.69–8.70 (m, 2H), 8.78 (d, J = 4.1 Hz, 1H), 8.86 (d, J = 4.1 Hz, 1H), 9.09 (d, J = 4.1 Hz, 1H), 9.62 (s, 1H); 13C NMR δ 31.0, 45.4, 50.4, 94.3, 97.0, 109.4, 123.8, 126.79, 126.84, 127.3, 127.6, 128.2, 129.2, 132.9, 133.3, 133.9, 142.6, 145.86, 145.97, 146.5, 147.4, 153.1, 154.0, 159.2, 171.0; LD-MS obsd 477.8; ESI-MS obsd 478.1154, calcd 478.1136 (C28H22N4Zn); λabs 405 (log ε = 5.44), 605 (4.83) nm; λem 608 nm.
4.7.3. Streamlined route II (ZnC)
A solution of 7a (344 mg, 2.00 mmol) in THF (40 mL) was treated with NBS (356 mg, 2.00 mmol) at −78 °C. After 1 h the mixture was allowed to warm to −20 °C. A mixture of water (30 mL) and hexanes (30 mL) was added. Excess CH2Cl2 was added. The organic layer was separated, dried (Na2SO4) and concentrated. The resulting solid was dissolved in dry CH2Cl2 (43 mL), to which a sample of 5 (0.360 g, 2.00 mmol) and a solution of p-TsOH·H2O (1.92 g, 10.0 mmol) in MeOH (33 mL) were added. The red mixture was stirred for 30 min. A sample of 2,2,6,6-tetramethylpiperidine (2.52 mL, 14.8 mmol) was added. The mixture was concentrated. The residue was dissolved in CH3CN (200 mL). Anhydrous Zn(OAc)2 (5.52 g, 30.0 mmol), AgOTf (1.55 g, 6.00 mmol) and 2,2,6,6-tetramethylpiperidine (8.40 mL, 49.5 mmol) were added. The mixture was refluxed exposed to air until the absorption spectrum showed no further change (typically 5-10 h). The mixture was concentrated. The resulting black mixture was chromatographed [silica, hexanes/CH2Cl2 (1:1)] to afford a blue-green solid (0.145 g, 18%). The characterization data 1(H NMR, 13C NMR, LD-MS, FAB-MS, UV-Vis) were consistent with those reported previously.77
4.7.4. AgOTf-free Route II: Pd(II)-17,18-Dihydro-18,18-dimethylporphyrin (PdC)
Following the AgOTf-free route II, samples of 5 (95 mg, 0.50 mmol) and 8a (126 mg, 0.500 mmol) were dissolved in CH2Cl2 (15 mL). A solution of p-TsOH·H2O (475.0 mg, 2.50 mmol) in anhydrous MeOH (5 mL) was added. The resulting mixture was stirred for 30 min and then treated with 2,2,6,6-tetramethylpiperidine (2.50 mL). The mixture was concentrated. The resulting yellow solid was dissolved in CH3CN (50 mL). Samples of 2,2,6,6-tetramethylpiperidine (2.54 mL, 15.0 mmol) and Pd(OAc)2 (0.338 g, 1.50 mmol) were added. The resulting mixture was refluxed exposed to air for 1 h. The resulting dark-green mixture was concentrated and chromatographed [silica, hexanes/CH2Cl2 (1:1)] to afford a violet-green solid (7.5 mg, 3%): H NMR δ 2.02 (s, 6H), 4.62 (s, 2H), 8.98 (d, J = 4.4 Hz, 1H), 8.73 (s, 1H), 8.74 (d, J = 4.4 Hz, 1H), 8.81 (s, 1H), 8.95–8.96 (m, 2H), 8.98 (d, J = 4.4 Hz, 1H), 8.99 (d, J = 4.4 Hz, 1H), 9.72 (s, 1H), 9.74 (s, 1H); LD-MS obsd 443.7; FAB-MS obsd 444.0557, calcd 444.0566 (C22H18N4Pd); λabs 388, 585 nm.
4.7.5. Cu(II)-17,18-Dihydro-18,18-dimethylporphyrin (CuC)
Following the AgOTf-free route II, samples of 5 (19.0 mg, 100 μmol) and 8a (25.3 mg, 100 μmol) were dissolved in CH2Cl2 (3 mL). A solution of p-TsOH·H2O (95.0 mg, 500 μmol) in anhydrous MeOH (1 mL) was added. The resulting mixture was stirred for 30 min and then neutralized with 2,2,6,6-tetramethylpiperidine (500 μL). The mixture was concentrated. The resulting yellow solid was dissolved in EtOH (4 mL). Samples of Cu(OAc)2 (39.2 mg, 200 μmol) and KOH (42.0 mg, 750 μmol) were added. The resulting mixture was refluxed exposed to air for 21 h. The resulting mixture was concentrated and chromatographed (silica/CH2Cl2) to afford a green solid (2.2 mg, 5%): LD-MS obsd 400.6; FAB-MS obsd 401.0837, calcd 401.0827 (C22H18N4Cu); λabs 396, 598 nm.
4.7.6. Pd(II)-17,18-Dihydro-18,18-dimethyl-10-phenylporphyrin (PdC-P10)
Samples of 5 (19.0 mg, 100 μmol) and 8b (32.6 mg, 100 μmol) were dissolved in CH2Cl2 (3 mL). A solution of p-TsOH·H2O (95.0 mg, 500 μmol) in anhydrous MeOH (1 mL) was added. The resulting mixture was stirred for 30 min and then neutralized with 2,2,6,6-tetramethylpiperidine (500 μL). The mixture was concentrated. The resulting yellow solid was dissolved in CH3CN (10 mL). Samples of 2,2,6,6-tetramethylpiperidine (0.509 mL, 3.00 mmol) and Pd(OAc)2 (0.338 g, 1.50 mmol) were added. The resulting mixture was refluxed exposed to air for 1 h. The resulting dark-green mixture was concentrated and chromatographed (silica, CH2Cl2) to afford a violet solid (6.0 mg, 12%): 1H NMR δ 2.02 (s, 6H), 4.60 (s, 2H), 7.69–7.72 (m, 3H), 8.04–8.07 (m, 2H), 8.53 (d, J = 4.8 Hz, 1H), 8.57–8.58 (m, 2H), 8.69 (s, 1H), 8.72 d, J = 4.8 Hz, 1H), 8.79 (s, 1H), 8.84 d, J = 4.8 Hz, 1H), 8.96 d, J = 4.8 Hz, 1H), 9.69 (s, 1H); LD-MS obsd 519.9; FAB-MS obsd 520.0901, calcd 520.0879 (C28H22N4Pd); λabs 394, 587 nm.
4.7.7. Cu(II)-17,18-Dihydro-18,18-dimethyl-10-phenylporphyrin (CuC-P10)
Following the AgOTf-free route II, samples of 5 (19.0 mg, 100 μmol) and 8b (32.9 mg, 100 μmol) were dissolved in CH2Cl2 (3 mL). A solution of p-TsOH·H2O (95.0 mg, 500 μmol) in anhydrous MeOH (1 mL) was added. The resulting mixture was stirred for 30 min and then neutralized with 2,2,6,6-tetramethylpiperidine (500 μL). The mixture was concentrated. The resulting yellow solid was dissolved in EtOH (5 mL). Samples of KOH (42 mg, 0.75 mmol) and Cu(OAc)2 (37.2 mg, 0.200 mmol) were added. The resulting mixture was refluxed exposed to air for 21 h. The resulting dark-green mixture was concentrated and chromatographed (silica, CH2Cl2) to afford a green solid (7.3 mg, 15%): LD-MS 476.9; FAB-MS obsd 477.1153, calcd 477.1140 (C28H22N4Cu); λabs 394, 587 nm.
4.8. Routes III and IV
4.8.1. General procedure for attempted chlorin formation
Samples of an Eastern half (0.10 mmol) and a Western half (0.10 mmol) were dissolved in anhydrous CH2Cl2 (4 mL) and treated with p-TsOH·H2O (0.095 g, 0.50 mmol). The resulting mixture was stirred for 15 min at room temperature. The reaction was quenched by addition of 10% aqueous NaHCO3. The resulting mixture was extracted with CH2Cl2. The organic phase was washed with water and brine, dried (Na2SO4), and concentrated. The residue was dissolved in CH3CN (10 mL) and treated with anhydrous Zn(OAc)2 (0.276 g, 1.50 mmol), AgOTf (0.077 g, 0.30 mmol) and 2,2,6,6-tetramethylpiperidine (0.504 mL, 3.00 mmol). The reaction mixture was refluxed for 20 h exposed to air, during which time the spectroscopic yield of chlorin was checked. This procedure was applied for the reaction of 12c + 11 to give ZnC-M10 (8% yield), and 10b + 5 to give ZnC-P10 (<1% yield).
4.9. Route V
4.9.1. Zn(II)-17,18-Dihydro-18,18-dimethyl-10-phenylporphyrin (ZnC-P10)
A solution of 1b (0.111 g, 0.500 mmol) and 11 (0.109 g, 0.500 mmol) in CH2Cl2 (15 mL) was treated with a solution of p-TsOH·H2O (0.478 g, 2.5 mmol) in MeOH (5 mL). The resulting mixture was stirred at room temperature for 30 min. The reaction mixture was neutralized with excess triethylamine, washed with water and brine, dried (Na2SO4), and concentrated to afford crude 9b as an orange solid. Crude 9b was dissolved in dry THF (5 mL) and treated with NBS (0.089 g, 0.50 mmol) at −78 °C. The reaction mixture was stirred at −78 °C for 1 h, allowed to warm to ∼ −20 °C, and then quenched by addition of a mixture of hexanes (5 mL) and water (5 mL). The resulting mixture was extracted with CH2Cl2. The organic extract was washed with water and brine, dried (Na2SO4) and concentrated to afford the crude 19-bromotetrahydrobiladiene (9b-Br) as a brown solid. The crude 9b-Br was dissolved in CH3CN (50 mL), to which anhydrous Zn(OAc)2 (1.38 g, 7.50 mmol), AgOTf (0.385 g, 1.50 mmol) and 2,2,6,6-tetramethylpiperidine (2.52 mL, 15.0 mmol) were added. The reaction mixture was refluxed for 20 h. Standard workup and chromatography [silica, hexanes/CH2Cl2 (1:1)] gave a bluish-green solid (15 mg). The 1H NMR and LD-MS data showed the presence of a significant amount of bromochlorins.
Acknowledgment
This work was supported by the NIH (GM36238). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the NSF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith JHC, Benitez A. In: Modern Methods of Plant Analysis. Paech K, Tracey MV, editors. IV. Springer-Verlag; Berlin: 1955. pp. 142–196. [Google Scholar]
- 2.Strain HH, Thomas MR, Katz JJ. Biochim. Biophys. Acta. 1963;75:306–311. doi: 10.1016/0006-3002(63)90617-6. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wang J-J. Chin. J. Org. Chem. 2005;25:1353–1371. [Google Scholar]; (b) Pavlov VY, Ponomarev GV. Chem. Heterocyclic Compounds. 2004;40:393–425. [Google Scholar]; (c) Pandey RK, Zheng G. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. Academic Press; San Diego, CA: 2000. pp. 157–230. [Google Scholar]; (d) Hynninen PH. In: Chlorophylls. Scheer H, editor. CRC Press; Boca Raton, FL, USA: 1991. pp. 145–209. [Google Scholar]
- 4.(a) Vicente MGH. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego, CA: 2000. pp. 149–199. [Google Scholar]; (b) Jaquinod L. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego, CA: 2000. pp. 201–237. [Google Scholar]
- 5.Dorough GD, Huennekens FM. J. Am. Chem. Soc. 1952;74:3974–3976. [Google Scholar]
- 6.Eisner U. J. Chem. Soc. 1957:3461–3469. [Google Scholar]
- 7.(a) Wang TY, Chen JR, Ma JS. Dyes Pigments. 2002;52:199–208. [Google Scholar]; (b) Shan X, Wang T, Li S, Yang L, Fu L, Yang G, Wang Z, Ma JS. J. Photochem. Photobiol. B: Biol. 2006;82:140–145. doi: 10.1016/j.jphotobiol.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Bonnett R, Djelal BD, Nguyen A. J. Porphyrins Phthalocyanines. 2001;5:652–661. [Google Scholar]
- 9.Brückner C, Dolphin D. Tetrahedron Lett. 1995;36:3295–3298. [Google Scholar]
- 10.(a) Sutton JM, Fernandez N, Boyle RW. J. Porphyrins Phthalocyanines. 2000;4:655–658. [Google Scholar]; (b) Wang TY, Liu HL, Chen JR, Liu FG, Gu Y, Ma JS. Bioorg. Med. Chem. Lett. 2001;11:2049–2052. doi: 10.1016/s0960-894x(01)00363-8. [DOI] [PubMed] [Google Scholar]; (c) Sutton JM, Clarke OJ, Fernandez N, Boyle RW. Bioconjugate Chem. 2002;13:249–263. doi: 10.1021/bc015547x. [DOI] [PubMed] [Google Scholar]; (d) Ferrand Y, Bourré L, Simonneaux G, Thibaut S, Odobel F, Lajat Y, Patrice T. Bioorg. Med. Chem. Lett. 2003;13:833–835. doi: 10.1016/s0960-894x(03)00002-7. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kuz'mitskii VA. J. Appl. Spectrosc. 2005;72:360–370. [Google Scholar]; (b) Linnanto J, Korppi-Tommola J. Phys. Chem. Chem. Phys. 2000;2:4962–4970. [Google Scholar]; (c) Almlöf J, Fischer TH, Gassman PG, Ghosh A, Häser M. J. Phys. Chem. 1993;97:10964–10970. [Google Scholar]
- 12.Sevchenko AN, Solov'ev KN, Shkirman SF, Sarzhevskaya MV. Dokl. Akad. Nauk SSSR. 1963:1391–1394. (Eng. Translation pp 1151–1155) [Google Scholar]
- 13.Sevchenko AN, Solov'ev KN, Mashenkov VA, Shkirman SF. Sov. Phys.–Dokl. 1966;10:778–780. [Google Scholar]
- 14.Gradyushko AT, Sevchenko AN, Solovyov KN, Tsvirko MP. Photochem. Photobiol. 1970;11:387–400. doi: 10.1111/j.1751-1097.1970.tb06011.x. [DOI] [PubMed] [Google Scholar]
- 15.Solov'ev KN, Mashenkov VA, Gradyushko AT, Turkova AE, Lezina VP. Zh. Prikl. Specktr. 1970;13:339–345. (Eng. Translation pp 1106–1111) [Google Scholar]
- 16.Gurinovich GP, Sevchenko AN, Solov'ev KN, editors. Spectroscopy of Chlorophyll and Related Compounds. National Technical Information Service, U.S. Department of Commerce; Springfield, VA: 1971. pp. 330–332. [Google Scholar]
- 17.Sinyakov GN, Poznyak AL, Gurinovich GP. Zh. Prikl. Specktr. 1972;16:732–734. (Eng. Translation pp 543–545) [Google Scholar]
- 18.van der Bent SJ, de Jager A, Schaafsma TJ. Rev. Sci. Instrum. 1976;47:117–121. [Google Scholar]
- 19.van der Bent SJ, Schaafsma TJ. J. Chem. Phys. 1978;68:1857–1861. [Google Scholar]
- 20.Völker S, Macfarlane RM. J. Chem. Phys. 1980;73:4476–4482. [Google Scholar]
- 21.Dicker AIM, Noort M, Thijssen HPH, Volker S, van der Waals JH. Chem. Phys. Lett. 1981;78:212–218. [Google Scholar]
- 22.Dicker AIM, Dobkowski J, Noort M, Völker S, van der Waals JH. Chem. Phys. Lett. 1982;88:135–137. [Google Scholar]
- 23.Keegan JD, Stolzenberg AM, Lu Y-C, Linder RE, Barth G, Moscowitz A, Bunnenberg E, Djerassi C. J. Am. Chem. Soc. 1982;104:4317–4329. [Google Scholar]
- 24.Dicker AIM, Johnson LW, Noort M, van der Waals JH. Chem. Phys. Lett. 1983;94:14–20. [Google Scholar]
- 25.Burkhalter FA, Suter GW, Wild UP, Samoilenko VD, Rasumova NV, Personov RI. Chem. Phys. Lett. 1983;94:483–487. [Google Scholar]
- 26.Gladkov LL, Ksenofontova NM, Solov'ev KN, Starukhin AS, Shul'ga AM, Gradyushko AT. Zh. Prikl. Specktr. 1983;38:598–605. (Eng. Translation pp 435–440) [Google Scholar]
- 27.Arabei SM, Egorova GD, Solov'ev KN, Shkirman SF. Zh. Prikl. Specktr. 1986;44:117–123. (Eng. Translation pp 96–100) [Google Scholar]
- 28.Avarmaa RA, Rebane KK. Spectrochim. Acta. 1985;41A:1365–1380. [Google Scholar]
- 29.Dzilinski K, Sinyakov GN, Shul'ga AM, Zotov NI. J. Structural Chem. 1990;31:227–231. [Google Scholar]
- 30.Ellervee A, Jaaniso R, Kikas J, Laisaar A, Suisalu A, Shcherbakov V. Chem. Phys. Lett. 1991;176:472–476. [Google Scholar]
- 31.De Caro C, Renn A, Wild UP. Appl. Optics. 1991;30:2890–2898. doi: 10.1364/AO.30.002890. [DOI] [PubMed] [Google Scholar]
- 32.Ellervee A, Hizhnyakov VV, Kikas J, Laisaar A, Suisalu A. J. Luminescence. 1992;53:223–226. [Google Scholar]
- 33.Kulikov S, Galaup JP. J. Luminescence. 1992;53:239–243. [Google Scholar]
- 34.Al'shits EI, Kharlamov BM, Ulitsky NI. J. Opt. Soc. Am. B. 1992;9:950–955. [Google Scholar]
- 35.Ellervee A, Kikas J, Laisaar A, Shcherbakov V, Suisalu A. J. Opt. Soc. Am. B. 1992;9:972–977. [Google Scholar]
- 36.Al'shits EI, Kharlamov BM, Ulitsky NI, Nekhaev DV. Chem. Phys. 1992;163:405–411. [Google Scholar]
- 37.Miles JR, Sarre PJ. J. Chem. Soc. Faraday Trans. 1992;88:1075–1076. [Google Scholar]
- 38.Miles JR, Sarre PJ. J. Chem. Soc. Faraday Trans. 1993;89:2269–2276. [Google Scholar]
- 39.Ellervee A, Kikas J, Laisaar A, Suisalu A. J. Luminescence. 1993;56:151–156. [Google Scholar]
- 40.Kikas J, Schellenberg P, Friedrich J. Chem. Phys. Lett. 1993;207:143–147. [Google Scholar]
- 41.Schellenberg P, Friedrich J, Kikas J. J. Chem. Phys. 1994;100:5501–5507. [Google Scholar]
- 42.Shulga AM, Sinyakov GN, Filatov IV, Gurinovich GP, Dzilinski K. J. Mol. Struct. 1995;348:65–68. [Google Scholar]
- 43.Dzilinski K, Synyakov GN, Shulga AM, Filatov IV, Gurinovich GP. Radiat. Phys. Chem. 1995;45:923–928. [Google Scholar]
- 44.Shulga AM, Sinyakov GN, Filatov IV, Gurinovich GP, Dzilinski K. Biospectroscopy. 1995;1:223–234. [Google Scholar]
- 45.Maniloff ES, Graf FR, Gygax H, Altner SB, Bernet S, Renn A, Wild UP. Chem. Phys. 1995;193:173–180. [Google Scholar]
- 46.Müschenborn H-J, Wild UP. Optoelectronics–Dev. Technol. 1995;10:311–332. [Google Scholar]
- 47.Huang W-Y, Riper EV, Johnson LW. Spectrochim. Acta Part A. 1996;52:761–769. [Google Scholar]
- 48.Kikas J, Suisalu A, Zazubovich V. Mol. Cryst. Liq. Cryst. 1996;291:215–222. [Google Scholar]
- 49.den Hartog FTH, Bakker MP, Koedijk JMA, Creemers TMH, Völker S. J. Luminescence. 1996;66 & 67:1–7. [Google Scholar]
- 50.Altmann RB, Kador L, Haarer D. Chem. Phys. 1996;202:167–174. [Google Scholar]
- 51.Kikas J, Suisalu A, Zazubovich V, Vois P. J. Chem. Phys. 1996;104:4434–4440. [Google Scholar]
- 52.Huang W-Y, Rebane A, Wild UP, Johnson LW. J. Luminescence. 1997;71:237–243. [Google Scholar]
- 53.Zazubovich V, Suisalu A, Kikas J. J. Luminescence. 1998;76 & 77:615–618. [Google Scholar]
- 54.den Hartog FTH, Bakker MP, Silbey RJ, Völker S. Chem. Phys. Lett. 1998;297:314–320. [Google Scholar]
- 55.Suisalu A, Zazubovich V, Kikas J, Friebel J, Friedrich J. Europhys. Lett. 1998;44:613–619. [Google Scholar]
- 56.Kikas J, Laisaar A, Suisalu A, Kuznetsov A, Ellervee A. Phys. Rev. B. 1998;57:14–17. [Google Scholar]
- 57.den Hartog FTH, van Papendrecht C, Silbey RJ, Völker S. J. Chem. Phys. 1999;110:1010–1016. [Google Scholar]
- 58.Singh A, Huang W-Y, Johnson LW. J. Phys. Chem. A. 2000;104:894–898. [Google Scholar]
- 59.Drobizhev M, Karotki A, Rebane A. Chem. Phys. Lett. 2001;334:76–82. [Google Scholar]
- 60.Singh A, Huang W-Y, Egbujor R, Johnson LW. J. Phys. Chem. A. 2001;105:5778–5784. [Google Scholar]
- 61.Zazubovich V, Suisalu A, Kikas J. Phys. Rev. B. 2001;64(104203):1–7. [Google Scholar]
- 62.Singh A, Johnson LW. Spectrochim. Acta Part A. 2002;58:1573–1576. doi: 10.1016/s1386-1425(01)00618-7. [DOI] [PubMed] [Google Scholar]
- 63.Singh A, Huang W-Y, Johnson LW. Spectrochim. Acta Part A. 2002;58:2177–2183. doi: 10.1016/s1386-1425(01)00688-6. [DOI] [PubMed] [Google Scholar]
- 64.Renge I. J. Luminescence. 2002;98:213–220. [Google Scholar]
- 65.Karotki A, Kruk M, Drobizhev M, Rebane A. J. Mod. Optics. 2002;49:379–390. [Google Scholar]
- 66.Renn A, Wild UP, Rebane A. J. Phys. Chem. A. 2002;106:3045–3060. [Google Scholar]
- 67.Berezin KV, Nechaev VV. Chem. Natural Compounds. 2003;39:540–548. [Google Scholar]
- 68.Renge I. Chem. Phys. 2003;295:255–268. [Google Scholar]
- 69.Berezin KV, Nechaev VV. Opt. Spectros. 2004;97:707–713. [Google Scholar]
- 70.Eisner U, Linstead RP. J. Chem. Soc. 1955:3749–3754. [Google Scholar]
- 71.Eisner U, Linstead RP. J. Chem. Soc. 1955:3742–3749. [Google Scholar]
- 72.Egorova GD, Solov'ev KN, Shul'ga AM. J. Gen. Chem. USSR. 1967;37:333–336. [Google Scholar]
- 73.Seely GR, Talmadge K. Photochem. Photobiol. 1964;3:195–206. [Google Scholar]
- 74.Montforts F-P, Gerlach B, Höper F. Chem. Rev. 1994;94:327–347. [Google Scholar]
- 75.Strachan J-P, O'Shea DF, Balasubramanian T, Lindsey JS. J. Org. Chem. 2000;65:3160–3172. doi: 10.1021/jo991942t. [DOI] [PubMed] [Google Scholar]
- 76.Taniguchi M, Ra D, Mo G, Balasubramanian T, Lindsey JS. J. Org. Chem. 2001;66:7342–7354. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]
- 77.Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M, Lindsey JS. J. Org. Chem. 2006;71:4092–4102. doi: 10.1021/jo060208o. [DOI] [PubMed] [Google Scholar]
- 78.O'Neal WG, Roberts WP, Ghosh I, Wang H, Jacobi PA. J. Org. Chem. 2006;71:3472–3480. doi: 10.1021/jo060041z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Balasubramanian T, Strachan JP, Boyle PD, Lindsey JS. J. Org. Chem. 2000;65:7919–7929. doi: 10.1021/jo000913b. [DOI] [PubMed] [Google Scholar]
- 80.Taniguchi M, Kim MN, Ra D, Lindsey JS. J. Org. Chem. 2005;70:275–285. doi: 10.1021/jo048440m. [DOI] [PubMed] [Google Scholar]
- 81.Taniguchi M, Ptaszek M, McDowell BE, Lindsey JS. Tetrahedron. 2007;63 doi: 10.1016/j.tet.2007.02.076. companion paper (second paper in series of three) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taniguchi M, Ptaszek M, McDowell BE, Boyle PD, Lindsey JS. Tetrahedron. 2007;63 doi: 10.1016/j.tet.2007.02.040. companion paper (third paper in series of three) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rao PD, Dhanalekshmi S, Littler BJ, Lindsey JS. J. Org. Chem. 2000;65:7323–7344. doi: 10.1021/jo000882k. [DOI] [PubMed] [Google Scholar]
- 84.Laha JK, Dhanalekshmi S, Taniguchi M, Ambroise A, Lindsey JS. Org. Process Res. Dev. 2003;7:799–812. [Google Scholar]
- 85.Zaidi SHH, Muthukumaran K, Tamaru S-I, Lindsey JS. J. Org. Chem. 2004;69:8356–8365. doi: 10.1021/jo048587d. [DOI] [PubMed] [Google Scholar]
- 86.Geier GR, III, Littler BJ, Lindsey JS. J. Chem. Soc., Perkin Trans. 2. 2001:701–711. [Google Scholar]
- 87.Ptaszek M, McDowell BE, Lindsey JS. J. Org. Chem. 2006;71:4328–4331. doi: 10.1021/jo060119b. [DOI] [PubMed] [Google Scholar]
- 88.Brückner C, Posakony JJ, Johnson CK, Boyle RW, James BR, Dolphin D. J. Porphyrins Phthalocyanines. 1998;2:455–465. [Google Scholar]
- 89.Battersby AR, Fookes CJR, Snow RJ. J. Chem. Soc. Perkin Trans. 1. 1984:2725–2732. [Google Scholar]
- 90.Sharada DS, Muresan AZ, Muthukumaran K, Lindsey JS. J. Org. Chem. 2005;70:3500–3510. doi: 10.1021/jo050120v. [DOI] [PubMed] [Google Scholar]
- 91.Rosenfeld A, Morgan J, Goswami LN, Ohulchanskyy T, Zheng X, Prasad PN, Oseroff A, Pandey RK. Photochem. Photobiol. 2006;82:626–634. doi: 10.1562/2005-09-29-RA-704. [DOI] [PubMed] [Google Scholar]
- 92.Taniguchi M, Balakumar A, Fan D, McDowell BE, Lindsey JS. J. Porphyrins Phthalocyanines. 2005;9:554–574. [Google Scholar]
- 93.(a) Sapunov VV, Egorova GD. J. Appl. Spectroscopy. 1987;46:519–522. [Google Scholar]; (b) Stolzenberg AM, Schussel LJ, Summers JS, Foxman BM, Petersen JL. Inorg. Chem. 1992;31:1678–1686. [Google Scholar]; (c) Singh A, Johnson LW. Spectrochim. Acta Part A. 2003;59:905–908. doi: 10.1016/s1386-1425(02)00258-5. [DOI] [PubMed] [Google Scholar]
- 94.(a) Borisevich EA, Egorova GD, Knyukshto VN, Solovev KN. Opt. Spectrosc. (USSR) 1984;56:255–258. [Google Scholar]; (b) Lomova TN, Mozhzhukhina EG, Shormanova LP, Berezin BD. J. Gen. Chem. USSR. 1989;54:2077–2084. [Google Scholar]; (c) Sengupta D, Robinson BC. Tetrahedron. 2002;58:5497–5502. [Google Scholar]; (d) Ciulla TA, Criswell MH, Danis RP, Snyder WJ, Small W., IV Retina. 2004;24:521–529. doi: 10.1097/00006982-200408000-00004. [DOI] [PubMed] [Google Scholar]; (e) Ciulla TA, Criswell MH, Snyder WJ, Small W., IV Br. J. Ophthalmol. 2005;89:113–119. doi: 10.1136/bjo.2004.043075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu L, Muthukumaran K, Sazanovich IV, Kirmaier C, Hindin E, Diers JR, Boyle PD, Bocian DF, Holten D, Lindsey JS. Inorg. Chem. 2003;42:6629–6647. doi: 10.1021/ic034559m. [DOI] [PubMed] [Google Scholar]
- 96.Kim H-J, Dogutan DK, Ptaszek M, Lindsey JS. Tetrahedron. 2007;63:37–55. doi: 10.1016/j.tet.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Srinivasan N, Haney CA, Lindsey JS, Zhang W, Chait BT. J. Porphyrins Phthalocyanines. 1999;3:283–291. [Google Scholar]
- 98.Ptaszek M, Bhaumik J, Kim H-J, Taniguchi M, Lindsey JS. Org. Process Res. Dev. 2005;9:651–659. doi: 10.1021/op050087e. [DOI] [PMC free article] [PubMed] [Google Scholar]