

Abstract
Because of their ecological functions, natural products have been optimized in evolution for interaction with biological systems and receptors. However, they have not necessarily been optimized for other desirable drug properties and thus can often be improved by structural modification. Using examples from the literature, this paper reviews the opportunities for increasing structural diversity among natural products by combinatorial biosynthesis, i.e., the genetic manipulation of biosynthetic pathways. It distinguishes between combinatorial biosynthesis in a narrower sense to generate libraries of modified structures, and metabolic engineering for the targeted formation of specific structural analogs. Some of the problems and limitations encountered with these approaches are also discussed. Work from the author’s laboratory on ansamycin antibiotics is presented which illustrates some of the opportunities and limitations.
Keywords: Combinatorial biosynthesis, metabolic engineering, natural products, rifamycin, maytansinoids, polyketides
Combinatorial biosynthesis can be defined as the application of genetic engineering to modify biosynthetic pathways to natural products in order to produce new and altered structures using nature’s biosynthetic machinery. The feasibility of this approach was first demonstrated in work by David Hopwood and colleagues (Hopwood et al., 1985). Following the cloning of the biosynthetic genes for the antibiotic actinorhodin from Streptomyces coelicolor (Malpartida & Hopwood, 1984), Hopwood and coworkers cloned some or all of these genes into the producers of the antibiotics medermycin (Takano et al., 1976) and dihydrogranaticin (Corbaz et al., 1957), respectively. The transformant of the medermycin producer, Streptomyces sp. AM-7161 produced, in addition to medermycin, large amounts of a new compound, mederrhodin A, which carried an additional hydroxy group characteristic of actinorhodin (Figure 1). The transformant of the dihydrogranaticin producer, Streptomyces violaceoruber Tü 22 produced a new compound, dihydrogranatirhodin, which has the actinorhodin configuration at one and the dihydrogranaticin configuration at the other stereocenter of the isochromane quinone system. This work represents the first, albeit modest, implementation of the concept of combinatorial biosynthesis of natural products. Since then, a large number of examples encompassing a wide range of natural product classes have appeared in the literature, and this approach has been accepted as a useful tool to increase the chemical diversity of natural products (Staunton and Wilkinson, 2001; Walsh, 2002; Rix et al., 2002; Reeves, 2003). Following the author’s primary interests, this article focuses on the application of the combinatorial biosynthesis approach to microbial natural products and within these to polyketides. However, it should be emphasized that it certainly can be and has been applied to many other compound classes and also to natural products from other biota, although the more complicated genetic makeup of higher organisms presents greater technical challenges.
Figure 1.
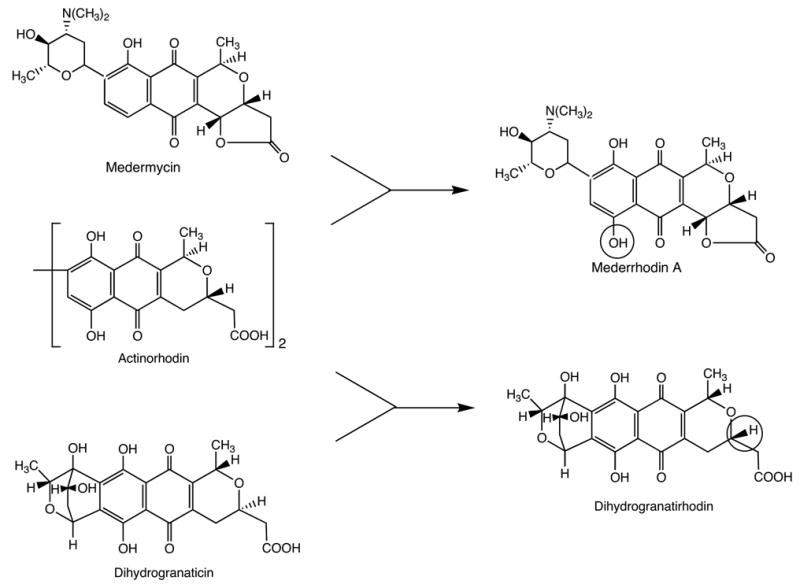
First demonstration of genetic engineering of hybrid natural products, mederrhodin A and dihydrogranatirhodin (Hopwood et al., 1985).
Natural products have long been recognized as an important source of clinical drugs, drug leads and biochemical tools. Natural products have provided or inspired a substantial fraction of the currently used clinical drugs . The screening of natural product libraries and extracts usually yields a substantially higher percentage of bioactive hits than that of chemical libraries. It is difficult to come up with good numbers for the magnitude of this advantage, but a recent review (Berdy, 2005) estimates an approximately hundred-fold higher hit rate for natural products. One of the characteristics of natural product libraries compared to chemical ones is that the input is not limited by human imagination. Another important role of natural products is to reveal novel structure - bioactivity relationships. As new biochemical targets arise, the screening of natural products libraries is often the best way to identify agents acting on this target. Even if the natural product itself is not suitable as a drug, its structure can be the starting point for drug development. Finally, the study of the mode of action of natural products can identify novel biochemical mechanisms. Natural product inhibitors of chemical processes allow the dissection of signaling pathways, they can reveal molecular disease mechanisms and suggest new biochemical targets.
Why is it that the screening of natural products libraries and extracts yields more bioactive hits than that of chemical libraries? The answer lies in the ecologic role of natural products. Their formation represents a selectable evolutionary advantage to the producing organism. Their role is to help the producing organism to maintain or improve its position in its ecological environment. In fulfilling this role they can have a multitude of different functions. Some are defense compounds, which deter predators, others are signal compounds, which attract other organisms, others inhibit the growth or proliferation of competitors, to give just a few examples. Natural products have been optimized through evolution for interaction with biological systems and receptors, many of which are common to a wide range of living organisms. For this reason natural products are superior sources of drug candidates or biologically active lead compounds compared to random chemical libraries.
However, natural products have not been optimized in evolution as clinical drugs, but rather for the biological functions useful to the producing organism. Their broader potential, beyond such obvious features as antibiotic activity in microbial natural products or CNS and cardiac activity in higher plant natural products, is due to the generality of biological receptors and mechanisms, which allows them to affect processes not relevant to the producing organism. The intrinsic potency of natural products often cannot be improved by structure modification, but other properties important for a clinical drug can be. This is illustrated by the examples in Figure 2. The pharmacokinetic properties of a drug typically are not optimized in nature and can often be improved by structure modification. For example, the antibiotic rifamycin B, the main product of the fermentation by Amycolatopsis mediterranei, is not orally active, but chemical structure modification led to orally active compounds, such as rifampicin with, in this case, also increased potency (Floss and Yu, 2005). Toxicity to humans is not relevant to the intrinsic function of a natural product and may thus be reduced by structure modification. For example, the antibiotic geldanamycin (DeBoer et al., 1970) has potent antitumor activity, but its hepatotoxicity prevented introduction into clinical use (Supko et al., 1995) . Replacement of the aromatic methoxy group by a nitrogen substituent led to a compound, 17-DMAG, which is currently undergoing clinical trials (Egorin et al., 2002). Finally, structure modification of a natural product may be carried out to strengthen a proprietary position by generating a patentable structure from a non-patented natural product, such as the generation of the semi-synthetic taxotere (Guéritte-Voegelein et al., 1986) from the non-patented natural product, the antitumor agent taxol (Wani et al., 1971). For all these reasons, the structure modification of natural products is an important component of the natural product drug development process. Wherever possible, such structure modifications are carried out by chemical means, but in many cases, the complexity of natural product structures makes chemical approaches difficult or impossible, providing the rationale for biological approaches, including combinatorial biosynthesis, as an alternative strategy.
Figure 2.
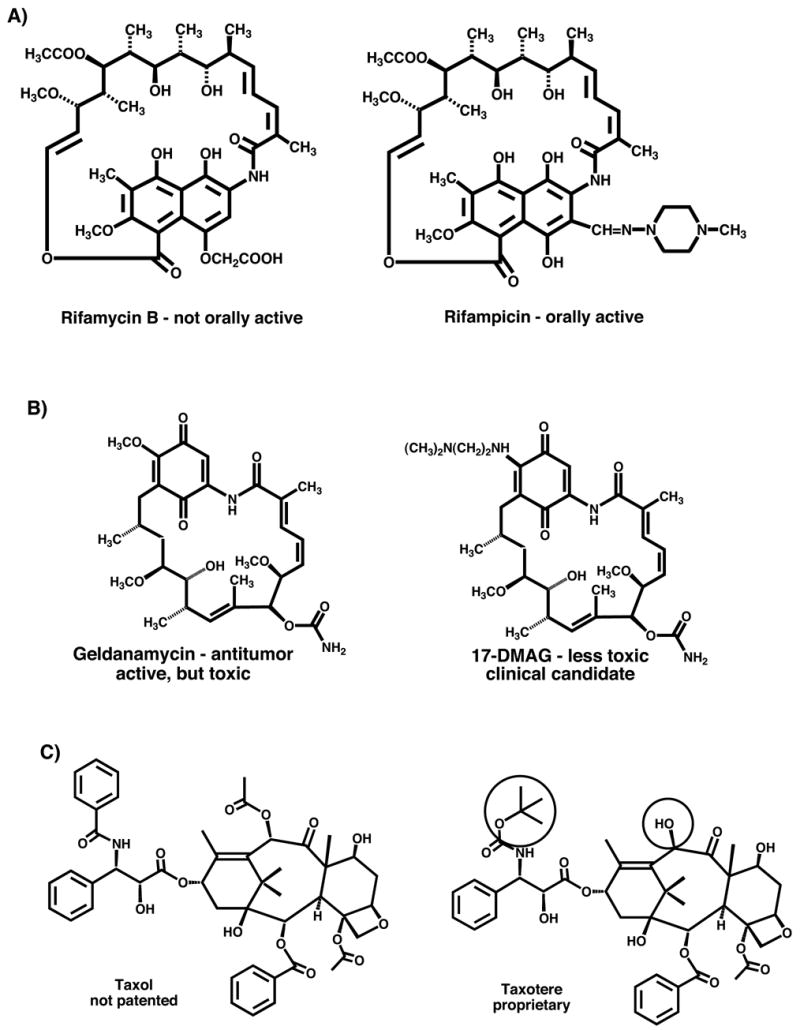
Examples of structure modification of natural products to improve utility as clinical drugs. (A) Modification of rifamycin B to rifampicin to improve pharmacokinetic properties, (B) modification of geldanamycin to 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin to reduce human toxicity and (C) modification of taxol to taxotere to generate a proprietary molecule.
As commonly applied, the term “combinatorial biosynthesis” actually encompasses two different approaches with somewhat different, albeit overlapping goals. Metabolic engineering to improve the drug properties of a natural product involves the targeted modification of a biosynthetic pathway to generate a single or limited number of modified structures. It can be applied in the same way as chemical structure modification but with different limitations. It is particularly suitable for the construction of molecules which are chemically inaccessible, usually complex structures. This has been pursued particularly intensely in polyketides. One can introduce backbone structural changes, e.g., modify ring systems, chain lengths or incorporate different building blocks, or one can introduce peripheral changes, such as different sugars, methyl groups, oxygen functions or halogens. An elegant example is shown in Figure 3 which summarizes work by Hutchinson and coworkers, genetically modifying the biosynthetic pathway for daunorubicin and doxorubicin to produce the new clinical agent epirubicin (Madduri et al., 1998). In the normal biosynthetic pathway to daunorubicin and doxorubicin in Streptomyces peucetius, the dnmV gene encodes a ketoreductase which produces a sugar moiety with 4S configuration. Inactivation of this gene and introduction of either the avrR or the eryBIV gene from the avermectin or erythromycin pathway, which encode ketoreductases of opposite stereospecificity, gave mutant strains producing the 4’-epi compounds.
Figure 3.
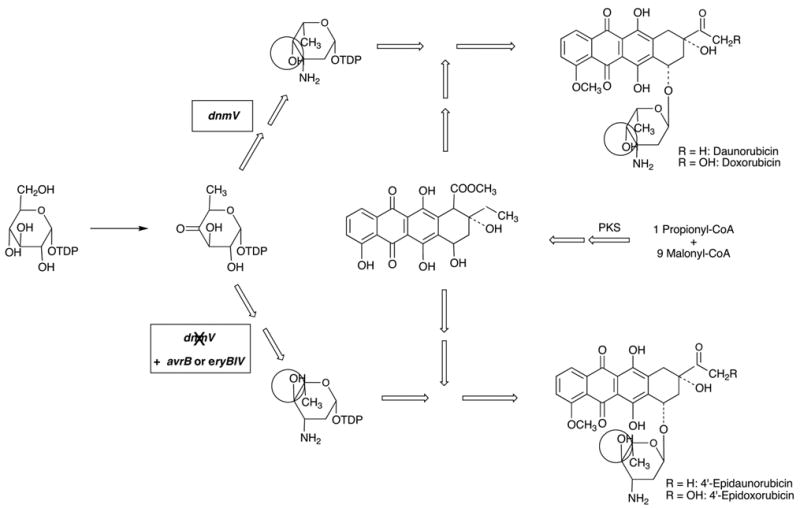
Genetic engineering of the doxorubicin producer, Streptomyces peucetius, for production of 4’-epidoxorubicin (epirubicin) (Madduri et al., 1998).
Combinatorial biosynthesis proper, in a narrower sense of the term, uses sets of genes from different biosynthetic pathways in different combinations to generate libraries of hybrid structures. This is illustrated by work published by Kosan Biosciences (McDaniel et al., 1999). on 6-deoxyerythronolide B (6-DEB), the alycone of erythromycin (Wiley et al., 1957; Harris et al., 1965), which is assembled on a type I modular polyketide synthase (PKS) (Figure 4) (Donadio et al., 1991). These large, multifunctional enzymes contain one module for each cycle of chain elongation in the polyketide assembly process which also includes the domains catalyzing the respective modification reactions associated with this reaction cycle. These polyketide synthases thus operate as large assembly lines in which the structure of the final product is largely programmed as a result of the domain structure of the enzyme. McDaniel and coworkers genetically engineered a number of changes in the domain structure of this PKS which they then used combinatorially to generate a library of 61 different 6-DEB analogs (McDaniel et al., 1999). This latter approach truly generates structural diversity. It has, however, the drawback that the compounds generated are no longer optimized by evolution for biological activity. It is likely that the screening of such libraries still gives higher hit rates for bioactive compounds than that of random chemical libraries, since the components are pharmacophoric substructures. Based on the ecological rationale, one might expect that the hit rates will drop, the further away from bioactive natural parent structures one moves, but there do not seem to be any data available to support or reject this notion.
Figure 4.
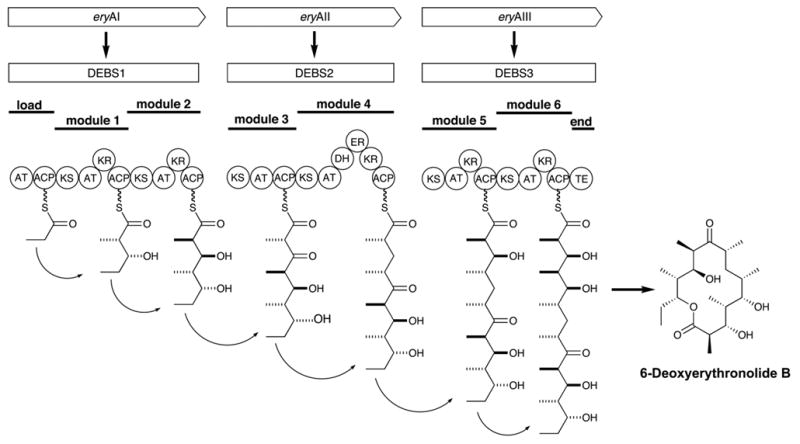
Assembly of 6-deoxyerythronolide B (6-DEB), the aglycone of erythromycin, on a type I modular polyketide synthase. (AT = adenyltransferase, KS = ketosynthase, ACP = acyl carrier protein, KR = ketoreductase, DH = dehydratase, ER = enoate reductase, TE = thioesterase).
It is fair to say that the great majority of applications of combinatorial biosynthesis in the broader sense published to date actually fall into the category of metabolic engineering. The strategies to achieve modifications by combinatorial biosynthesis/metabolic engineering depend somewhat on whether the product is made by a linear or a branched biosynthetic pathway. In linear biosynthetic pathways, as exemplified by erythromycin, one can introduce backbone modifications as demonstrated in the work mentioned above (McDaniel et al., 1999), or one can introduce alternative decorations onto the backbone of the molecule. As shown in Figure 5, the further conversion of 6-DEB into erythromycin A involves a series of oxidations, glycosylations and a methylation (Staunton and Wilkinson, 1999). All these steps are potentially amenable to modification, and a number of publications report the generation of peripherally modified erythromycins by genetic engineering (Rawlings, 2001). In a branched biosynthetic pathway, in which several components are biosynthesized separately and then assembled, as exemplified by novobiocin, one can also swap biosynthetic building blocks. This approach, called mutasynthesis (Shier et al., 1969; Rinehart, 1981) predates the advent of genetic engineering, but it can be practiced in more sophisticated form using genetic tools. This is illustrated in Figure 6 with work by Heide and coworkers on the genetic engineering of aminocoumarin antibiotics. Novobiocin and chlorobiocin, the products of two different streptomycetes, differ in the substitution patterns of their aminocoumarin and sugar moieties. Exchange of the appropriate biosynthetic pathway genes between the two organisms led to the formation of hybrid products, such as novclobiocin 102 or novclobiocin 114 (Eustaquio et al., 2004).
Figure 5.
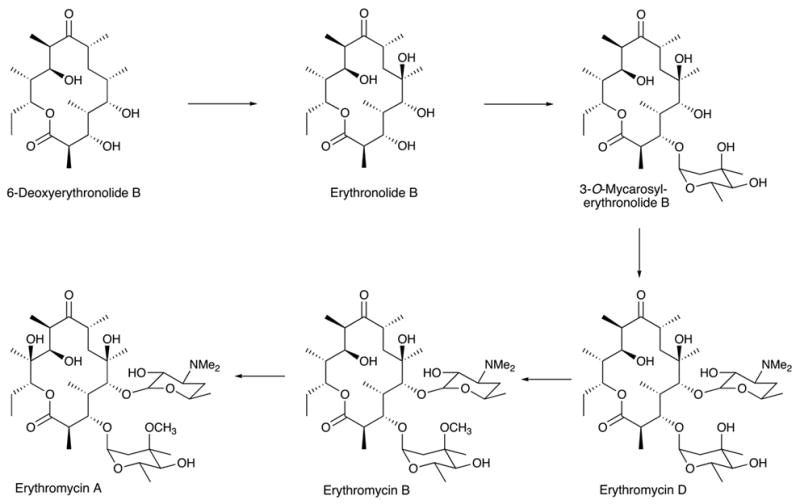
Post-PKS reactions of erythromycin biosynthesis from 6-DEB.
Figure 6.
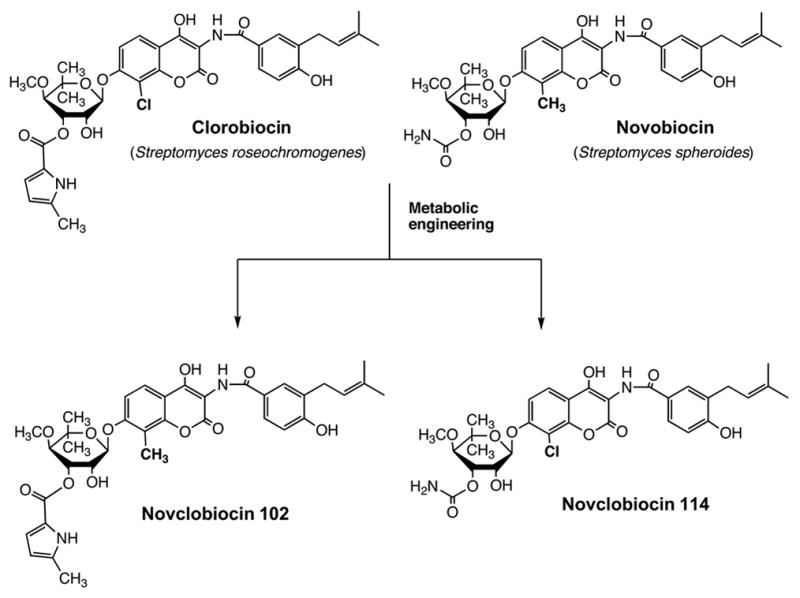
Genetic engineering of the formation of novclobiocins, structural hybrids of novobiocin and clorobiocin (Eustaquio et al., 2004).
The following are some examples of “decorating” reactions that are suitable for genetic engineering of natural products:
Glycosylation. This can involve different sugar nucleotides or different aglycones in vivo or in vitro, and is probably the process most suitable for truly combinatorial applications (Luzhetskyy and Bechthold, 2005; Yang et al., 2004).
Oxidations and reductions. This includes hydroxylations, epoxidations, reductions of carbonyl groups or double bonds, and oxidations of alcohols.
Methylations. On oxygen, nitrogen or carbon.
Isoprenylations. On carbon, oxygen or nitrogen.
Halogenations.
Acylations. Either on oxygen to form esters or on nitrogen to form amides.
As the above list indicates, the range of possible structural modifications of natural products is extensive, making combinatorial biosynthesis/metabolic engineering potentially an extremely powerful tool. In practice, however, a number of problems are encountered in the application of this tool. First of all, most applications are not truly “combinatorial”; rather, the generation of each new structures involves the, often elaborate, construction of a mutant strain of an organism. This is labor intensive and costly. Secondly, the process relies on relaxed substrate specificity of biosynthetic pathway enzymes, but many enzymes are fairly specific. As a result, many predicted transformations do not take place and the outcomes are not always predictable. Furthermore, the yields of engineered new metabolites are often much lower than those of the parent compound produced by the wild-type. Genetic constructs may not be optimally expressed and pathway enzymes often work less efficiently with structurally altered substrates. In a number of cases where this has been tried, yield optimization by classical or genetic approaches has been challenging. Another problem involves the question whether to express altered pathways in heterologous hosts, whose regulatory elements may not work well with the foreign genes and which may lack precursor supply routes, or to genetically modify the pathway in the parent organism, for which often efficient genetic systems are not available. This points to a need for suitable engineered universal expression hosts. Finally, the more modified structures differ from the bioactive parent molecule, the less likely they probably are to have potent bioactivity, i.e., hit rates will probably be lower.
Following these general remarks, let us now turn to some work from the authors laboratory which illustrates some of the potential and limitations of combinatorial biosynthesis. These studies deal with two groups of ansamycin antibiotics, the rifamycins and the ansamitocins. The rifamycins were isolated in 1959 by Sensi and coworkers (Sensi et al., 1959) from the actinomycete, Amycolatopsis mediterranei. They have strong antibacterial activity, particularly against mycobacteria, including Mycobacterium tuberculosis. They block protein synthesis by inhibiting DNA-dependent RNA polymerase due to binding to the B subunit. Semisynthetic derivatives, such as rifampicin (Figure 2), are used clinically for the treatment of tuberculosis, leprosy, and AIDS-related mycobacterial infections. Resistance to these antibiotics develops easily due to mutations in the rpoB gene, calling for the development of next generation drugs to overcome this problem (Floss and Yu, 2005). The ansamitocins (Higashide et al., 1977), produced by the actinomycete, Actinosynnema pretiosum, belong to the family of the maytansinoids first isolated from higher plants (Kupchan et al., 1972). They are notable for their extremely potent antitumor activity which is due to binding to the tubulin β-subunit, blocking the assembly of functional microtubules (Cassady et al., 2004). Both the rifamycins and ansamitocins are assembled by a type I polyketide synthase, using an aromatic starter unit, 3-amino-5-hydroxybenzoic acid (AHBA) which is biosynthesized by a branch of the shikimate pathway. Cloning by reverse genetics of the gene encoding AHBA synthase (Kim et al., 1998) provided a probe for the identification of the rifamycin B (August et al., 1998) (Figure 7) and ansamitocin (Yu et al., 2002) (Figure 8) biosynthetic gene clusters. These were isolated from cosmid libraries of their respective genomic DNAs, sequenced and their identities confirmed by appropriate gene inactivations. Each cluster contains candidate genes for all the reactions necessary to assemble the respective final product, including genes for the formation of the AHBA starter unit, a set of PKS genes (rifA,B,C,D,E for rifamycin {see also Schupp et al., 1998} and asmA,B,C,D for ansamitocin) with a loading module (adenylation domain plus acyl carrier protein) “borrowed” from non-ribosomal peptide synthases (Admiraal et al., 2001), followed by a gene coding for a downloading enzyme, an amide synthase (rifF and asm9), as well as candidate genes for the various post-PKS modification reactions. The ansamitocin biosynthesis genes notably are contained not in one, but two clusters in the A. pretiosum genome. While the majority of these genes are present in cluster I, cluster II contains some of the AHBA biosynthesis genes and is thus also essential (Figure 8). Clusters I and II are separated by 30 kb of DNA which is not required for either growth or ansamitocin production (Yu et al., 2002).
Figure 7.
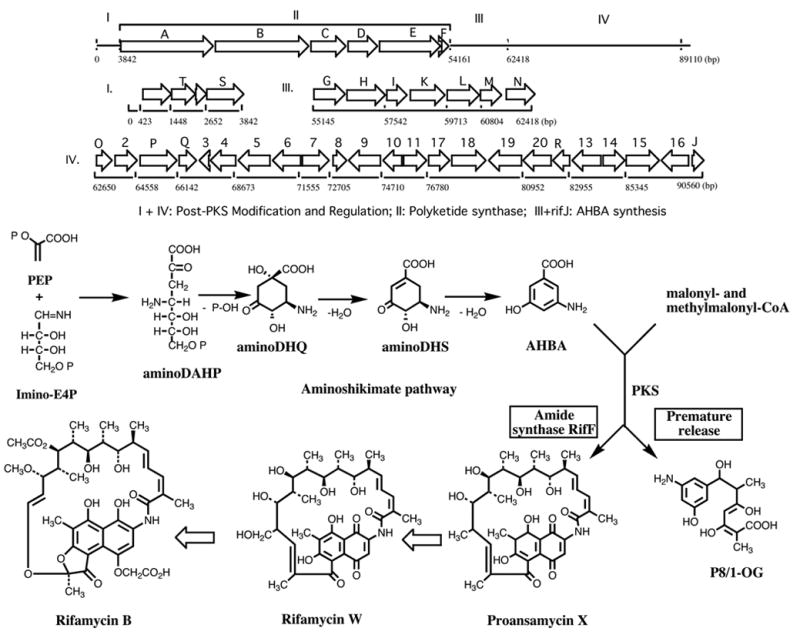
The rifamycin biosynthetic gene cluster from Amycolatopsis mediterranei and the pathway of rifamycin B biosynthesis (August et al., 1998)
Figure 8.
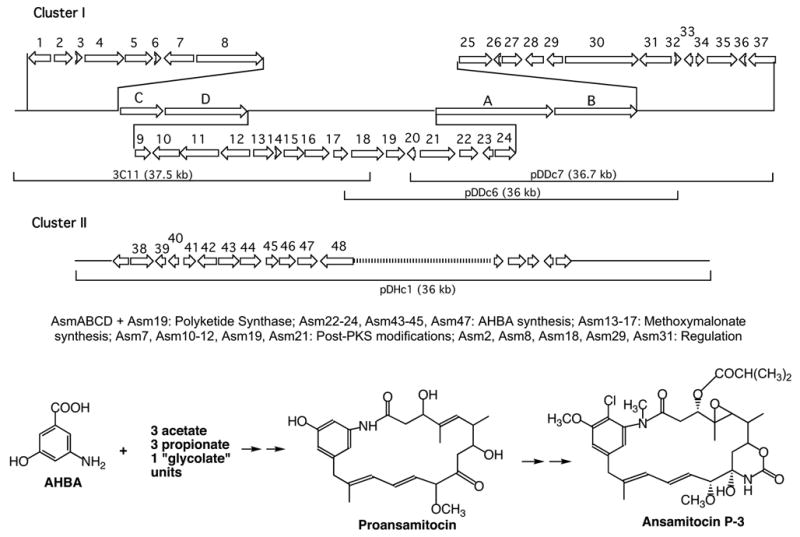
The ansamitocin biosynthetic gene cluster from Actinosynnema pretiosum and the structure and biosynthesis of ansamitocin P-3 (Yu et al., 2002).
In early attempts to modify the aromatic moiety of these antibiotics, we used a mutant in which the AHBA synthase gene in the genome of the rifamycin producer had been inactivated (Kim et al., 1998). Feeding of AHBA to this mutant restored full rifamycin B production, but feeding of two analogs gave copious amounts of the corresponding tetraketides (Figure 9). This result, confirmed subsequently by in vitro studies (Admiraal et al., 2001), showed that the loading module of the rif PKS is rather promiscuous, but that a down-stream step, apparently after the third chain elongation, discriminates against the structurally altered intermediates (Hunziker et al., 1998). An explanation for the latter phenomenon was revealed when rifF, the gene encoding the downloading enzyme, was inactivated. This resulted in the accumulation of the complete series of open chain ketides from the tetra- to the undecaketide, rather than just of the product immediately before the blocked reaction, the undecaketide (Figure 10) (Yu et al., 1999; Stratmann et al., 1999; Kubota, T., Yu, T. – W. and Floss, H. G., unpublished results). This provided unequivocal proof for the processive nature of the polyketide assembly process and showed that the enzyme can process multiple ketide units simultaneously. Also, the tetraketide had the unmodified benzenoid starter unit, whereas from the pentaketide on this unit had been modified to the naphthalenic structure. It follows that the naphthalene ring closure occurs between the third and fourth chain extension step, that it is necessary for further processing of the ketide, and that it is sensitive to structural changes in the aromatic moiety. We have recently identified the rif orf 19 gene as being essential for this ring closure (Xu et al., 2005); the encoded enzyme must presumably dock with the PKS in order to transform its enzyme-bound substrate. We also found subsequently that the undecaketide is released from the PKS by a different mechanism than the other ketides and that its terminal double bond, contrary to the earlier report (Stratmann et al., 1999), has Z configuration as in rifamycin (Kubota, T., Yu, T. – W. and Floss, H. G., unpublished results). These results with the rifF(−) mutant suggest that backbone modifications of the rifamycins by genetic engineering may be rather difficult to implement, because the rif PKS is, at least at some point, quite sensitive to the structure of the intermediates coming along the assembly line. It may, however, be possible to engineer modified rifamycins by manipulating the post-PKS modification reactions and, perhaps, also some of the late steps of polyketide assembly.
Figure 9.
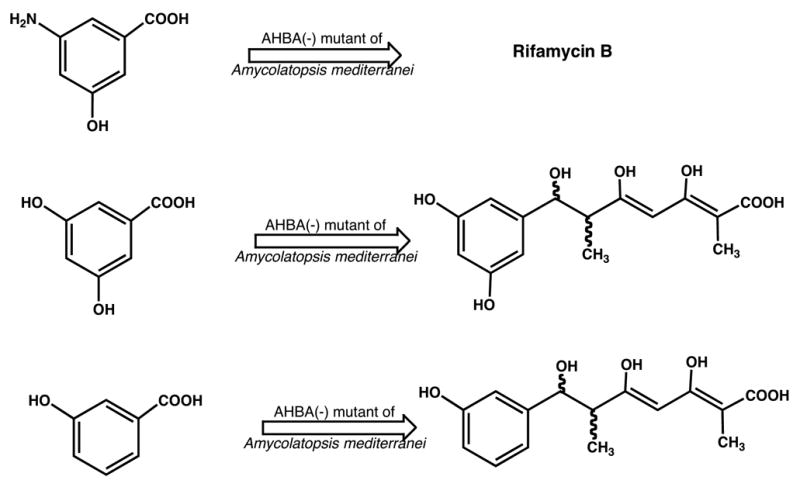
Production of tetraketides upon feeding of AHBA analogs to an AHBA(−) mutant of the rifamycin producer, Amycolatopsis mediterranei (Hunziker et al., 1998).
Figure 10.
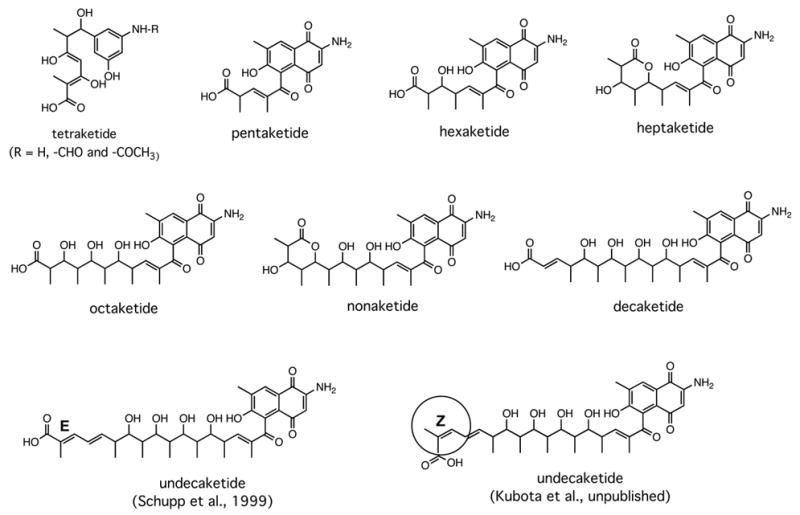
Ketides isolated from mutants of the rifamycin producer, Amycolatopsis mediterranei, carrying an inactivated rifF gene (Yu et al., 1999; Stratmann et al., 1999).
Similar attempts to modify the aromatic moiety of ansamitocins also gave disappointing results, but for different reasons. In this case, the loading domain of the PKS seems to be much more specific, i.e., it does not readily accept analogs of AHBA (Yu, T. – W., Müller, M., Grond, S. and Floss, H. G., unpublished results). Attempts to circumvent this loading domain by directly loading preformed di- or triketides, activated as thioesters the coenzyme A mimic, N-acetylcysteamine (Yue et al., 1987; Cane and Yang, 1987), onto the cognate ACP of the PKS were only partially successful. Only the R enantiomer of the diketide complemented an AHBA(−) mutant of Actinosynnema presiosum to restore some ansamitocin production, but with rather low efficiency (Figure 11). While this result reveals the cryptic stereochemistry of the diketide intermediate in the assembly process, the yield of the final product is too low to make this a practical alternative for the modification of the polyketide starter unit. We also demonstrated that the inefficient utilization of the diketide-SNAC is not due to enzyme inactivation, but rather reflects inefficient loading onto the PKS, by showing that cells pretreated with the diketide SNAC still were fully complemented by AHBA (M. Brünjes, T. Frenzel, Kubota, T., Floss, H. G. and Kirschning, A., unpublished work).
Figure 11.
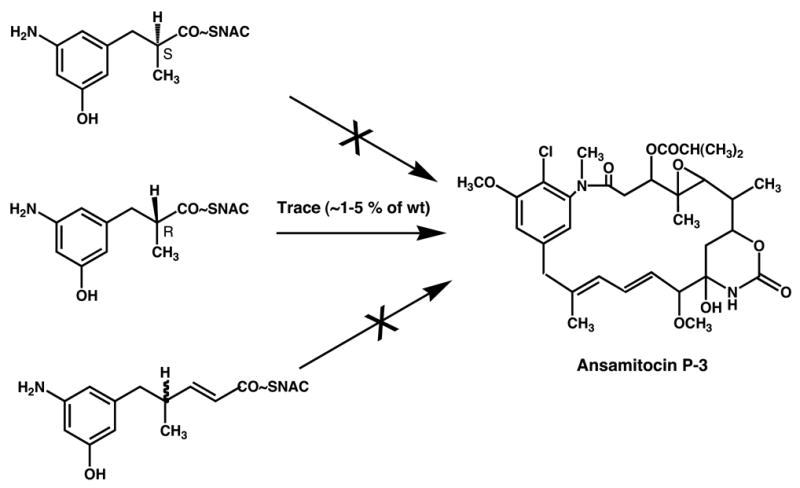
Feeding of diketide and triketide N-acetylcysteamine (SNAC) thioesters to an AHBA(−) mutant of the ansamitocin producer, Actinosynnema pretiosum.
One unusual feature of the ansamitocin polyketide assembly is the incorporation, in the third chain elongation step, of a “glycolate” extender unit. In analogy to the use of malonyl-CoA to incorporate acetate and methylmalonyl-CoA to incorporate propionate units, one would expect the substrate for the incorporation of such a “glycolate” unit to be 2-hydroxy- or 2-methoxymalonyl-CoA. Feeding experiments with the corresponding SNAC thioesters, however, showed that this is not the case (Carroll et al., 2002). Instead, biosynthetic gene clusters for antibiotics containing “glycolate” units contain subclusters of genes (Wu et al., 2000), in this case asm13 – 17, containing a dedicated acyl carrier protein (ACP). By individually inactivating each of these genes we showed that each was necessary for formation of the chain extension substrate (Carroll et al., 2002; Bai, L., Moss, S. J., Kato, Y., Yu, T. – W. and Floss, H. G., unpublished results). By expressing all five genes as a cassette, it was also demonstrated that they were sufficient to provide the substrate, 2-methoxymalonyl-ACP (Kato et al., 2002). Co-expression of this cassette with a DEB synthase modified in the last acyltransferase domain to select for methoxymalonate rather than methylmalonate (Reeves et al., 2002) resulted in the efficient production of 2-methoxy-6-DEB, whereas in the absence of the methoxymalonate cassette, malonate or methylmalonate was utilized inefficiently as the default mode (Kato et al., 2002). The starting material for the formation of methoxymalonyl-ACP is still unknown, but is speculated to be 3-phosphoglycerate, and the biosynthetic pathway to this extender unit is as shown in Figure 12. The methoxymalonate cassette thus represents a new addition to the toolkit for the combinatorial biosynthesis of type I polyketides.
Figure 12.
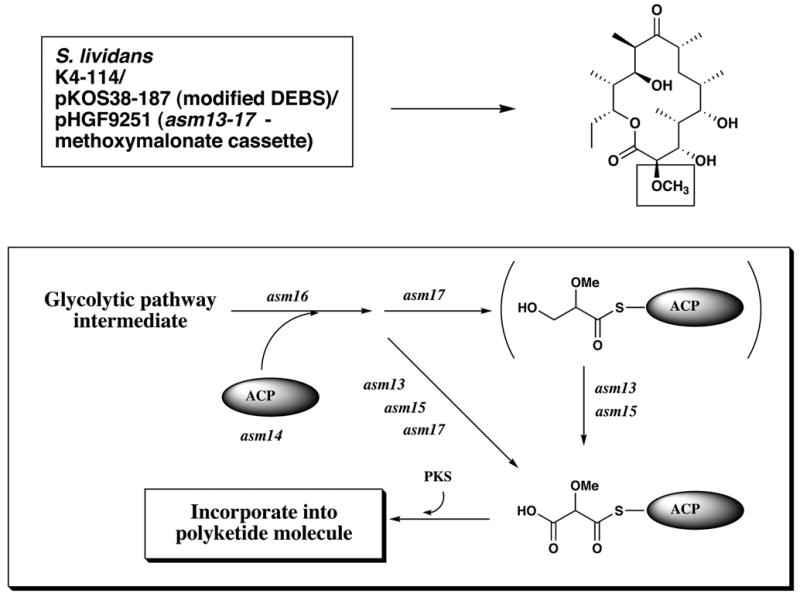
Genetic engineering of the formation of 2-desmethyl-2-methoxy-6-deoxyerythronolide B in a heterologous host, and the pathway of formation of methoxymalonyl-ACP, the precursor of “glycolate” units (Kato et al., 2002).
Following the assembly and release of the first cyclic polyketide from the asm-PKS, a series of six post-PKS modification reactions installs the peripheral decorations which render the ansamitocin molecule biologically active. The genes/enzymes responsible for each of these steps, and the order in which these reactions occur, were determined by individual inactivation experiments (Moss et al., 2002; Spiteller et al., 2003). For practical reasons, we were particularly interested in the attachment of the ester side chain, catalyzed by Asm19.
Despite its high antitumor activity in vivo and in animal models, maytansine failed in phase II clinical trials against a wide range of human cancers (Cassady et al. 2004). This may well be due, at least in part, to dose-limiting toxicity. Because of their potency, however, maytansinoids are now of high interest as “warheads” in immunoconjugates with tumor specific antibodies (Chari et al., 1992; Liu et al., 1996), several of which are in clinical trials (Cassady et al., 2004). The most prominent of these “warheads” is a compound called DM1, developed by Immunogen, which is prepared from ansamitocin P3 by a series of chemical steps (Figure 13) (Chari et al., 1993; Chari and Widdison, 2001). However, the preparation of DM1 is very costly, not only because the starting material is expensive, but also the process is inefficient. The chemical esterification of maytansinol proceeds only in modest yield and under forcing conditions which result in racemization of the alanine moiety.
Figure 13.
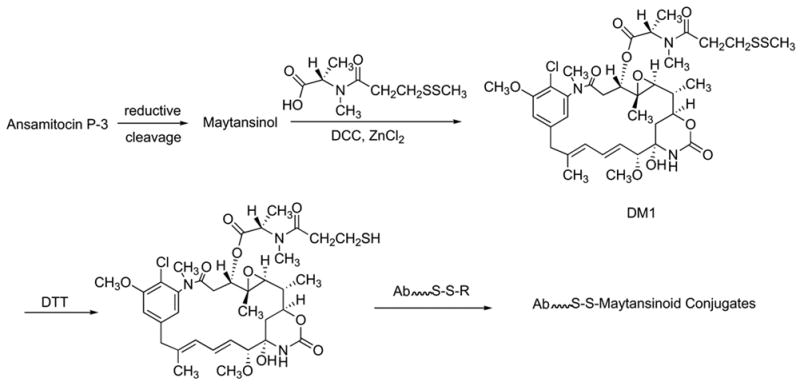
Synthesis of DM1 from ansamitocin P-3 and its conjugation to tumor-specific antibodies through disulfide linkages (Chari et al., 1993).
Since maytansinol is a minor constituent of the ansamitocin P-3 fermentation, it was thought that its esterification might be the terminal step in the biosynthesis, making Asm19 a candidate as catalyst for the enzymatic attachment of the DM1 side chain. Unfortunately, however, it was found that inactivation of asm19, led to the accumulation not of maytansinol, but of its N-demethyl-desepoxy analog (Moss et al., 2002). Exploration of the substrate specificity of Asm19 showed that it is rather promiscuous as far as the acyl moiety is concerned, confirming earlier observations (Hatano et al., 1984). However, neither alanine nor the entire maytansine side chain were attached to the ansamitocin backbone (Mahmud, T. and Floss, H. G., unpublished results). Furthermore, the enzyme will utilize a variety of pathway intermediates as substrate for the attachment of ester side chains, but not maytansinol (Moss et al., 2002). Thus, this enzyme in its native form is probably not useful for the preparation of DM1.
The analysis of the downstream processing reactions of ansamitocin biosynthesis showed that the first step is the introduction of the chlorine into proansamitocin, followed by the transfer of the carbamate group, then methylation of the aromatic hydroxyl group followed by attachment of the ester side chain. Epoxidation followed by N-methylation then complete the biosynthesis. However, many of the downstream processing enzymes show relaxed substrate specificity which results in a metabolic grid with, in addition to the major pathway, several minor alternative routes to the final product. Also as a result, many of the mutants accumulate not just one product but a series of products. It was also observed that the yields of intermediates accumulated by the various mutants were roughly an order of magnitude lower than the yields of ansamitocin P3 in the wild-type. Thus, structure modification of ansamitocins by manipulation of post-PKS modification reactions is a somewhat challenging prospect (Moss et al., 2002; Spiteller et al., 2003).
In conclusion, combinatorial biosynthesis in principle has considerable potential for drug discovery, optimization and production. However, as the above examples and many others illustrate, there are substantial problems to be overcome and much more work needs to be done to derive practical applications from this approach. Secondly, truly combinatorial applications of this technology will probably be less prominent than applications towards metabolic engineering of production of specific compounds. Finally, combinatorial biosynthesis/metabolic engineering should be viewed as a complement to, not a substitute for, the screening for natural product libraries and extracts.
Acknowledgments
This paper is based on a lecture presented at Bioperspectives 2005 in Wiesbaden, Germany, May 10, 2005. The author is greatly indebted to the many dedicated coworkers who carried out the experimental work from our laboratory. Their names are listed in the references cited. He also wishes to thank Mrs. Kay B. Kampsen for expertly transcribing the text of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Admiraal SJ, Walsh CT, Khosla C. The loading module of rifamycin synthase is an adenylation-thiolation didomain with substrate tolerance for substituted benzoates. Biochemistry. 2001;40:6116–6123. doi: 10.1021/bi010080z. [DOI] [PubMed] [Google Scholar]
- August PR, Tang L, Yoon YJ, Ning S, Müller R, Yu T-W, Taylor M, Hoffmann D, Kim C-G, Zhang X, Hutchinson CR, Floss HG. Biosynthesis of the ansamycin antibiotic rifamycin. Deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei. Chem Biol. 1998;5:69–79. doi: 10.1016/s1074-5521(98)90141-7. [DOI] [PubMed] [Google Scholar]
- Berdy J. Bioactive microbial metabolites – A personal view. J Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- Cane DE, Yang CC. Macrolide biosynthesis. 4 Intact incorporation of a chain-elongation intermediate into erythromycin. J Am Chem Soc. 1987;109:1255–1257. [Google Scholar]
- Carroll BJ, Moss SJ, Bai L, Kato Y, Toelzer S, Yu T-W, Floss HG. Identification of a set of genes involved in the formation of the substrate for the incorporation of the unusual “glycolate” chain extension unit in ansamitocin biosynthesis. J Am Chem Soc. 2002;124:4176–4177. doi: 10.1021/ja0124764. [DOI] [PubMed] [Google Scholar]
- Cassady JM, Chan KK, Floss HG, Leistner E. Recent developments in the maytansinoid antitumor agents. Chem Pharm Bull. 2004;51:1–26. doi: 10.1248/cpb.52.1. [DOI] [PubMed] [Google Scholar]
- Chari RJ, Widdison WC. Process for the preparation and purification of thiol-containing maytansinoids. 6333410 US Patent. 2001
- Chari RJ, Goldmacher VS, Lambert JM, Blättler WA. Cytotoxic agents comprising maytansinoids and their therapeutic use. 5208020 US Patent. 1993
- Chari RJ, Martell BA, Gross JL, Cook SB, Shah SA, Blättler WA, McKenzie SJ, Goldmacher VS. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res. 1992;52:127–131. [PubMed] [Google Scholar]
- Corbaz R, Ettlinger L, Gäumann E, Kalvoda J, Keller-Schierlein W, Kradolfer F, Manukian BK, Neipp L, Prelog V, Reusser P, Zähner H. Stoffwechselprodukte von Aktinomyceten. Granaticin Helv Chim Acta. 1957;40:1262–1269. [Google Scholar]
- DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J Antibiot. 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- Donadio S, Staver MJ, McAlpine JB, Swanson S, Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- Egorin MJ, Lagattuta TF, Hamburger DR, Covey JM, White KD, Musser SM, Eiseman JL. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother Pharmacol. 2002;49:7–19. doi: 10.1007/s00280-001-0380-8. [DOI] [PubMed] [Google Scholar]
- Eustaquio AS, Gust B, Li S-M, Pelzer S, Wohlleben W, Chater KF, Heide L. Production of 8’-halogenated and 8’-unsubstituted novobiocin derivatives in genetically engineered Streptomyces coelicolor strains. Chem Biol. 2004;11:1561–1572. doi: 10.1016/j.chembiol.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Floss HG, Yu T-W. Rifamycin – Mode of action, resistance, and biosynthesis. Chem Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- Guéritte-Voegelein F, Sénilh V, David B, Guénard D, Potier P. Chemical studies of 10-deacetyl baccatin III: Hemisynthesis of taxol derivatives. Tetrahedron. 1986;42:4451–4460. [Google Scholar]
- Harris DR, McGeachin SG, Mills HH. The structure and stereochemistry of erythromycin A. Tetrahedron Lett. 1965;11:679–685. doi: 10.1016/s0040-4039(00)90018-2. [DOI] [PubMed] [Google Scholar]
- Hatano K, Higashide E, Akiyama S-I, Yoneda M. Fermentative production and biosynthesis of ansamitocins. Part I Selective accumulation of ansamitocins P-2, P-3 and P-4, and biosynthetic origins of their acyl moieties. Agric Biol Chem. 1984;48:1721–1729. [Google Scholar]
- Higashide E, Asai M, Ootsu K, Tanida S, Kozai Y, Hasegawa T, Kishi T, Sugino Y, Yoneda M. Ansamitocin, a group of novel maytansinoid antibiotics with antitumor properties from Nocardia. Nature. 1977;270:721–722. doi: 10.1038/270721a0. [DOI] [PubMed] [Google Scholar]
- Hopwood DA, Malpartida F, Kieser HM, Ikeda H, Duncan J, Fujii I, Rudd BAM, Floss HG, Omura S. Production of “hybrid” antibiotics by genetic engineering. Nature. 1985;314:642–644. doi: 10.1038/314642a0. [DOI] [PubMed] [Google Scholar]
- Hunziker D, Yu T-W, Hutchinson CR, Floss HG, Khosla C. Primer unit specificity in rifamycin biosynthesis principally resides in the later stages of the biosynthetic pathway. J Am Chem Soc. 1998;120:1092–1093. [Google Scholar]
- Kato Y, Bai L, Xue Q, Revill WP, Yu T-W, Floss HG. Functional expression of genes involved in the biosynthesis of the novel polyketide chain extension unit, methoxymalonylacyl carrier protein, and engineered biosynthesis of 2-desmethyl-2-methoxy-6-deoxyerythronolide B. J Am Chem Soc. 2002;124:5268–5269. doi: 10.1021/ja0127483. [DOI] [PubMed] [Google Scholar]
- Kim C-G, Yu T-W, Fryhle CB, Handa S, Floss HG. 3-Amino-5-hydroxybenzoic acid (AHBA) synthase, the terminal enzyme in the formation of the precursor of mC7N units in rifamycin and related antibiotics. J Biol Chem. 1998;273:6030–6040. doi: 10.1074/jbc.273.11.6030. [DOI] [PubMed] [Google Scholar]
- Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, Gilmore CJ, Haltiwanger RC, Bryan RF. Tumor inhibitors. LXXIII Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc. 1972;94:1354–1356. doi: 10.1021/ja00759a054. [DOI] [PubMed] [Google Scholar]
- Liu C, Tadayoni BM, Bourret LA, Mattocks KM, Derr SM, Widdison WC, Kedersha NL, Ariniello PD, Goldmacher VS, Lambert JM, Blättler WA, Chari RVJ. Eradication of large colon tumor xenografts by targeted delivery of maytansinoids. Proc Nat Acad Sci USA. 1996;93:8618–8623. doi: 10.1073/pnas.93.16.8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzhetskyy A, Bechthold A. It works: combinatorial biosynthesis for generating novel glycosylated compounds. Mol Microbiol. 2005;58:3–5. doi: 10.1111/j.1365-2958.2005.04815.x. [DOI] [PubMed] [Google Scholar]
- Madduri K, Kennedy J, Rivola G, Inventi-Solari A, Filippini S, Zanuso G, Colombo AL, Gewain KM, Occi JL, MacNeil DL, Hutchinson CR. Production of the antitumor drug epirubicin (4’-epidoxorubicin) and its precursor by a genetically engineered strain of Streptomyces peucetius. Nature Biotechnol. 1998;16:69–74. doi: 10.1038/nbt0198-69. [DOI] [PubMed] [Google Scholar]
- Malpartida F, Hopwood DA. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature. 1984;309:462–464. doi: 10.1038/309462a0. [DOI] [PubMed] [Google Scholar]
- McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci USA. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Bai L, Toelzer S, Carroll BJ, Mahmud T, Yu T-W, Floss HG. Identification of Asm19 as an acyltransferase attaching the biologically essential ester side chain of ansamitocins using N-desmethyl-4,5-desepoxymaytansinol, not maytansinol, as its substrate. J Am Chem Soc. 2002;124:6544–6545. doi: 10.1021/ja020214b. [DOI] [PubMed] [Google Scholar]
- Rawlings BJ. Type I polyketide biosynthesis in bacteria (Part A – Erythromycin biosynthesis) Nat Prod Rep. 2001:190–227. doi: 10.1039/b009329g. [DOI] [PubMed] [Google Scholar]
- Reeves CD. The enzymology of combinatorial biosynthesis. Rev Biotechnol. 2003;23:95–147. doi: 10.1080/713609311. [DOI] [PubMed] [Google Scholar]
- Reeves CD, Chung LM, Liu Y, Xue Q, Carney JR, Revill WP, Katz L. A new substrate specificity for acyl transferase domains of the ascomycin polyketide synthase in Streptomyces hygroscopicus. J Biol Chem. 2002;277:9155–9159. doi: 10.1074/jbc.M111915200. [DOI] [PubMed] [Google Scholar]
- Rinehart KL., Jr Mutasynthesis of antibiotics. Develop in Biochem. 1981;19:71–91. [Google Scholar]
- Rix U, Fischer C, Remsing LL, Rohr J. Modification of post-PKS tayloring steps through combinatorial biosynthesis. Nat Prod Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- Schupp T, Toupet C, Engel N, Goff S. Cloning and sequence analysis of the putative rifamycin polyketide synthase gene cluster from Amycolatopsis mediterranei. FEMS Microbiol Lett. 1998;159:201–207. doi: 10.1111/j.1574-6968.1998.tb12861.x. [DOI] [PubMed] [Google Scholar]
- Sensi P, Margalith P, Timbal MT. Rifomycin, a new antibiotic; preliminary report. Farmaco, Ed Pr. 1959;14:146–147. [PubMed] [Google Scholar]
- Shier WT, Rinehart KL, Jr, Gottlieb D. Preparation of four new antibiotics from a mutant of Streptomyces fradiae. Proc Natl Acad Sci USA. 1969;63:198–204. doi: 10.1073/pnas.63.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteller P, Bai L, Shang G, Carroll BJ, Yu T-W, Floss HG. The post-polyketide synthase modification steps in the biosynthesis of the antitumor agent ansamitocin by Actinosynnema pretiosum. J Am Chem Soc. 2003;125:14236–14237. doi: 10.1021/ja038166y. [DOI] [PubMed] [Google Scholar]
- Stratmann A, Toupet C, Schilling W, Traber R, Oberer L, Schupp T. Intermediates of rifamycin polyketide synthase produced by an Amycolatopsis mediterranei mutant with inactivated rifF gene. Microbiol. 1999;145:3365–3375. doi: 10.1099/00221287-145-12-3365. [DOI] [PubMed] [Google Scholar]
- Staunton J, Wilkinson B. Biosynthesis of erythromycin and related macrolides. Compr Nat Prod Chem. 1999;1:495–532. [Google Scholar]
- Staunton J, Wilkinson B. Combinatorial biosynthesis of polyketides and nonribosomal peptides. Curr Opin Chem Biol. 2001;5:159–164. doi: 10.1016/s1367-5931(00)00185-x. [DOI] [PubMed] [Google Scholar]
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacological evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- Takano S, Hasuda K, Ito A, Koide Y, Ishii F. A new antibiotic, medermycin. J Antibiot. 1976;29:765–768. doi: 10.7164/antibiotics.29.765. [DOI] [PubMed] [Google Scholar]
- Walsh CT. Combinatorial biosynthesis of antibiotics: Challenges and opportunities. Chem Bio Chem. 2002;3:124–134. doi: 10.1002/1439-7633(20020301)3:2/3<124::AID-CBIC124>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- Wiley PF, Gerzon K, Flynn EH, Sigal MV, Jr, Weaver O, Quarck UC, Chauvette RR, Monahan R. Erythromycin, X Structure of erythromycin. J Am Chem Soc. 1957;79:6062–6070. [Google Scholar]
- Wu K, Chung L, Revill WP, Katz L, Reeves CD. The FK520 gene cluster of Streptomyces hygroscopicus var. ascomyceticus (ATCC 14891) contains genes for biosynthesis of unusual polyketide extender units. Gene. 2000;251:81–90. doi: 10.1016/s0378-1119(00)00171-2. [DOI] [PubMed] [Google Scholar]
- Xu J, Wan E, Kim C-J, Floss HG, Mahmud T. Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei. Microbiol. 2005;151:2515–2528. doi: 10.1099/mic.0.28138-0. [DOI] [PubMed] [Google Scholar]
- Yang J, Hoffmeister D, Liu L, Fu X, Thorson JS. Natural product glycorandomization. Bioorg Med Chem. 2004;12:1577–1588. doi: 10.1016/j.bmc.2003.12.046. [DOI] [PubMed] [Google Scholar]
- Yue S, Duncan JS, Yamamoto Y, Hutchinson CR. Macrolide biosynthesis. Tylactone formation involves the processive addition of three carbon units. J Am Chem Soc. 1987;109:1253–1255. [Google Scholar]
- Yu T-W, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Nat Acad Sci USA. 2002;99:7968–7973. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T-W, Shen Y, Doi-Katayama Y, Tang L, Park C, Moore BS, Hutchinson CR, Floss HG. Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc Nat Acad Sci USA. 1999;96:9051–9056. doi: 10.1073/pnas.96.16.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]