

Abstract
Reduced supply of glucose involves in many pathological conditions such as stroke and contributes to ischemic injuries. In contrast, hyperglycemia has also been regarded as an important factor in causing and exaggerating stroke damage. Although the molecular mechanism(s) of imbalanced glucose-induced cellular injuries under low oxygen conditions are not clear, oxidative stress has been implicated in both hypo- and hypeglycemic damage. Redox status is critical for the regulation of cellular signaling and cell survival. The effects of glucose levels on redox status are not well understood in neurons under hypoxia. The purpose of this study was to determine the effects of glucose concentration on the redox status of rat primary neurons under hypoxia. The cellular redox status was determined from GSH/GSSG ratios, and oxidation of 2,3-dichlorofluorscein diacetate was used to assess levels of reactive oxygen species (ROS). We found that glucose levels were critical in regulating redox state in these neurons under hypoxia. The results showed that under hypoxic conditions: 1) there was an optimal glucose concentration (25mM) at which neurons maintained a reducing environment and showed the lowest levels of ROS and cell death; 2) in the concentration range of 0–25 mM, the presence of glucose increased cellular GSH/GSSG ratio and reduced ROS and cell death; and 3) over-supply of glucose (25–100 mM) elevated ROS levels, produced an oxidizing oxidizing environment, and increased cell death. These results suggest that cellular redox status regulated by glucose may play an important role in glucose-mediated cellular responses in hypoxia.
Keywords: high glucose, glucose deprivation, redox status, neuron, stroke
Glucose is essential for neuronal function and metabolism. Interestingly, both glucose deprivation and exposure to elevated glucose levels have been shown to be toxic to cells. For example, simply removing glucose from cell culture media can induce cytotoxicity in human breast cancer cells [13] and cultured rat cortical neurons [26]. However, incubating with elevated glucose levels (45–100 mM) can also induce neuronal death [18, 20]. Besides glucose, oxygen is critical to the brain. Although the brain is only 2% of the body's weight, it uses 20% of the oxygen supply and accounts for approximately 20% of aerobic metabolism. There are pathological conditions that involve both abnormal oxygen and glucose levels resulting from their abnormal supplies and/or abnormal metabolism, such as ischemic stroke. Cerebral ischemia causes dynamic and heterogeneous alterations of oxygen and glucose supplies and cellular metabolism [6, 25]. Decreased level of glucose in ischemia is believed to play an important role in ischemic injury. In contrast to ischemic conditions where glucose concentration decreases, other pathophysiological conditions such as diabetes have elevated glucose concentrations. Hyperglycemia and diabetes are conditions known to aggravate neuronal damage in stroke patients and in animals subjected to transient ischemia [7, 27]. Thus, dysregulated glucose levels may act in concert with hypoxia to produce neuronal injuries. The mechanism(s) responsible for deleterious effects of abnormal levels of glucose in hypoxia such as in an ischemic brain are not fully understood.
Recently, cellular injury induced by both low and high glucose has been linked to the generation of reactive oxygen species (ROS) and oxidative stress. It has been reported that glucose deprivation causes perturbations in cellular sensitivity to oxidative stress, which is mediated by increased generation of pro-oxidants and decreased scavenging of free radicals [13]. Formation of hydrogen peroxide [10] as well as more general measurements of ROS generation [24] have been observed under high glucose conditions and are suggested to be responsible for high glucose-induced cell injury and/or death in non-neuronal cells. In primary dorsal root ganglion neurons, glucose (45 mM) rapidly induces a rise in ROS, and the toxicity induced by high glucose is blocked by inhibiting ROS induction [18]. In the studies described above, all measurements were made in normoxic conditions. ROS generation and oxidative stress has also been implicated in toxicity induced by low or high glucose under hypoxia in cerebral ischemia and diabetic and hyperglycemic mellitus [4, 15, 17].
The redox environment refers to the reduction potential or reducing capacity in cellular or other biological systems (see reference[19] for review), which is often represented by the term “redox status”. The redox environment plays a critical role in normal cellular function, including regulation of proliferation and differentiation, as well as processes such as apoptosis [9, 19]. However, the mechanism(s) by which the glucose concentration may affect cellular redox status and ROS generation under hypoxia has not been systematically investigated in neurons. Understanding the redox status of neurons in low and high glucose under hypoxic conditions should provide new insights into the mechanism of cell injuries induced by low or high glucose under hypoxic conditions such as ischemic stroke, which in turn may assist in designing effective and safe stroke treatment strategies. The objectives of the present study were to determine the effects of a wide range of glucose concentrations on cellular redox status in neurons under hypoxic conditions.
Cortical neurons from whole cerebral cortex of fetal rats (E=17 d) were prepared according to a published procedure [3] with modifications. Protocols involving the use of animals complied with the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee. Cells were seeded out in Becton Dickinson poly-L-lysine coated plates or dishes at the density of 1000 cells/mm2 and grown in Neurobasal medium (GibcoBRL, CA) supplemented with B27 (GibcoBRL, CA) in a humidified normoxic atmosphere containing 5% CO2 at 37 °C. The content of astroglial cells in the culture was less than 10%. Twice a week half of the medium was changed from the culture. For glucose and hypoxic treatments, primary cultured cells (DIV 12) were gently washed twice with pre-warmed phosphate buffered saline (PBS, pH 7.4), and then placed in DMEM with a range of glucose concentrations (0, 0.5, 5.5, 25, 50, and 100 mM), which had been gassed with nitrogen for 10 min. Cells were incubated at 37 °C for the desired time in a humidified hypoxia chamber (Billups-Rothenberg inc. Del Mar, CA) with 1% oxygen, 5% CO2 and balance nitrogen. The time points (3 and 5 hrs) were selected based on both our research data and others’ reports on in vitro hypoxia/ischemic studies. In the literature, many in vitro hypoxic/ischemic experiments were carried out with 2–8 hr treatments. Our previous study has shown cell death and GSH decrease in glioma cells after 3 hr treatment under in vitro ischemic condition. The test time point(s) were designed to be in the common tested-time frame and with the possibility to observe changes in reducing capacity, ROS level and cell death. The 50 mM and 100 mM concentrations were selected as high glucose concentrations for primary neurons, based on previous reports [18, 20–22], which have shown that these concentrations do not themselves have osmotic effects on neurons.
A widely-used indicator of cellular redox environment (status) (glutathione (GSH) and glutathione disulfide (GSSG) ratio) [19] was used to determine the effect of glucose levels on cellular redox status under low oxygen conditions. The ratio was measured with a kit from Cayman (kit Cat# 703002, Ann Arbor, MI), which uses a spectrophotometeric recycling assay to measure cellular levels of GSH and GSSG based on previous reports [5, 13]. Briefly, following treatment, neurons were scrape-harvested in cold PBS on ice and centrifuged. The cell pellets were frozen at −80 °C. Samples were thawed and whole homogenates were prepared as described [13]. Total glutathione (GSH + 2GSSG) and GSSG were measured, and then GSH levels and the ratios of GSH/GSSG were determined. All the determinations were normalized to protein content using the method of Lowry et al. [16].
Oxidative stress was monitored using the cell-permeable probe 2,3-dichlorofluorscein diacetate (DCFH-DA). The probe has been widely used to measure cellular H2O2 levels. However, it also reacts with other reactive species, such as hydroxyl radical and peroxynitrite, to generate the fluorescent molecule DCF. Thus, it has been suggested as a marker for overall reactive species level [12]. During hypoxia, primary neurons were exposed to 5 μM DCFH-DA. At the end of hypoxic exposures, neurons were washed twice with cold PBS and lysed with 0.2% Triton x-100 on ice. DCF fluorescence in the cellular lysates was measured with a Wallac Victor2 1420 multilabel counter (ex 488 nm; em 525 nm).
As a marker of cell viability, lactate dehydrogenase (LDH) levels were measured in the media of primary neurons submitted to the different experimental conditions. The measurements were conducted at 490 nm spectrophotometrically using a LDH detection kit (Takara Bio Inc., Shiga, Japan). Three sets of experiments were carried out, unless otherwise specified.
Results were expressed as mean ± SD. The statistical analysis was evaluated by unpaired Student's t-tests, with the level of significance chosen at p<0.05.
Figure 1 shows the effect of glucose concentration on cellular GSH level, GSSG level, and GSH/GSSG ratio (redox status) in neurons under hypoxic conditions. As shown in Figure 1A, glucose did not cause statistically significant changes in cellular GSH level in neurons cultured under hypoxia for 3 or 5 hrs. In contrast, glucose had a significant effect on cellular GSSG level in neurons under these culture conditions (Figure 1B). The effect was glucose concentration dependent. The lowest GSSG level was observed in neurons cultured with 25 mM glucose. In the range of 0–25 mM, the presence of glucose decreased cellular GSSG levels, while in the range 25–100 mM, increasing glucose concentration elevated GSSG levels. Figure 1C shows the effect of glucose on cellular GSH/GSSG ratio in neurons under hypoxia. The highest GSH/GSSG ratio was observed in neurons with 25 mM glucose after either 3 or 5 hr exposure to hypoxic condition. The presence of glucose increased the GSH/GSSG ratio in the range of 0–25 mM while it decreased the ratio in the range of 25–100 mM. These results indicate that exposure to both low and high glucose conditions can induce an oxidizing environment, and there is an optimal glucose concentration (25 mM under these experimental conditions) at which neurons maintain their reducing environment under hypoxia. It is noteworthy that a glucose concentration of 25 mM has been reported as an optimal concentration for normal primary neuron culture [3, 18]. In addition, the possible explanations for the relative constant GSH level and significantly altered GSSG level are: 1) GSSG level is relatively low, compare to GSH level, and 2) the amount of oxidized GSH was not great enough to result in statistically significant difference in GSH level, but was great enough to alter GSSG level significantly. This indicates that the ratio of GSH/GSSG is a better measure of cellular reducing capacity than the GSH level alone. While many investigators have used GSH level as an end point for cellular oxidative stress studies, if we had relied on GSH measures in the present study, the results would have suggested no effect of glucose on cellular reducing capacity under hypoxic conditions.
Figure 1.
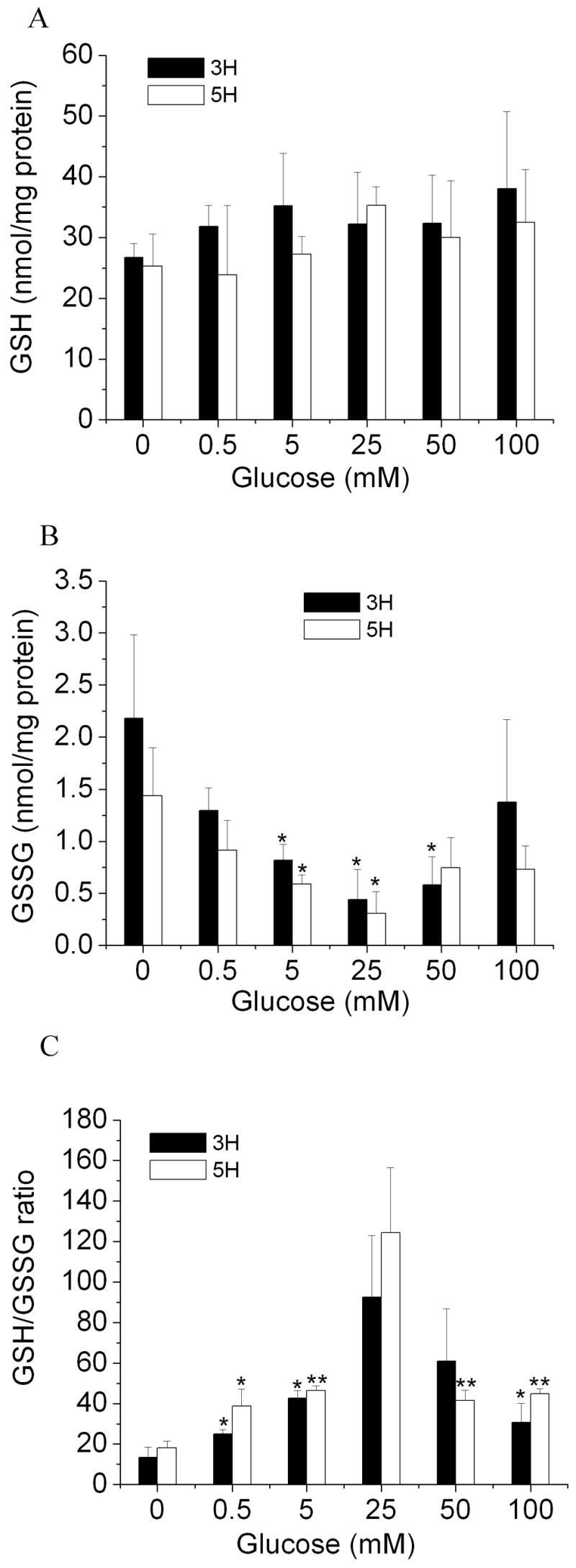
Effects of glucose on redox status (GSH/GSSG ratio) in rat primary cortical neurons under hypoxia. Neurons were exposed to various glucose concentrations under low oxygen condition (1%) for 3 and 5 hrs. A: cellular GSH level, B: cellular GSSG level, C: cellular GSH/GSSG ratio. All data are expressed as means ± SD of three independent determinations. *p < 0.05, **p < 0.01 compared with glucose at 0 mM.
To confirm the results describe above, we studied effect of glucose on cellular oxidative stress or ROS levels. The ROS levels in primary neurons were assessed with the fluorescent probe DCFH-DA. As shown in Figure 2, after 3 hrs of incubation, the glucose level had a substantial impact on intracellular ROS levels in neurons under these hypoxic conditions. Specifically, neurons cultured with 25 mM glucose had the lowest level of ROS. In the range of 0–25 mM, the presence of glucose caused a concentration-dependent decrease in ROS generation, suggesting that the presence of glucose over this concentration range is needed to maintain a more reducing environment. However, in the range of 25–100 mM, glucose elevated ROS level in neurons under hypoxia. These results suggest that either deprivation of glucose or exposure to elevated glucose can increase cellular ROS level in neurons under hypoxia, which is in line with the GSH/GSSG ratio observations described above (Figure 1C).
Figure 2.
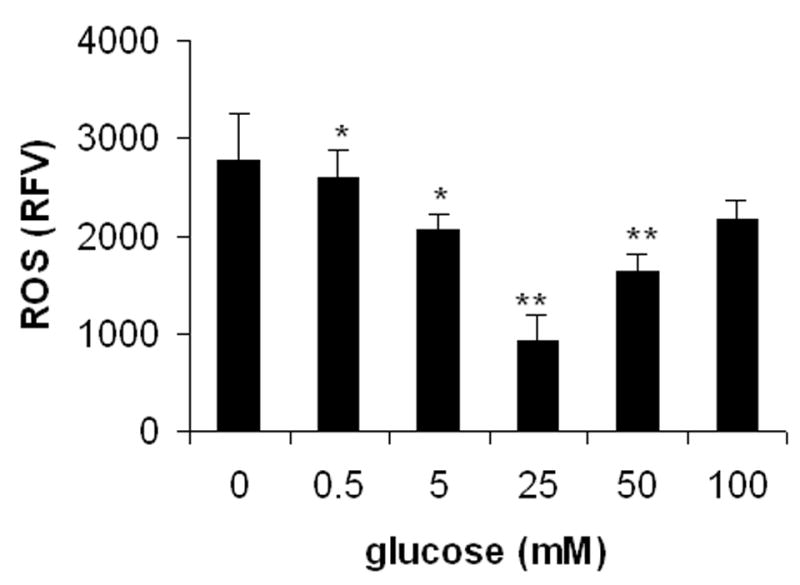
Effects of glucose on reactive oxygen species (ROS) levels in primary neurons under hypoxia. Neurons were exposed to various glucose concentrations under low oxygen condition (1%) for 3 hrs. ROS levels were evaluated by the measurement of oxidation of the probe 2,3-dichlorofluorscein diacetate (DCFH-DA). All data are expressed as means ± SD of four determinations. RFV: relative fluorescence value. *p < 0.05, **p < 0.01 compared with glucose at 0 mM.
To examine the effects of glucose concentration on the viability of primary cultured neurons, LDH release measurements were used. As shown in Figure 3, glucose induced a dose-dependent LDH release from neurons under hypoxic conditions. The lowest level of LDH release (indicating the highest rate of cell survival) was observed with 25 mM glucose. Cell survival decreased when glucose was either elevated or decreased from 25 mM.
Figure 3.
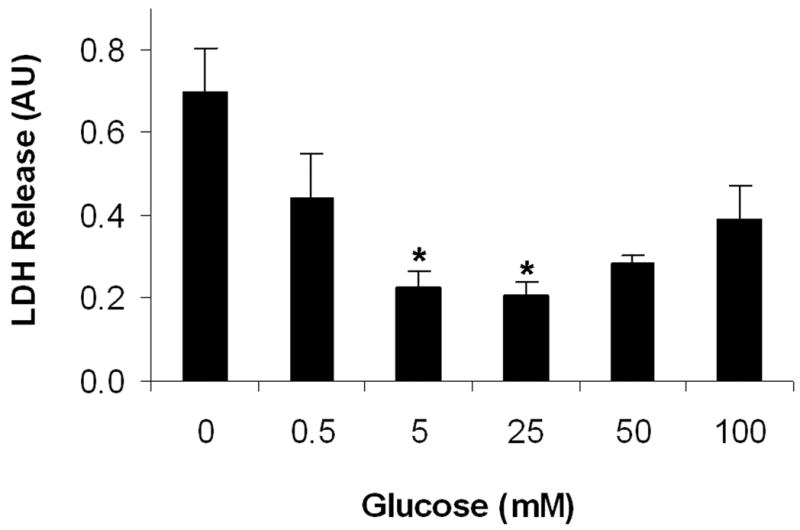
Effects of glucose on cell viability of primary neurons under hypoxia. Relative cell viability was measured by cellular LDH release, a commonly-used indicator for cell death. Neurons were exposed to various glucose concentrations under low oxygen condition (1%) for 3 hrs and an extended 24 hr culture. All data are expressed as means ± SD of three determinations. AU: arbitrary unite. *p < 0.05, compared with glucose at 0 mM.
The present study demonstrates that glucose levels have a great impact on cellular redox status, ROS levels, and cell viability in primary neurons under hypoxic conditions. Specifically, glucose has opposing effect on cellular redox status, depending on its concentration. There appear to be an optimal glucose concentration (~25 mM under these experimental conditions), which maintains an appropriate reducing environment, low ROS levels, and high cell viability in neurons under hypoxic condition. At concentrations below this optimal concentration, increasing glucose elevates cellular reducing capacity (GSH/GSSG ratio) and lowers ROS level and cellular damage, while beyond the optimal point, further increasing glucose concentration significantly decreases cellular reducing capacity (GSH/GSSG ratio) and increases intracellular ROS level and cell damage in neurons under hypoxic conditions.
Many studies have shown that primary neurons require a higher basal concentration of glucose (~25 mm) than non-neuronal cells (~5 mM), mainly due to the high metabolic rate and energy requirements of active neurons, when compared to non-neuronal cells [18]. Russell and coworkers showed that, under normoxia, glucose at concentrations either lower or higher than 25 mM induced programmed cell death though the induction of caspase 3 [18]. Thus, glucose at 25 mM appears an optimal condition for primary neuronal culture. Our results indicate that elevated level of the ROS and oxidative stress may contribute to the cell death in non-optimal conditions of glucose in normal cultured neurons. It also suggests that glucose at 25 mM is optimal for neurons under hypoxia.
The cellular redox environment is mainly determined by redox-regulated gene expression, free radical generation, and especially by the levels of antioxidants/reducing agents. Glucose is an important source of cellular reductants. Glucose provides the principal intracellular reductant NADPH through the pentose phosphate pathways (PPP), which is especially important in the brain [2, 11]. NADPH is essential in recycling GSSG to GSH. Thus, glucose appears to play a critical role in maintaining cellular redox environment under normoxic conditions, and the current result shows that glucose (in the concentration range 0–25mM) essentially plays the same role in maintaining cellular reducing environment in neurons subjected to hypoxia. In fact, it has been reported that glucose is being actively utilized through the PPP during hypoxia [1, 28], and that the PPP pathway is more activated under hypoxic than normoxic conditions [8]. This may explains, at least in part, the observation that increasing glucose in the range of 0–25 mM increases GSH/GSSG ratio and lowers ROS levels. Cerebral ischemia results in a central region, which has little or no blood flow (ischemic core), and a peripheral region (penumbra), which experience only a restriction in flow [14]. It has been demonstrated that ROS levels in the ischemic core (glucose deprivation) is greater than that in penumbra (glucose still available but at decreased level) [15, 26]. This in vivo result is consistent to our findings that under low oxygen, glucose (not at high concentration) is critical in maintaining cellular reducing environment and reduces ROS level.
In contrast to the effects of glucose at low concentrations, exposure to high glucose concentrations can transform neuronal redox state to a more oxidizing environment, and elevate ROS levels during hypoxia. Many previous studies have shown increased oxidative status in various cells submitted to hyperglycemic conditions under normoxia, based either on direct quantifications of ROS levels or determinations of oxidative end products [10, 23]. Our observations suggest that high glucose also significantly increases neuronal oxidative stress under hypoxic conditions. Many factors have been suggested to contribute to ROS generation mediated by high glucose in normoxic conditions, including glucose autoxidation, advanced glycation end products formation, PKC-dependent activation of NADPH oxidase, and abnormal mitochondrial metabolism. Although the mechanism (s) of high glucose-induced oxidative stress under hypoxia is not yet known, it may involve similar pathways to those described previously in normoxia. For example, PKC, which can increase superoxide anion radical generation through activation of NADPH oxidase, is highly activated in high glucose with hypoxic condition [29]. In addition, our observation is in agreement to results from a previous in vivo study, which showed that hyperglycemia increases superoxide anion radical and peroxynitrite productions in global cerebral ischemia [17].
The effects of glucose concentration on neuronal injury in cerebral ischemia are complex. On the one hand, glucose deprivation is harmful to brain cells, but it is also clear that glucose can contribute to neuronal injury in cerebral ischemia due to acidosis resulting from glucose metabolism. On the other hand, hyperglycemia apparently increases stroke occurrence and exaggerates stroke outcomes. Some experimental evidence has further complicated this issue, by demonstrating a neuroprotective effect of glucose. For example, either normal glucose or elevated glucose concentrations have been found to be neuroprotective in neuronal cultures [21, 22]. Although a single mechanism to explain these complex effects of glucose has been lacking, changes in antioxidant and ROS levels have been implicated in the harmful and beneficial effect of glucose in cerebral ischemia. The current observation, using a wide range of glucose concentrations, suggests a potential underlying mechanism to explain how glucose may regulate redox status in neurons under hypoxia. The result suggests that glucose concentration may be a key player in determining its effect on cells and tissues under hypoxia by the regulation of cellular redox status.
Taken together, these data present the first direct evidence that glucose concentration is critical for the regulation of redox status in neurons under hypoxia. Depending on its concentration, glucose can be either an important contributor to the maintenance of cellular redox status or a factor that transforms neurons to an oxidizing environment under hypoxic conditions.
Acknowledgments
This research was supported in part by grants from NIH (P20 RR15636) and AHA (0565598Z). We thank Dr. William Shuttleworth of Department of Neurosciences, UNM, for editing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002;81:207–17. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Yoseph O, Boxer PA, Ross BD. Oxidative stress in the central nervous system: monitoring the metabolic response using the pentose phosphate pathway. Dev Neurosci. 1994;16:328–36. doi: 10.1159/000112127. [DOI] [PubMed] [Google Scholar]
- 3.Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–83. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- 4.Cao W, Carney JM, Duchon A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett. 1988;88:233–8. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- 5.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–9. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furlan M, Marchal G, Viader F, Derlon JM, Baron JC. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–26. doi: 10.1002/ana.410400213. [DOI] [PubMed] [Google Scholar]
- 7.T.D.C.a.C.T.R. Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 8.Gupte SA, Okada T, McMurtry IF, Oka M. Role of pentose phosphate pathway-derived NADPH in hypoxic pulmonary vasoconstriction. Pulm Pharmacol Ther. 2006;19:303–9. doi: 10.1016/j.pupt.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Hiroi M, Ogihara T, Hirano K, Hasegawa M, Morinobu T, Tamai H, Niki E. Regulation of apoptosis by glutathione redox state in PC12 cells exposed simultaneously to iron and ascorbic acid. Free Radic Biol Med. 2005;38:1057–72. doi: 10.1016/j.freeradbiomed.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation. 2000;101:2618–24. doi: 10.1161/01.cir.101.22.2618. [DOI] [PubMed] [Google Scholar]
- 11.Hotta SS. Glucose metabolism in brain tissue: the hexosemonophosphate shunt and its role in glutathione reduction. J Neurochem. 1962;9:43–51. doi: 10.1111/j.1471-4159.1962.tb07491.x. [DOI] [PubMed] [Google Scholar]
- 12.Huang HM, Zhang H, Ou HC, Chen HL, Gibson GE. alpha-keto-beta-methyl-n-valeric acid diminishes reactive oxygen species and alters endoplasmic reticulum Ca(2+) stores. Free Radic Biol Med. 2004;37:1779–89. doi: 10.1016/j.freeradbiomed.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Jacob C, Maret W, Vallee BL. Ebselen, a selenium-containing redox drug, releases zinc from metallothionein. Biochem Biophys Res Commun. 1998;248:569–73. doi: 10.1006/bbrc.1998.9026. [DOI] [PubMed] [Google Scholar]
- 14.Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–53. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Liu M, Peterson S, Miyake M, Vallyathan V, Liu KJ. Hydroxyl radical formation is greater in striatal core than in penumbra in a rat model of ischemic stroke. J Neurosci Res. 2003;71:882–8. doi: 10.1002/jnr.10534. [DOI] [PubMed] [Google Scholar]
- 16.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 17.Muranyi M, Li PA. Hyperglycemia increases superoxide production in the CA1 pyramidal neurons after global cerebral ischemia. Neurosci Lett. 2006;393:119–21. doi: 10.1016/j.neulet.2005.09.079. [DOI] [PubMed] [Google Scholar]
- 18.Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. Faseb J. 2002;16:1738–48. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- 19.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 20.Semra YK, Smith NC, Lincoln J. Comparative effects of high glucose on different adult sympathetic neurons in culture. Neuroreport. 2004;15:2321–5. doi: 10.1097/00001756-200410250-00004. [DOI] [PubMed] [Google Scholar]
- 21.Seo SY, Kim EY, Kim H, Gwag BJ. Neuroprotective effect of high glucose against NMDA, free radical, and oxygen-glucose deprivation through enhanced mitochondrial potentials. J Neurosci. 1999;19:8849–55. doi: 10.1523/JNEUROSCI.19-20-08849.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo SY, Kim EY, Kim H, Jou I, Gwag BJ. Attenuation of neuronal death by NMDA and oxygen-glucose deprivation in cortical neurons maintained in high glucose. Ann N Y Acad Sci. 1999;893:396–9. doi: 10.1111/j.1749-6632.1999.tb07864.x. [DOI] [PubMed] [Google Scholar]
- 23.Sharpe PC, Liu WH, Yue KK, McMaster D, Catherwood MA, McGinty AM, Trimble ER. Glucose-induced oxidative stress in vascular contractile cells: comparison of aortic smooth muscle cells and retinal pericytes. Diabetes. 1998;47:801–9. doi: 10.2337/diabetes.47.5.801. [DOI] [PubMed] [Google Scholar]
- 24.Sharpe PC, Yue KK, Catherwood MA, McMaster D, Trimble ER. The effects of glucose-induced oxidative stress on growth and extracellular matrix gene expression of vascular smooth muscle cells. Diabetologia. 1998;41:1210–9. doi: 10.1007/s001250051054. [DOI] [PubMed] [Google Scholar]
- 25.H Shi, K Liu. Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Frontiers Biosci. doi: 10.2741/2150. in press. [DOI] [PubMed] [Google Scholar]
- 26.Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA. Hypoglycemic Neuronal Death and Cognitive Impairment Are Prevented by Poly(ADP-Ribose) Polymerase Inhibitors Administered after Hypoglycemia. J Neurosci. 2003;23:10681–10690. doi: 10.1523/JNEUROSCI.23-33-10681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warner DS, Todd MM, Dexter F, Ludwig P, McAllister AM. Temporal thresholds for hyperglycemia-augmented ischemic brain damage in rats. Stroke. 1995;26:655–60. doi: 10.1161/01.str.26.4.655. [DOI] [PubMed] [Google Scholar]
- 28.White CW, Jackson JH, McMurtry IF, Repine JE. Hypoxia increases glutathione redox cycle and protects rat lungs against oxidants. J Appl Physiol. 1988;65:2607–16. doi: 10.1152/jappl.1988.65.6.2607. [DOI] [PubMed] [Google Scholar]
- 29.Young TA, Wang H, Munk S, Hammoudi DS, Young DS, Mandelcorn MS, Whiteside CI. Vascular endothelial growth factor expression and secretion by retinal pigment epithelial cells in high glucose and hypoxia is protein kinase C-dependent. Exp Eye Res. 2005;80:651–62. doi: 10.1016/j.exer.2004.11.015. [DOI] [PubMed] [Google Scholar]