

Abstract
Endothelial cells regulate vascular integrity and express complement binding proteins including gC1qR/p33 (gC1qR), which recognize C1q, a subunit of the first component of the classical complement pathway. Experiments were performed to investigate classical complement pathway activation on resting endothelial cells and endothelial cells exposed to shear stress. C1q deposition and C4 activation (C4d) were demonstrated by solid phase ELISA and flow cytometry on human microvascular and umbilical vein endothelial cells after exposure to serum or plasma. C4d deposition was accompanied by downstream complement activation including C3b and C5b-9 deposition. C4 activation failed to occur in C1q depleted serum, but was not affected by Factor B depleted serum, confirming classical complement pathway activation. Moreover, C4 activation occurred following exposure of endothelial cells to purified C1 and C4, in the absence of other plasma proteins, and in the absence of detectable cell surface IgG and IgM. Shear stress (18 dynes/cm2) increased C1q (n=9, p<0.05) and C4d (n=9, p<0.05) deposition approximately 2-fold, and enhanced endothelial cell gC1qR expression (n=7, p<0.05). Treatment of endothelial cells with anti gC1qR monoclonal antibody F(ab’)2 fragments reduced C4d deposition by approximately 20% (n=5, p<0.05). These data demonstrate direct classical complement pathway activation on endothelial cells. gC1qR appears to play a minor but definable role, whereas cell surface IgG or IgM are not required.
Keywords: complement, gC1qR, classical pathway, shear stress
Introduction
Atherosclerosis and its related thrombotic complications are leading causes of cardiac death and stroke in developed countries. Recent studies have shown that the development of atherosclerotic lesions is associated with inflammation (Peerschke, Minta et al. 2004; Acosta, Qin et al. 2004;Ross 1999;Tiong and Brieger 2005) and related thrombosis (Libby, Ridker et al. 2002). Complement activation has been demonstrated in atherosclerotic lesions, and is considered an important contributor in the development of pathological plaques (Bhakdi 1998;Niculescu, Niculescu et al. 2004;Torzewski, Bowyer et al. 1997). A number of complement proteins have been identified in atherosclerotic lesions, including C1q (Niculescu and Rus 1999;Yasojima, Schwab et al. 2001), the recognition unit of the classical complement pathway.
Vascular endothelial cells express several C1q binding sites, including cC1qR/calreticulin and gC1qR/p33 (gC1qR) (Eggleton, Tenner et al. 2000;Ghebrehiwet and Peerschke 2004). Among these, gC1qR is the most versatile. gC1qR binds C1q via its globular head domain, and also recognizes high molecular weight kininogen and factor XII, with the capacity to activate the kinin system (Joseph, Tholanikunnel et al. 2004;Reddigari, Shibayama et al. 1993) and coagulation cascade (Peerschke and Ghebrehiwet 1998). Recently purified gC1qR was found to directly activate the classical complement pathway (Ghebrehiwet, CebadaMora et al. 2006).
Vascular endothelial cells (EC) play an important role in regulating local hemostasis and thrombosis (Saadi, Holzknecht et al. 2000). Given the ability of gC1qR to interact with components of complement, coagulation, and kinin systems, endothelial cell gC1qR may play a crucial role in local inflammatory reactions and thrombotic complications related to atherosclerosis (Ghebrehiwet and Peerschke 2004;Guo, Ghebrehiwet et al. 1999).
Complement activation and endothelial cell damage are involved in the pathogenesis of atherosclerosis. However, the mechanism of complement activation in atherosclerotic lesions, the effect of blood flow/shear stress on this process, and the participation of gC1qR have not been investigated. The present study was designed to evaluate direct classical complement pathway activation on endothelial cells and access the role of gC1qR.
Methods
Cell Culture
Bone marrow (BMEC) (Schweitzer, Vicart et al. 1997) and brain (Weksler, Subileau et al. 2005) microvascular endothelial cell lines were used between passage 14 – 30. BMEC were grown to confluence on 0.2% gelatin (Sigma-Aldrich Corp., St. Louis, MO), and maintained in Dulbecco’s Modified Eagle Media (DMEM), supplemented with 5% fetal bovine serum (FBS), 10 mM HEPES (hydroxyethyl piperazine ethanesulfonic acid), and 1:100 Penicillin/Streptomycin (10,000 unit Penicillin and 10,000 μg/ml Streptomycin) (Invitrogen Corp, Carlsbad, CA). Human brain microvascular endothelial cells were grown to confluence on 1:20 collagen type I (Rat Tail collagen I, Becton Dickinson, Lincoln Park, NJ), and maintained in Endothelial Cell Basal Medium-2 (EBM-2) (Cambrex Corporation, NJ) supplemented with 5% FBS, 1:1000 human fibroblast growth factor (hFGF), 1:1000 vascular endothelial cell growth factor (VEGF), 1:1000 Ascorbic Acid, 1:1000 h-epidermal growth factor and 50μg/ml Gentamicin Sulfate (Cambrex Corporation, NJ). Primary human umbilical vein endothelial cells (HUVEC) (ScienCell Research Laboratories, San Diego CA) (passage 2 and 3) were grown to confluence on 1% gelatin, and maintained in endothelial cell medium (ECM) (ScienCell Research Laboratory) supplemented with 5% FBS, 1:5 Penicillin/Streptomycin (ScienCell Research Laboratory) and 1:5 endothelial cell growth supplement (ECGS) (ScienCell Research Laboratory).
All endothelial cells were detached with 1X trypsin/EDTA (Invitrogen Corp, Carlsbad, CA). Trypsin was neutralized with excess culture medium. Cells were washed by centrifugation at 150g for 5 minutes, and the cell pellet was suspended in HEPES buffered modified Tyrode’s solution (HMBT) containing 1 mM CaCl2 and 2 mM MgCl2 (Peerschke, Smyth et al. 1996), or gelatin containing Veronal buffer with magnesium and calcium (GVB++) (Boston BioProducts Inc., Worcester, MA).
Plasma/Serum
Platelet poor plasma (PPP), anticoagulated with 0.32% sodium citrate and normal human serum (NHS) were prepared as described previously (Peerschke, Yin et al. 2006). Since activation of complement by thrombin and plasmin, two enzymes present in serum, has been described (Schaiff and Eisenberg 1997;Huber-Lang, Sarma et al. 2006), preliminary studies compared complement activation on endothelial cells following incubation with plasma or serum. Dilution (1/10) of anticoagulated plasma in divalent cation containing buffers (HBMT or GVB++) assured the presence of sufficient calcium and magnesium to support complement activation.
Antibodies
A murine monoclonal antibody, designated 74.5.2 (Ghebrehiwet, Lu et al. 1996;Peerschke, Yin et al. 2006), against gC1qR was used at 5 – 100 μg/ml throughout the study. Biotinylated monoclonal anti-human C1q, and C4d antibodies, as well as monoclonal anti-human iC3b and scC5b-9 were obtained from Quidel Corporation (San Diego, CA), and used at dilutions of 1:200 (~ 1 – 5 μg/ml) in HBMT. Polyclonal anti-human C1q and C4 antibodies were raised in our laboratory. These antibodies were biotinylated (Nguyen, Ghebrehiwet et al. 2000), and used at dilutions of 1:200 (about 1–2 μg/ml) in GVB++.
Classical pathway complement activation on endothelial cells
Deposition of C1q and C4d, a degradation product of C4b, on endothelial cells was measured by solid phase ELISA or flow cytometry. For ELISA, endothelial cell suspensions were immobilized on poly-L-lysine (Sigma-Aldrich Corp., St. Louis, MO) (10 μg/ml) coated 96-well microtiter plates (Becton Dickinson, Lincoln Park, NJ) by centrifugation (1000 g, 5 minutes) (~ 2 × 104 cells/well). Adherent cells were fixed with 0.5% glutaraldehyde, followed by neutralization with 100 mM glycine-0.1% bovine serum albumin (BSA, 99%, fatty acid free, Sigma-Aldrich). After washing, endothelial cells were exposed (1 hour, 37°C) to PPP, NHS, or C1q- (classical pathway) or factor B-(alternative pathway) depleted serum (Quidel Corporation, San Diego, CA) diluted (1:10) in HBMT. In separate experiments, BMEC were exposed to the combination of purified C1 (5 μg/ml) (The Binding Site, Inc.) and C4 (5 μg/ml) (Quidel Corporation) in GVB++ for 60 min at 37°C. Bound complement components were detected by addition of biotinylated anti-C1q, biotinylated anti C4d, anti iC3b, or anti scC5b-9 antibodies, as needed. Primary antibody binding was detected using alkaline phosphatase conjugated streptavidin (Immunopure Streptavidin, Pierce Biotechnology Inc.) or an alkaline phosphatase conjugated goat anti mouse antibody, followed by addition of 1 mg/ml p-nitrophenyl phosphate substrate (pNPP) (Pierce Biotechnology, Inc.). Color development was quantified at 405 nm (reference at 490 nm) using a Thermomax microplate reader (Molecular Devices Corp, Palo, Alto, CA). Background complement deposition was measured on endothelial cells exposed to HBMT instead of plasma or serum.
To rule out artifacts of endothelial cell fixation, complement activation was evaluated by flow cytometry of endothelial cell suspensions in HBMT incubated with PPP (1:10) at 37 °C for 45 minutes. After washing, classical pathway complement activation was measured using biotinylated anti-human C4d (30 min at 25°C), detected with Delight 647 conjugated streptavidin (Pierce Biotechnology Inc.). MOPC 21 (IgG1k) (Sigma-Aldrich) was used as a nonimmune primary antibody control. Background C4d deposition was assessed on endothelial cells exposed to HMBT instead of plasma.
gC1qR expression on endothelial cells
gC1qR expression on endothelial cells was measured by solid phase ELISA or flow cytometry, using murine monoclonal antibody, 74.5.2 (Ghebrehiwet, Lu et al. 1996).
Effects of gC1qR blockade on endothelial cell surface complement activation
C1q and C4d deposition were measured by ELISA, after preincubation of endothelial cells (1 hour, 37°C) with 74.5.2 F(ab’)2 fragments (5 μg/ml). Antibody fragments were prepared with ImmunoPure IgG Fab and F(ab’)2 preparation Kit from Pierce Biotechnology Inc.
Endothelial cell surface IgG/IgM measurement
Rabbit anti human IgG (Sigma Aldrich) and monoclonal anti human IgM (Sigma Aldrich) were used to measure endothelial cell surface IgG and IgM deposition by solid phase ELISA before and after endothelial cell exposure to plasma.
Immunohistochemical localization of gC1qR and C activation products on endothelial cells
BMEC were grown to confluence on Type I collagen (0.013 mg/cm2) (Dardik, Chen et al. 2005) (Sigma-Aldrich), to maintain anchorage on the culture plate during staining. After fixation, confluent cells were incubated (1 hour at 37°C) with 74.5.2 (1:100 in HBMT) for gC1qR detection. MOPC-21 was used as a control antibody. After 1-hour incubation with PPP, complement deposition on EC was evaluated using murine anti-human C1q, C4d or iC3b antibodies. Primary antibody binding was detected using a biotinylated horse anti mouse antibody followed by streptavidin-peroxidase reagent (Vecta stain Elite ABC kit; Vector Laboratories Inc., Burlingame, CA). The reaction was developed with 3,3’-diaminobenzidine solution. The cells were counterstained with Hematoxylin and examined by light microscopy.
Endothelial cell exposure to elevated shear stress
Endothelial cells were grown to confluence on Type I collagen. Endothelial cell monolayers were placed in a cone-and-plate shearing device (Matise Medical, Israel) and exposed to constant shear stress of 18 dyne/cm2 for 1 hour in culture medium.
Statistics
The paired student t-test was used to analyze data obtained from all experiments.
Results
Complement activation on endothelial cells
Complement activation occurred on endothelial cells exposed to either normal human plasma or serum. Significant deposition of C1q (n=4, P<0.05) and C4d (n=4, P<0.05) was noted using bone marrow (BMEC) and brain microvascular endothelial cell lines, as well as primary HUVEC by solid phase ELISA (Figure 1, Table 1) or flow cytometry (Figure 2). Complement activation proceeded to completion with evidence of C3b and C5b-9 formation (Table 1). Classical complement activation failed to occur in C1q- but not Factor B-deficient serum (Table 1). Although C1q binding was not affected by divalent cation chelation, no C4d deposition occurred in the presence of 13 mM EDTA (n=4, P<0.05) (Figure 1). C1q-dependent C4d deposition on endothelial cells was partially inhibited by gC1qR blockade with 74.5.2 F(ab’)2 fragments (n=5, p< 0.05) (Table 1).
Figure 1.
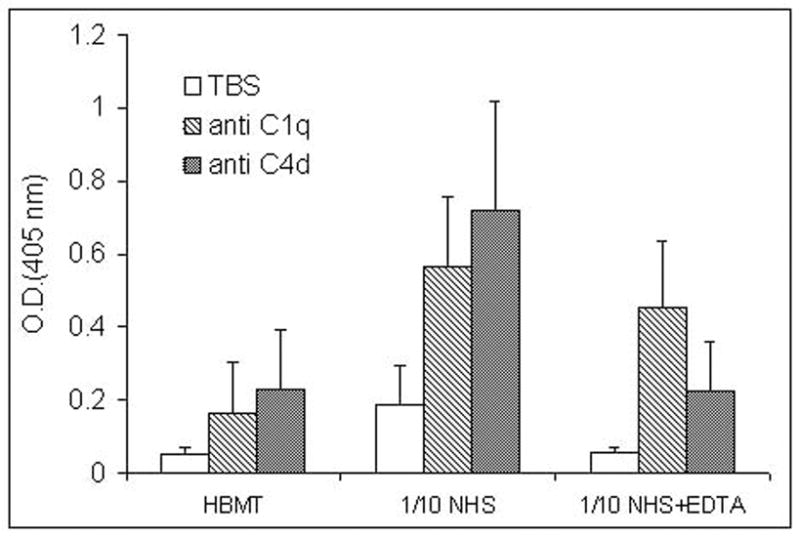
Complement activation on BMEC assessed by solid phase ELISA. BMEC were exposed to NHS (1/10) with or without 13 mM EDTA for 60 min at 37°C. Complement activation was measured using antibodies against C1q and C4d. The data represent O.D. at 405 nm (mean ± S.D., n=4).
Table 1.
Complement activation on human endothelial cells.
| Cells/Conditions | Antibody Binding (Ratio Relative to Control) | |||
|---|---|---|---|---|
| Anti C1q | Anti C4d/C4 | Anti iC3b | Anti scC5b-9 | |
| BMEC/Plasma | 1 | 1 | 1 | 1 |
| HUVEC/Plasma (n=3) | 0.80 ± 0.16 | 0.90 ± 0.28 | 0.92 ± 0.22 | 0.84 ± 0.32 |
| Brain micro-vascular EC/Plasma (n=3) | 1.18 ± 0.20 | 0.89 ± 0.36 | 1.27 ± 0.20 | 0.85 ± 0.19 |
| BMEC/74.5.2 F(ab’)2 Plasma (n=6) | NA | 0.77 ± 0.15* | NA | NA |
| BMEC/C1q depleted Serum (n=3) | 0.012 ± 0.03* | 0.006 ± 0.04* | 0.238 ± 0.12* | −0.016 ± 0.20* |
| BMEC/Factor B depleted Serum (n=3) | 0.93 ± 0.24 | 0.83 ± 0.17 | 1.01 ± 0.19 | 0.79 ± 0.33 |
| BMEC/NHS (n=4) | 1.35 ± 0.65 | 1.36 ± 0.56 | 0.88 ± 0.09 | 0.76 ± 0.29 |
Complement activation on cultured endothelial cells derived from different vascular beds was investigated using plasma or serum to assess the involvement of classical pathway (C1q-deficient plasma), alternative pathway (Factor B depleted serum) and gC1qR (74.5.2). The data are expressed as mean ± S.D., reflecting changes in antigen expression relative to resting BMEC (control).
denotes statistical significance p<0.05
NA – study not performed.
Figure 2.
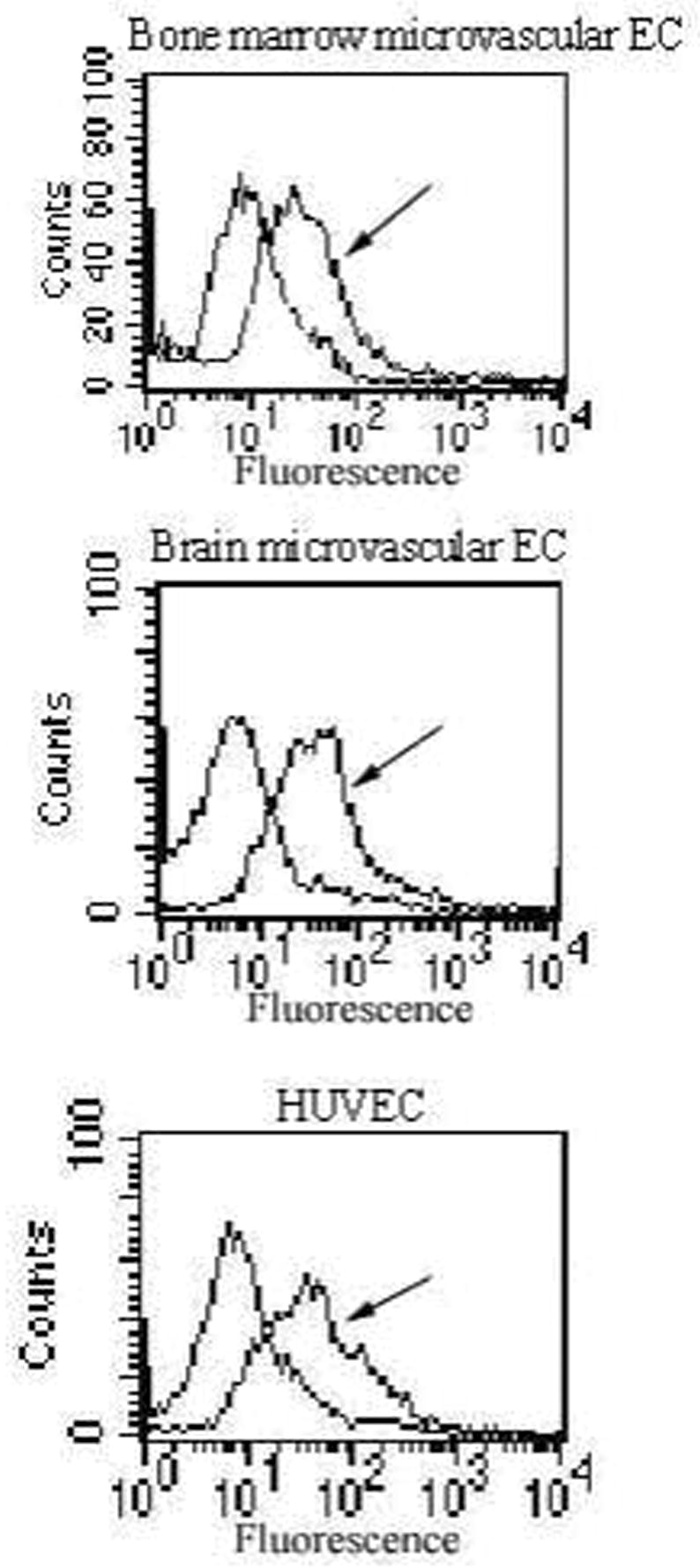
Flow cytometry measurement of C1q-dependent C4d deposition on endothelial cells (EC) in suspension. After exposure to human plasma or buffer, C1q-dependent C4 activation was quantified using a monoclonal antibody against C4d. Results demonstrate a shift in fluorescence following endothelial cell (BMEC, brain microvascular EC and HUVEC) exposure to plasma (arrow).
Endothelial cell ace IgG or IgM was not required for classical complement activation. surf No surface IgG or IgM expression was detected over background on endothelial cells before incubation with plasma. Under these conditions, the addition of purified C1 and C4 to endothelial cells, in the absence of plasma or serum, resulted in marked C1q binding (O.D. at 405 nm: 0.65 ± 0.25, n=3) and C4d deposition (O.D. at 405 nm: 2.21 ± 0.19, n=3).
Endothelial cell exposure to plasma resulted in low levels of IgG but no IgM binding, as reported by others (Devaraj, Du Clos et al. 2005). However, corresponding levels of purified immobilized IgG failed to activate complement. Moreover, the amount of endothelial cell surface associated IgG post plasma exposure was approximately 4-fold lower than the lowest amount of immobilized IgG required to activate C4, and was equivalent to only 50% of that seen on endothelial cells.
Complement activation on endothelial cell exposed to shear stress
A moderate but detectable level of gC1qR expression was described previously on resting BMEC (Guo, Ghebrehiwet et al. 1999) and HUVEC (Mahdi, Madar et al. 2002;Peerschke, Smyth et al. 1996). The present study demonstrates a significant increase in gC1qR expression following endothelial cell exposure to shear stress (n=7, p<0.05). This was accompanied by increased deposition of C1q (n=9, p<0.05) and C4d (n=9, p<0.05) (Figure 3).
Figure 3.
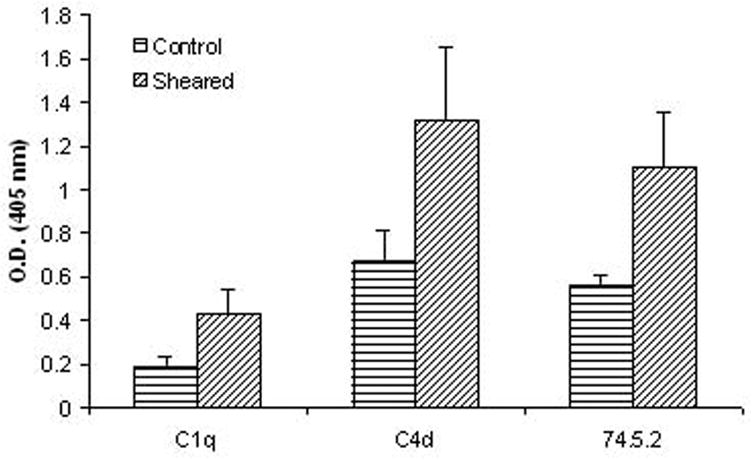
Effect of shear stress (18 dynes/cm2 for 1 hour) on complement activation and BMEC surface gC1qR expression. Significant increases in endothelial cell surface gC1qR expression (n=7, p<0.05), C1q (n=9, p<0.05) and C4d (n=9, p<0.05) deposition were noted after cell exposure to shear stress.
Cellular distribution of gC1qR and activated complement components on endothelial cells
Homogeneous endothelial cell staining for gC1qR was noted following immunohistochemical analysis (Figure 4A). Interestingly, focal staining for C4d and iC3b (Figure 4 C, D, as indicated by arrows) was seen. Staining of EC for C1q showed a mixed pattern of predominantly focal deposition with fainter homogenous staining in the background (Figure 4B). Control endothelial cells treated with buffer (HBMT) instead of plasma (Figure 4E) are depicted for reference.
Figure 4.
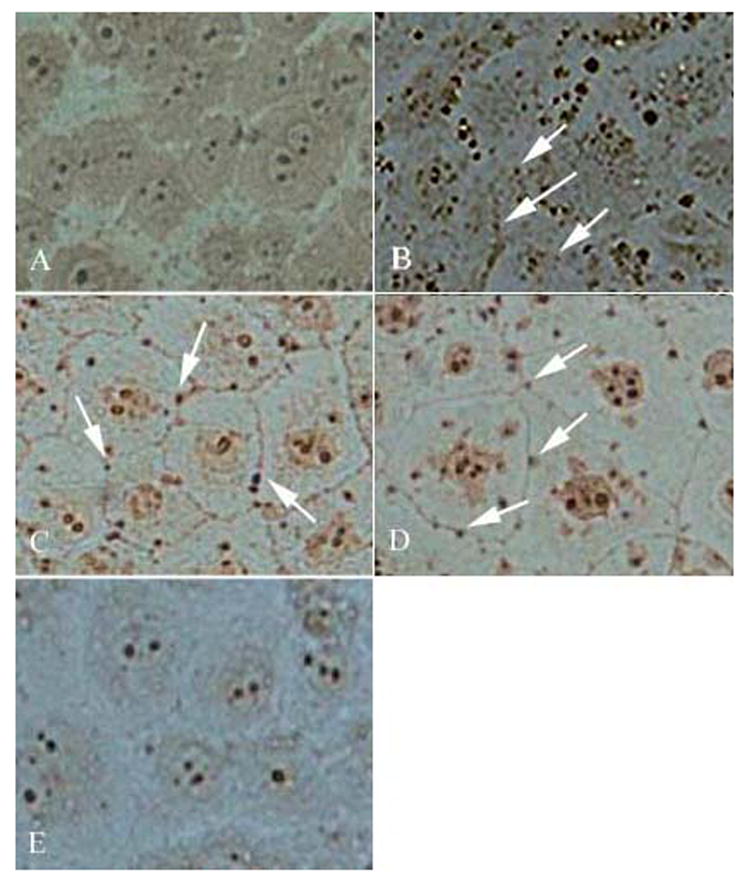
Immunohistochemical staining of BMEC, comparing the distribution of gC1qR (A), C1q (B), C4d (C) and iC3b (D) following exposure to plasma (1/10) (60 min at 37°C). Control BMEC staining with a nonimmune murine control antibody, is shown in panel E. Brown/black stain precipitate is indicative of antibody binding. Arrows denote examples of focal complement deposition along cell boundaries.
Discussion
The present study provides evidence of direct classical pathway complement activation on human endothelial cells. Low levels of complement activation were detected on endothelial cells that were not deliberately stimulated beyond trypsinization and/or immobilization on poly-L-lysine surfaces. However, significant increases in C4d activation occurred following endothelial cell exposure to elevated shear stress. These findings suggest that low level complement activation may occur on endothelial cells with regulation by cell surface and plasma inhibitors (Acosta, Qin, and Halperin 2004), and that enhanced complement activation beyond the control of natural inhibitors may be induced following endothelial cell activation or injury by pathological blood flow.
C1q-dependent complement activation on endothelial cells proceeded to completion with deposition of C3b and C5b-9. Activation of C4 and particularly C3 and C5 leads to the release of highly active inflammatory peptides, such as anaphylatoxins C3a and C5a, which induce local chemotactic and pro-inflammatory responses (Jagels, Daffern et al. 2000). In addition, formation of C5b-9 on the cell surface may lead to cell activation and/or lysis (Fosbrink, Niculescu et al. 2005). Thus, complement activation can contribute directly to local tissue destruction, inflammation, and atherogenesis. In this regard, Speidl et al. (Speidl, Exner et al. 2005) recently reported that plasma C5a levels were predictive of cardiovascular risk in patients with advanced atherosclerosis.
Further studies investigated the mechanism of complement activation on endothelial cells. gC1qR, an endothelial cell C1q binding protein, recognizes the globular domains of C1q (Ghebrehiwet, Lu et al. 1996), and was found recently to directly activate the classical complement pathway (Ghebrehiwet, CebadaMora et al. 2006). gC1qR expression on endothelial cells was previously shown to be upregulated by inflammatory cytokines (Guo, Ghebrehiwet et al. 1999), and in the present study by shear stress, parallelling increased C4 activation. Interestingly, immunohistochemical analyses demonstrated uniform gC1qR distribution on fixed endothelial cells compared to focal C4d and iC3b deposition. The distribution of C1q was mixed, with evidence for both homogenous staining and focal deposition. These findings suggest that complement activation may occur in permissive membrane microenvironments on the endothelial cell surface depending on the presence of cofactors and/or the absence of cell surface regulators.
gC1qR blockade by monoclonal antibody 74.5.2 F(ab’)2 fragments, resulted in consistent but only partial inhibition of C4d deposition on endothelial cells. 74.5.2 F(ab’)2 fragments also failed to fully inhibit C4 activation on purified gC1qR, suggesting either potential low binding efficiency of 74.5.2 F(ab’)2 fragments, or that additional C1q binding domains may reside within gC1qR and/or on the endothelial cell membrane, providing additional triggers for classical complement pathway activation. Alternatively, endothelial cell mediated complement activation may occur via expression of other C1q recognition motifs, such as membrane phosphatidyl serine (Botto and Walport 2002;Ogden and Elkon 2006). Failure to detect endothelial surface IgG or IgM, and the ability of endothelial cells to activate C4 in the absence of plasma, following addition of purified C1 and C4, demonstrate that IgG/IgM is not required for endothelial cell mediated classical complement pathway activation.
In conclusion, results from the present study provide evidence that endothelial cells support IgG/IgM independent classical complement pathway activation, in part via gC1qR. These findings extend previous observations, describing the presence of gC1qR (Peerschke, Minta et al. 2004), C1q and other activated complement products (Niculescu and Rus 1999;Yasojima, Schwab et al. 2001) in atherosclerotic lesions. Regulation of classical pathway complement activation may constitute a potential therapeutic adjunct in patients with vascular injury and/or atherosclerosis.
Acknowledgments
This work was supported in part by grants HL67211from the National Institutes of Health, Heart Lung and Blood Institute (EIP), and an American Heart Association Heritage Affiliate post doctoral award # 0625900T (WY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Acosta J, Qin X, Halperin J. Complement and complement regulatory proteins as potential molecular targets for vascular diseases. Curr Pharm Des. 2004;10:203–211. doi: 10.2174/1381612043453441. [DOI] [PubMed] [Google Scholar]
- 2.Bhakdi S. Complement and atherogenesis: the unknown connection. Ann Med. 1998;30:503–507. doi: 10.3109/07853899809002596. [DOI] [PubMed] [Google Scholar]
- 3.Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205:395–406. doi: 10.1078/0171-2985-00141. [DOI] [PubMed] [Google Scholar]
- 4.Dardik A, Chen L, Frattini J, Asada H, Aziz F, Kudo FA, Sumpio BE. Differential effects of orbital and laminar shear stress on endothelial cells. J Vasc Surg. 2005;41:869–880. doi: 10.1016/j.jvs.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359–1363. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- 6.Eggleton P, Tenner AJ, Reid KB. C1q receptors. Clin Exp Immunol. 2000;120:406–412. doi: 10.1046/j.1365-2249.2000.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fosbrink M, Niculescu F, Rus H. The role of c5b-9 terminal complement complex in activation of the cell cycle and transcription. Immunol Res. 2005;31:37–46. doi: 10.1385/IR:31:1:37. [DOI] [PubMed] [Google Scholar]
- 8.Ghebrehiwet B, CebadaMora C, Tantral L, Jesty J, Peerschke EI. gC1qR/p33 serves as a molecular bridge between the complement and contact activation systems and is an important catalyst in inflammation. Adv Exp Med Biol. 2006;586:95–105. doi: 10.1007/0-387-34134-X_7. [DOI] [PubMed] [Google Scholar]
- 9.Ghebrehiwet B, Lu PD, Zhang W, Lim BL, Eggleton P, Leigh LE, Reid KB, Peerschke EI. Identification of functional domains on gC1Q-R, a cell surface protein that binds to the globular “heads” of C1Q, using monoclonal antibodies and synthetic peptides. Hybridoma. 1996;15:333–342. doi: 10.1089/hyb.1996.15.333. [DOI] [PubMed] [Google Scholar]
- 10.Ghebrehiwet B, Peerschke EI. cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol. 2004;41:173–183. doi: 10.1016/j.molimm.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Guo WX, Ghebrehiwet B, Weksler B, Schweitzer K, Peerschke EI. Up-regulation of endothelial cell binding proteins/receptors for complement component C1q by inflammatory cytokines. J Lab Clin Med. 1999;133:541–550. doi: 10.1016/s0022-2143(99)90183-x. [DOI] [PubMed] [Google Scholar]
- 12.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 13.Jagels MA, Daffern PJ, Hugli TE. C3a and C5a enhance granulocyte adhesion to endothelial and epithelial cell monolayers: epithelial and endothelial priming is required for C3a-induced eosinophil adhesion. Immunopharmacology. 2000;46:209–222. doi: 10.1016/s0162-3109(99)00178-2. [DOI] [PubMed] [Google Scholar]
- 14.Joseph K, Tholanikunnel BG, Ghebrehiwet B, Kaplan AP. Interaction of high molecular weight kininogen binding proteins on endothelial cells. Thromb Haemost. 2004;91:61–70. doi: 10.1160/TH03-07-0471. [DOI] [PubMed] [Google Scholar]
- 15.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 16.Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002;99:3585–3596. doi: 10.1182/blood.v99.10.3585. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun. 2000;68:2061–2068. doi: 10.1128/iai.68.4.2061-2068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niculescu F, Rus H. Complement activation and atherosclerosis. Mol Immunol. 1999;36:949–955. doi: 10.1016/s0161-5890(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 19.Niculescu F, Niculescu T, Rus H. C5b-9 terminal complement complex assembly on apoptotic cells in human arterial wall with atherosclerosis. Experimental and Molecular Pathology. 2004;76:17–23. doi: 10.1016/j.yexmp.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr Dir Autoimmun. 2006;9:120–142. doi: 10.1159/000090776. [DOI] [PubMed] [Google Scholar]
- 21.Peerschke EI, Ghebrehiwet B. Platelet receptors for the complement component C1q: implications for hemostasis and thrombosis. Immunobiology. 1998;199:239–249. doi: 10.1016/S0171-2985(98)80030-2. [DOI] [PubMed] [Google Scholar]
- 22.Peerschke EI, Minta JO, Zhou SZ, Bini A, Gotlieb A, Colman RW, Ghebrehiwet B. Expression of gC1q-R/p33 and its major ligands in human atherosclerotic lesions. Mol Immunol. 2004;41:759–766. doi: 10.1016/j.molimm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 23.Peerschke EI, Smyth SS, Teng EI, Dalzell M, Ghebrehiwet B. Human umbilical vein endothelial cells possess binding sites for the globular domain of C1q. J Immunol. 1996;157:4154–4158. [PubMed] [Google Scholar]
- 24.Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. 2006;4:2035–2042. doi: 10.1111/j.1538-7836.2006.02065.x. [DOI] [PubMed] [Google Scholar]
- 25.Reddigari SR, Shibayama Y, Brunnee T, Kaplan AP. Human Hageman factor (factor XII) and high molecular weight kininogen compete for the same binding site on human umbilical vein endothelial cells. J Biol Chem. 1993;268:11982–11987. [PubMed] [Google Scholar]
- 26.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 27.Saadi S, Holzknecht RA, Patte CP, Platt JL. Endothelial cell activation by pore-forming structures: pivotal role for interleukin-1alpha. Circulation. 2000;101:1867–1873. doi: 10.1161/01.cir.101.15.1867. [DOI] [PubMed] [Google Scholar]
- 28.Schaiff WT, Eisenberg PR. Direct induction of complement activation by pharmacologic activation of plasminogen. Coron Artery Dis. 1997;8:9–18. doi: 10.1097/00019501-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Schweitzer KM, Vicart P, Delouis C, Paulin D, Drager AM, Langenhuijsen MM, Weksler BB. Characterization of a newly established human bone marrow endothelial cell line: distinct adhesive properties for hematopoietic progenitors compared with human umbilical vein endothelial cells. Lab Invest. 1997;76:25–36. [PubMed] [Google Scholar]
- 30.Speidl WS, Exner M, Amighi J, Kastl SP, Zorn G, Maurer G, Wagner O, Huber K, Minar E, Wojta J, Schillinger M. Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur Heart J. 2005;26:2294–2299. doi: 10.1093/eurheartj/ehi339. [DOI] [PubMed] [Google Scholar]
- 31.Tiong AY, Brieger D. Inflammation and coronary artery disease. American Heart Journal. 2005;150:11–18. doi: 10.1016/j.ahj.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Torzewski J, Bowyer DE, Waltenberger J, Fitzsimmons C. Processes in atherogenesis: complement activation. Atherosclerosis. 1997;132:131–138. doi: 10.1016/s0021-9150(97)00100-7. [DOI] [PubMed] [Google Scholar]
- 33.Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male DK, Roux F, Greenwood J, Romero IA, Couraud PO. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 34.Yasojima K, Schwab C, McGeer EG, McGeer PL. Complement components, but not complement inhibitors, are upregulated in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2001;21:1214–1219. doi: 10.1161/hq0701.092160. [DOI] [PubMed] [Google Scholar]