

Abstract
Splicing of eukaryal intron-containing tRNAs requires the action of the heterotetrameric splicing endonuclease, which is composed of two catalytic subunits, Sen34 and Sen2, and two structural subunits, Sen15 and Sen54. Here we report the solution structure of the human tRNA splicing endonuclease subunit HsSen15. To facilitate the structure determination, we removed the disordered 35 N-terminal and 15 C-terminal residues of the full-length protein to produce HsSen15(36–157). The structure of HsSen15(36–157), the first for a subunit of a eukaryal splicing endonuclease, revealed that the protein possesses a novel homodimeric fold. Each monomer consists of three α-helices and a mixed antiparallel/parallel β-sheet, arranged in a topology similar to that of the C-terminal domain of Methanocaldococcus jannaschii endonuclease. The dimeric interface is dominated by a β-barrel structure, formed by face-to-face packing of two, three-stranded β-sheets. Each of the β-sheets results from reciprocal parallel pairing of one β-strand from one subunit with two other β-strands from the symmetric subunit. The structural model provides insights into the functional assembly of the human tRNA splicing endonuclease.
Keywords: tRNA splicing endonuclease, NMR solution structure, homodimeric protein assembly beta barrel dimer interface
Introduction
The genes from all three taxonomic kingdoms that encode tRNA molecules contain intron sequences.1 These intron sequences must be removed from the precursor tRNAs (pre-tRNA) in order to generate mature and functional tRNAs. Despite the essentiality of this step, different splicing mechanisms have evolved. In bacteria, group I and group II introns are self spliced by a coordinated, auto-catalytic mechanism.2 In Archaea and Eukarya, the splicing of tRNA introns requires the serial actions of three protein-based enzymes: a splicing endonuclease that recognizes and cleaves the pre-tRNA,3–5 a ligase that joins the tRNA exons,6–8 and a 2′-phosphotransferase that transfers the residual 2′-phosphate group to nicotinamide adenine dinucleotide.9,10
Given the recent structural and biochemical evidence for conservation of their active sites,11,12 the eukaryal and archaeal splicing endonucleases likely use a similar mechanism to cleave their tRNA substrates.13 However, their substrate recognition properties are different.14,15 All archaeal endonucleases recognize the bulge-helix-bulge (BHB1) motif at the intron-exon junction of tRNA,4 rRNA,16,17 and mRNA.18 The BHB motif is two-fold pseudo-symmetric, with two three-nucleotide bulges separated by a four-base-pair helix.19 This local structure is essential for the recognition by archaeal endonucleases, whereas the presence of the mature tRNA domain is not required.20,21 By contrast, the eukaryal endonuclease appears to recognize the splice sites via the “ruler mechanism”,22 in which the endonuclease locates the cleavage sites by “measuring” the distance between an anchor site in the mature domain and the intron-exon junctions. Although the 3′ splice sites of eukaryal tRNA genes are invariably defined by bulge loops,23 similar to those in the archaeal pre-tRNAs, their splice sites exhibit little sequence conservation.
Much of our knowledge about the structural features of the tRNA splicing endonucleases has been gained from studies of archaeal endonucleases. In accordance with the pseudo-two-fold symmetry of the BHB substrates, all the archaeal splicing endonucleases identified to date adopt an architecture that places two catalytic sites in symmetric positions: as homodimers (α2) in some Euryarchaea (e.g. Archeoglobus fulgidus (AF));21,24 as homotetramers (α4) in other Euryarchaea (e.g. Methanocaldococcus jannaschii (MJ));25,26 and as heterodimeric homodimers (α2β2) in Crenarchaeota (e.g. Sulfolobus solfataricus (SS), and Sulfolobus tokodaii ( ST)) 27,28. Because each monomer of the AF endonuclease (AF_endo) actually contains two homologous repeats,24,26 the organization of all euryarchaeal endonucleases can be generalized into the “four-subunits” architecture represented by the MJ endonuclease (MJ_endo):25 this endonuclease is organized as a dimer of dimers that exhibits two-fold symmetry; each dimer contains one catalytic subunit and one structural subunit, associated by pairing of their last β-strands (β9); the two dimers further associate into a tetramer by electrostatic interactions. The detailed assembly of crenarchaeal endonucleases is yet to be determined. However, it has been demonstrated that the catalytic subunits of the SS endonuclease (SS_endo) and the ST endonuclease (ST_endo) (PDB accession number: 2CV8) both form a stable homodimer in solution,27 through pairing of their β9-strands. This implicated an asymmetric assembly mechanism between the catalytic subunits (endo1) and the structural subunits (endo2) of crenarchaeal endonucleases,27 different from what is observed for euryarchaeal endonucleases.24 Furthermore, the heterotetrameric SS_endo is also able to recognize a broader range of substrates (e.g. the noncanonical bulge-helix-loop (BHL) motif) than the homotetrameric enzymes, suggesting the coevolution of the endonuclease architecture and their RNA substrates.28,29
The eukaryal endonuclease consists of four different subunits (Sen2, Sen15, Sen34, and Sen54) with the likely stoichiometry 1:1:1:1.5,30 Sen2 and Sen34, which are responsible, respectively, for the 5′ and 3′ cleavage, show strong sequence similarity to MJ_endo in their active site domains.5,31 Sen15 and Sen54, which are otherwise non-homologous, contain C-terminal sequences similar to the stretch of MJ_endo that spans the last two β-strands (β8 and β9) and enclosed loop (l10) of its 3D structure.5,25 Yeast two-hybrid analysis further showed strong interactions between Sen2 and Sen54 and between Sen34 and Sen15; these results support the hypothesis that Sen15 and Sen54 play regulatory roles in tRNA splicing.5 On the basis of these observations, it was proposed that the eukaryal endonuclease assembles in a fashion similar to MJ_endo.25 In this assembly model, the interaction between each catalytic subunit and its corresponding structural subunit is mediated by pairing of the conserved last β-strands; the two resulting dimers further associate to form the heterotetramer through the electrostatic interactions contributed by the l10-equivalent loops of the structural subunits. Because the “ruler mechanism” is primarily seen in 5′ site recognition, it has been proposed that the highly basic subunit Sen54 defines the molecular ruler of the human endonuclease.5
Detailed structural elucidation of each subunit of the eukaryal endonuclease complex is crucial to testing models for its assembly and function. Here, we report the solution structure of human endonuclease Sen15 subunit, HsSen15(36–157). The structure reveals a novel dimeric fold, with a βbarrel dimer interface formed by two three-stranded β-sheets. The structure of the homodimeric HsSen15(36–157) implicates a distinct assembly mechanism for human endonuclease.
Results and discussion
Construct refinement
The 1H,15N-HSQC NMR spectrum of full-length [15N]-HsSen15, indicated that it contained many disordered residues. Analysis of the amino acid sequence of HsSen15 by the program PSIPRED,32 suggested that residues from both the N- and C-terminus would be disordered. To facilitate the NMR structural study, a construct was produced in which the 35-residue N-terminus and the 14-residue C-terminus were removed. 1H,15N-HSQC NMR spectrum of the truncated protein, HsSen15(36–157), exhibited structural features of the full-length protein, but without appreciable interference from the disordered residues. As noted above this construct contained an additional N-terminal serine residue as an artifact of the TEV cleavage site. Because of its improved NMR spectral properties, we chose HsSen15(36–157) for the solution structure determination.
Evidence for the homodimer
Four lines of experimental evidence indicated that HsSen15(36–157) is a homodimer. First, the gel filtration profiles of both full-length HsSen15 and HsSen15(36–157) indicated a dimer rather than a monomer. Second, the rotational correlation time (14.7 ns) estimated from backbone 15N relaxation measurements, was equivalent to that for a known 22 kD homodimeric protein.33 Third, the translational diffusion coefficient (0.85 x 10-6 cm2/s), measured by pulsed field gradient NMR experiments, fell in the range expected for a 17–37 kD protein.34 Fourth, NOE studies of the asymmetrically labeled protein revealed interactions between subunits and identified the interface (see below).
Structure determination
To determine the solution structure of the homodimeric HsSen15(36–157), a monomeric structural model of HsSen15(36–157) was generated first by using the iterative structure refinement module35 of the CYANA software package;36 these calculations also yielded an initial set of intramolecular NOE assignments. Unambiguous intermolecular distance constraints, derived from the 3D 13C,15N-filtered/13C-edited 1H,1H-NOESY spectrum of the 13C,15N/12C,14N mixed sample, were then added to define the homodimeric interface. Concomitant with manual spectral inspection, multiple rounds of NOE assignment corrections were performed, followed by structure recalculations. A final set of 100 conformers were calculated from 2,203 intramolecular NOEs (per monomer), 55 intermolecular NOEs (per monomer), 163 φ and Ψ dihedral angle constraints (per monomer) and 46 hydrogen bonds (per monomer). The 20 conformers with the lowest target functions were further refined in explicit solvent37 by using the XPLOR program38. At this stage of structural refinement, the set of 1DNH RDCs measured for HsSen15(36–157) were also added to the experimental constraints. In addition, non-crystallographic symmetry constraints were used for the residues in the α-helices or β-strands to ensure the molecular symmetry; these were omitted for regions within the homodimer manifesting internal dynamics. The final structural ensemble contained no NOE violation greater than 0.5Å and no dihedral angle violation greater than 5°. The 90 1DNH RDC values, which were fitted to the structural model of HsSen15(36–157) by the program PALES39, yielded a correlation coefficient R = 0.99 (Figure 1). One of the three principal axes of the alignment tensor was coincident with the two-fold symmetry axis of the dimeric structure; this further confirmed the orientation of the symmetry axis. PROCHECK 40 was used to analyze the quality of the structures, and MOLMOL41 was used to evaluate r.m.s.d. values. The structural statistics are summarized in Table 1.
Figure 1.
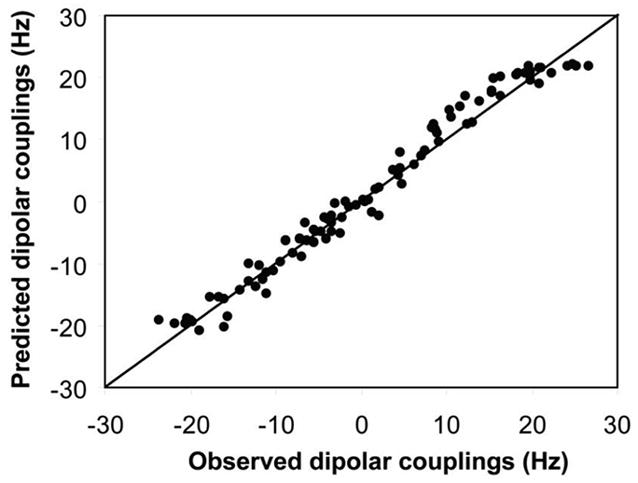
Correlation between the observed backbone NH dipolar couplings and couplings predicted from the alignment tensor obtained from the singular value decomposition (SVD) fit to the lowest-energy conformer of HsSen15(36–157) selected from the 20 conformers derived from the set of constraints that included residual dipolar couplings (RDCs).
Table 1.
Statistics for the 20 NMR-derived conformers that represent the solution structure of HsSen15(36–157)
| Experimental constraints (per monomer) | |
|---|---|
| Distance constraints | |
| Long (intramolecuar) | 431 |
| Long (intermolecular) | 55 |
| Medium [1<(i–j)≤5] | 401 |
| Sequential [2 (i–j)=1] | 519 |
| Intraresidue [i=j] | 852 |
| Dihedral angle constraints (ϕ and Ψ) | 163 |
| Hydrogen bonds | 46 |
| DNH residual dipolar couplings | 94 |
|
| |
| Target function ( Å2) | 2.68 ± 0.49 |
|
| |
| Average atomic R.M.S.D. to the mean structure (Å) | |
|
| |
| Dimer (residues 10–122 of the construct) | |
| Backbone ( Cα, C′, N, O) | 1.09 ± 0.19 |
| Heavy atoms | 1.45 ± 0.15 |
| Monomer I (residues 10–122 of the construct) | |
| Backbone ( Cα, C′, N, O) | 0.78 ± 0.12 |
| Heavy atoms | 1.21 ± 0.10 |
| Monomer II (residues 10–122 of the construct) | |
| Backbone ( Cα, C′, N, O) | 0.82 ± 0.12 |
| Heavy atoms | 1.25 ± 0.10 |
| Deviations from idealized covalent geometry | |
| Bond (Å) | 0.018 ± 0.001 |
| Angles (°) | 1.901 ± 0.089 |
| Impropers (°) | 1.744 ± 0.091 |
|
| |
| R.M.S.D. from experimental distance constraints (Å) | 0.019 ± 0.003 |
|
| |
| R.M.S.D. from experimental dihedral constraints (Å) | 0.468 ± 0.064 |
|
| |
| Ramachandran statistics (% of all residues) | |
|
| |
| Most favored | 84.23 ± 1.63 |
| Additionally allowed | 12.72 ± 1.61 |
| Generously allowed | 2.47 ± 0.90 |
| Disallowed | 0.58 ± 0.47 |
Description of the structure
The monomeric subunit of HsSen15(36–157) adopts a compact α/β fold, arranged in an α-α-β-β-β-β-αβ-β topology (Figure 2). The first eight residues in the N-terminus are disordered, as evidenced by both chemical shift and NOE analysis. The short α1-helix (P44–M50) is associated with the α2-helix (L57–T72) by antiparallel coiled-coiled interactions. The subsequent β1 (E78–L84), β2 (L89–T95), β3 (T104–P109), β5 (S138–V144) and β6 (I150–T156) fold into a mixed parallel/antiparallel β-sheet. In forming this, two orthogonal folding units, β1-β2 and β5-β6, are patched together by the tilted strand β3. Consequently, one face of the β-sheet curls into a shallow groove, cradling α2 as the result of hydrophobic interactions among A63, V66, Y67, and L70 from α2, and V79 and L92 from the β-sheet. The hairpin loop l7 (connecting β5 and β6) is pulled toward the C-terminus of α2 by a hydrogen bond between the carboxyl group of D69 and the hydroxyl group in the aromatic ring of Y152. The opposite face of the β-sheet is covered by the α3-helix (H116-L129). The short β4 (S113-S115), which bends away from the plane of the central β-sheet, has little to do with intramolecular interactions.
Figure 2.
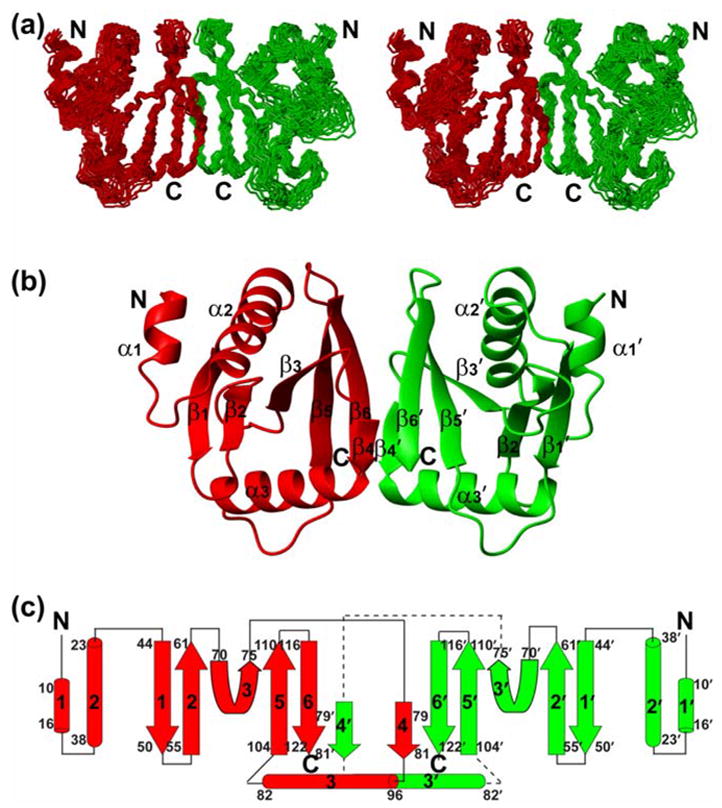
NMR solution structure of the homodimeric HsSen15(36–157). (a) Stereoscopic view of the backbone trace of the final family of 20 conformers representing the structure with individual subunits colored red and green. (b) Ribbon diagram of the HsSen15(36–157) structure, with individual elements of secondary structure labeled. For clarity, the disordered nine-residue N-terminal tag is not shown. (c) Schematic of the secondary structure topology. The color schemes in (b) and (c) are the same as in (a).
The dimer interface of HsSen15(36–157) is dominated by a six-stranded β-barrel structure (Figure 3(a)). The β6 from one monomer joins in parallel with the β4′ from the symmetric subunit, extending the mixed β-sheet in the monomer to six continuous strands across the dimer. The two β5-β6-β4′sheets then pack face-to-face to form a compact β-barrel stabilized predominantly by hydrophobic interactions, burying 717 Å2 of surface area. Inside this β-barrel, the hydrophobic core, centered on the pair of Y153 aromatic rings from the two subunits, is surrounded by the side chains of I110, A112, L114, I143, V151, and L155 from both subunits (Figure 3(b)). Analysis by VAST (http://www.ncbi.nlm.nih.gov/Structure/VAST/vast.shtml) failed to identify any suitable match between the dimeric fold of HsSen15(36–157) and structures in the Protein Data Bank. Thus, HsSen15(36–157) represents a novel dimeric fold.
Figure 3.
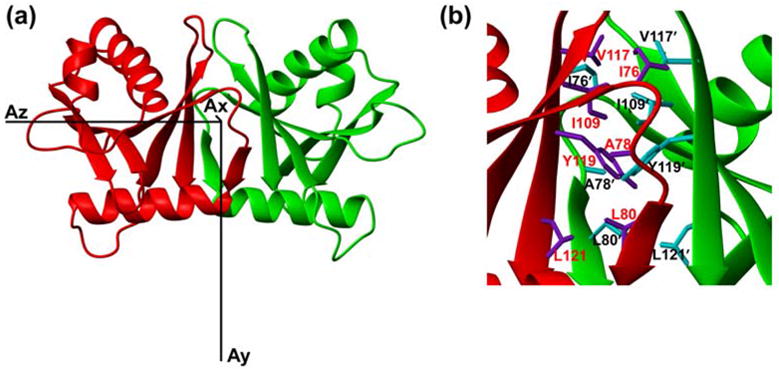
(a) Ribbon diagram of the HsSen15(36–157) structure oriented with regard to three principal axes of alignment tensor. (b) The dimer interfaces of HsSen15(36–157): the side chains of selected residues from one monomer are colored in purple and labeled in red, and those from the symmetric subunit are colored in cyan and labeled in black. Otherwise the color schemes are the same as in Figure 2.
Sequence conservation
The amino acid sequences of Sen15 vary significantly across species (Figure 4). Although HsSen15 shows a 91% sequence identity to mouse Sen15 (MmSen15), it shows only 23% and 20% sequence identities, respectively, to two yeast orthologs, SpSen15 from Schizosaccharomyces pombe and ScSen15 from Saccharomyces cerevisiae. HsSen15 shows even lower sequence identity to HsSen34 and to archaeal endonucleases. Structure-based sequence alignments shows that the most conserved residues are located in the regions equivalent to α2, β5 and β6 of HsSen15(36–157) (Figure 4). As revealed by the structures of both MJ_endo and HsSen15(36–157), these conserved structural elements are responsible for mediating either dimeric or tetrameric association of the protein subunits. Because these residues are also conserved in HsSen34, it can be further inferred that HsSen34 may also exist in solution as dimer (or higher oligomer). The N-terminal amino acid sequence exhibits the greatest divergence among endonuclease subunits. In particular, the N-terminal domain of MJ_endo (β1, β2, α1, β3 and β4) has been deleted in ScSen15 and ST_endo2, and it has been replaced by a structurally disordered segment in HsSen15 and SpSen15. Although strands β1 and β2 of HsSen15(36–157) appear in the same topological order as strands β4 and β5 of archaeal endonucleases, their sequences do not align well. In addition, an N-terminal stretch of 11–15 residues, corresponding to α3 of HsSen15(36–157), is also absent in ScSen15, SpSen15, and ST_endo2.
Figure 4.

Multiple sequence alignment of selected splicing endonucleases. Identical and homologous residues are highlighted in green and cyan, respectively. The completely conserved tyrosine is indicated by red lettering highlighted in yellow. The secondary structural elements determined for HsSen15(36–157) and MJ_endo are shown above and below the alignment respectively. Swiss-Prot ID: HsSen15, Q8WW01; ScSen15, Q04675; SpSen15, Q7LKV3; ST_endo2, Q975V0; HsSen34, Q9BSV6; ST_endo1, Q975R3 ; MJ_endo, Q58819. The two red asterisks represent the start and end of the protein product HsSen15(36–157).
Structure comparison to other endonuclease proteins
Crystallographic structures have been determined for four archaeal endonuclease family members: MJ_endo,25 AF_endo,24 the catalytic subunits of SS (SS_endo1),27 and ST (ST_endo1) (PDB accession number of ST_endo1: 2CV8). All of these structures contain the same dimeric architecture, except that the dimers in AF_endo are covalently “fused” by two monomers. For the sake of simplicity, we have limited our discussion to a structural comparison of HsSen15(36–157) and MJ_endo.
The monomeric fold of HsSen15(36–157) appears similar to that of the C-terminal domain of MJ_endo (Figure 5). Although with only 18% sequence identity, HsSen15(36–157) and MJ_endo share the same mixed α/β topology. Structural superposition of the HsSen15 monomer with the MJ_endo monomer yielded a Cα r.m.s.d. of 3.5 Å over 103 aligned residues. In particular, the equivalent hydrogen bond between the side chain hydroxyl group of Y152 and the carboxylate of D69 of HsSen15(36–157) is observed in both structures. The major difference between the two monomeric structures lies in the region equivalent to α3 of HsSen15(36–157). In HsSen15(36–157), the C-terminus of α3-helix tilts toward the outer edge of the β-sheet as the result of a hydrophobic interaction between the side chains of I122 from α3 and L89 from β2. By contrast, the equivalent helix in MJ_endo (α5) runs parallel to the last three β-strands, with the residue equivalent to I122 of HsSen15(36–157) interacting with a helical turn (equivalent to l3 of HsSen15(36–157), which connects β1 and β2). As a consequence, residues from α5 of MJ_endo (L144, G146, V148) are heavily involved in the formation of the hydrophobic core in the dimer interface 25, whereas α3 of HsSen15(36–157) is predominantly involved in intramolecular interactions. It appears that the difference in the orientation of this equivalent helix between the two structures may partially arise from the presence of a longer loop following α3 in HsSen15(36–157) (Figure 4).
Figure 5.
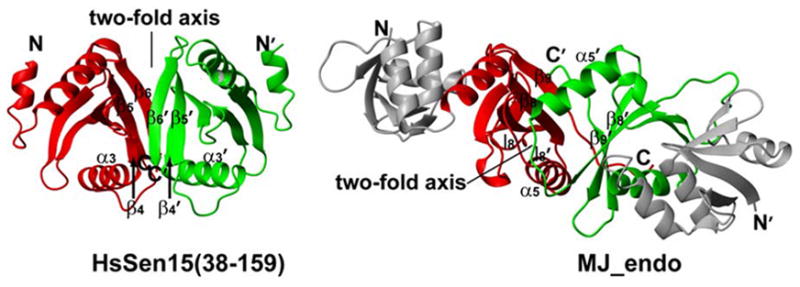
Comparison of the orientation of the two-fold symmetry axis between HsSen15(36–157) and MJ_endo. In MJ_endo, the C-terminal domains (residues 85–179) are painted red and green, while the N-terminal domains (residues 9–84) are painted grey. The two-fold axes and equivalent structural elements responsible for dimerization are labeled.
Despite their similar monomeric fold, the oligomeric structures of HsSen15(36–157) and MJ_endo are quite distinct. In MJ_endo, each structural subunit associates with one catalytic subunit by antiparallel matching of their last strands β9. The symmetry-related l8 loops then pair with each other on top of the β9 strands, enclosing a hydrophobic core in the dimer interface together with α5, β8, and β9 from both subunits.25 As a result, the relative orientation between the two symmetric subunits of MJ_endo differs from that in HsSen15(36–157) by a 180° rotation of one subunit around the axis orthogonal to the dimer interface (Figure 5). Accordingly, the two-fold symmetry axis is orthogonal to the orientation of β9 sheet in MJ_endo, whereas it is parallel to the β4′-β5-β6 sheets in HsSen15(36–157) (Figure 5). A number of residues involved in the hydrophobic core in the dimer interface, including L114, L141, I143, V151, Y153, and L155 of HsSen15(36–157), as well as their interactions (e.g. Y153–L155′, L141–L155′), are either fully or partially conserved in the two proteins. On the other hand, it is also evident that significant differences exist in structural elements of the dimer interface, including the differences in hydrophobicity at sites 110(E→I), 112(S→A) and 116(L→H) of HsSen15(36–157). These changes might lead to disengagement of α3 from the dimer interface and stabilization of the interaction between β6 and β4′ in HsSen15(36–157). Furthermore, the insertion of two hydrophilic residues, D157 and G158, in the C-terminus of HsSen15, divides the continuous hydrophobic stretch present in MJ_endo into two segments (Figure 4). This insertion at the C-terminus presumably disrupts the potential hydrophobic core, which is important for antiparallel matching of the two C-terminal β-strands in MJ_endo,25 and leads to the observed dimer interface of HsSen15. Two arguments suggest that the 14 C-terminal residues of HsSen15, which are deleted in HsSen15(36–157), play a negligible role in determining the dimeric fold of HsSen15: first, these residues are not evolutionarily conserved and are deleted in many endonuclease structural subunits;28 second, the dimeric fold of HsSen15(36–157) is preserved after deletion of these residues from HsSen15.
Functional and evolutionary implications
The work presented here provides the first experimental structural model for a eukaryal endonuclease subunit. The observed dimeric nature of HsSen15(36–157) reinforces the finding that all known endonuclease structures form a homodimer as their basic folding unit. In addition, the structure provides new insights into the functional assembly of eukaryal endonuclease. A prior assembly model for all endonucleases was derived from the structure of MJ_endo. In this assembly model, each catalytic subunit associates with one structural subunit by tail-to-tail pairing, and the acidic l10-equivalent loops from the structural subunits are inserted into the basic concavities of catalytic subunits through charge-charge interactions. Although this model is supported by the conservation of sequence motifs, it is limited by the fact that the catalytic and structural subunits of MJ_endo, despite their different functions, are part of the same polypeptide.
According to a proposed heterotetrameric model of eukaryal endonuclease,25 monomeric Sen15 interacts with Sen34 through pairing of β6 and associates with the monomeric Sen2 via loop l7. In the structure of HsSen15(36–157), the last strands β6 are involved in forming the β-barrel in the dimer interface. Thus, further pairing of β6 with the equivalent β-strand from HsSen34 will require significant structural rearrangement of HsSen15 in the dimer interface. Thermodynamically, this also requires that the heterodimeric association between HsSen15 and HsSen34 be stronger than the homodimeric association of HsSen15.
On the basis of the above considerations, we propose a speculative alternative model in which dimeric HsSen15 associates with dimeric HsSen34 to form a α2β2 tetramer, as implicated by recent structures of SS_endo and ST_endo. In support of this proposal, many of the hydrophobic residues in the dimer interfaces of both HsSen15 (36–157) and MJ_endo are also conserved in HsSen34, HsSen2, and HsSen54 (Figure 4);30 this would suggest that these subunits also exist in solution as homodimers.
The surface of HsSen15(36–157) is highly charged. The structural elements of MJ_endo that are responsible for its tetrameric association (l7 and α3) have homologs in HsSen15(36–157) at the two ends of the β-barrel (Figure 6). At one end of the barrel, the two negatively charged loops l7 define an acidic platform that constitutes a potential site of interaction between HsSen15 and its interaction partners. At the opposite end of the β-barrel, a cluster of basic residues (R118, R120, K124, R127, K128) from the two α3-helices form a concave surface that may provide an alternative protein interaction site.
Figure 6.
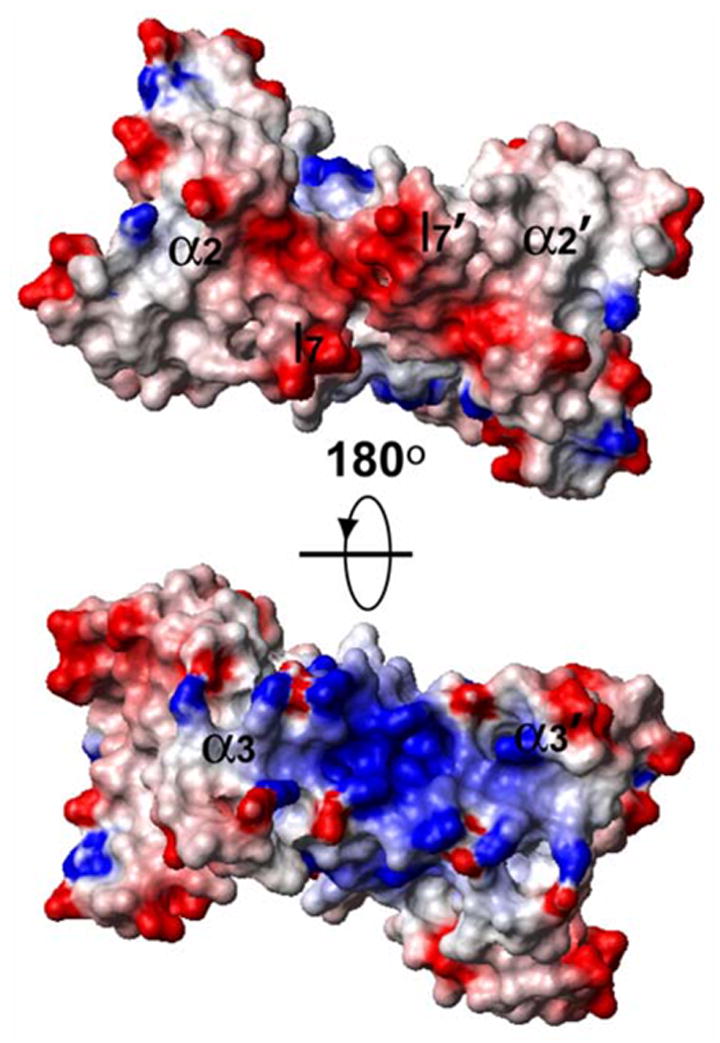
Representation of the surface electrostatic potential, with positive regions in blue and negative regions in red as calculated by the program MOLMOL.40
Sequence conservation across the archaeal and eukaryal kingdoms suggests that all the tRNA splicing endonucleases come from the same origin.5,42 It also appears that archaeal splicing endonucleases coevolved with their tRNA substrates by “subfunctionization”,28 a process in which mutations lead to specialization of protein functions. For example, the homotetrameric MJ_endo can only cleave the canonical BHB structure in its substrates, whereas the homodimeric AF_endo and the heterotetrameric SS_endo can also cleave variants of the BHL structure.43 With even greater diversity, the eukaryal endonculeases have developed distinct substrate recognition mechanisms and separate machinery for cleavage of 5′ and 3′ splice sites.
The solution structure of HsSen15(36–157) presented here adds a structural link between eukaryal endonucleases and archaeal endonucleases. Both have similar monomeric topology, but differ in their dimeric architecture. The high sequence conservation in the dimer interface highlights the importance of homodimerization or heterodimerization in regulating the functions of endonucleases. Evolutionary changes in structural elements forming the dimer interface appear to have altered the oligomeric topology and resulted in the observed functional diversity of endonucleases. To test the hypothetical dimer-of-dimers model advanced here, it will be important to obtain experimental information on the interactions among all subunits of the tetrameric human endonuclease.
Materials and Methods
Protein production and sample preparation
The cDNA for full-length HsSen15 was purchased from Open Biosystems Inc. and subcloned by restriction digestion into the pVP13 vector developed in house. The gene fragment (36–157) of HsSen15 was amplified by PCR from the plasmid containing the cDNA coding for full-length HsSen15. The PCR primers also encoded a 5′ BamHI and a 3′ AscI site used for ligation of the gene fragment into the pVP13 vector. Each expressed gene product contained an N-terminal His6 affinity tag on maltose binding protein (MBP) connected to the target protein by a linker containing a tobacco etch virus (TEV) protease cleavage site. Each construct was transformed into Rosetta(DE3)/pLysS (Novagen) host cells, which were then grown in a medium enriched with 15N or 15N/13C according to an established protocol.44 The cells were harvested by centrifugation, suspended in 50 ml buffer A (50 mM Na2HPO4, 300 mM NaCl, pH 7.5) and stored overnight at −20 °C. To purify the protein, the frozen cells were thawed at 37 °C and sonicated on ice until a very low viscosity of the suspension was achieved. The pellet in the mixture was removed by centrifugation, and the supernatant was loaded onto a precharged nickel nitrilotriacetic acid (Ni-NTA) affinity column (QIAGEN) with equilibrated resin. Following wash with buffer B (50 mM Na2HPO4, 300 mM NaCl, 25 mM imidazole, pH 7.5), the protein was eluted with buffer C (50 mM Na2HPO4, 300 mM NaCl, 250 mM imidazole, pH 7.5) and concentrated by using an Amicon ultrafiltration device (Amersham Pharmacia, Inc.). TEV protease prepared in house was mixed with the target protein (1/100, w/w), and the mixture was dialyzed against the TEV reaction buffer (50 mM Tris, 0.5 mM DTT, pH 8.0) overnight at 25 °C. After cleavage, the protein solution was exchanged back into buffer A by dialysis and then applied to a Ni-NTA column to remove the His-tagged MBP and uncleaved fusion protein. The target protein was collected from the column flow-through, and ultrafiltration was used to concentrate the protein and to exchange it into the NMR buffer (90% H2O/10% D2O, 20 mM Bis-Tris, 100 mM NaCl, 5 mM DTT and 0.02% w/v sodium azide, pH 6.0). The 123-residue protein product included a non-native N-terminal serine left after TEV cleavage. The numbering system used here is that of the full-length HsSen15 protein. SDS-PAGE was used to determine the purity of the protein, and the BCA method45 was used to determine the protein concentration.
To identify the dimer interface, an asymmetrically labeled sample was produced by mixing 0.5 mM [15N,13C]-HsSen15(36–157) with 0.5 mM [14N,12C]-HsSen15(36–157). The protein mixture was first denatured by 6 M urea to dissociate the protein dimers; then the urea was rapidly diluted into the NMR buffer to allow refolding of the protein. The sample used for measurements of residual dipolar couplings (RDCs) contained 0.5 mM [U-15N]-HsSen15(36–157) and 15 mg/ml Pf1 phage cosolvent (ALSA Biotech.) in the NMR buffer.
NMR spectroscopy
NMR spectra were recorded at the National Magnetic Resonance Facility at Madison (NMRFAM) on 600 MHz, 900 MHz Varian Inova and 500 MHz Bruker spectrometers equipped with 1H, 15N, 13C triple-resonance cryogenic. Unless stated otherwise, the temperature of each sample was held at 30 °C. A 2D 1H,15N-HSQC spectrum was acquired of the full-length nitrogen-15 labeled protein ([U-15N]-HsSen). The [U-13C,15N]-HsSen15(36–157) sample was used in acquiring a suite of NMR spectra for sequence specific chemical shift assignment and structural constraints: 2D 1H,15N-HSQC, 2D 1H,13C-HSQC, 2D (HB)CB(CGCD)HD, 2D (HB)CB(CGCDCE)HE, 3D HNCACB, HN(CO)CACB, HNCO, H(CCO)NH, C(CO)NH, HCCH-TOCSY, 15N edited 1H,1H-NOESY (τmix = 100 ms) and 13C edited 1H,1H-NOESY (τmix = 100 ms). A 3D 13C,15N-filtered/13C-edited 1H,1H-NOESY46 (τmix = 160 ms) spectrum was recorded on the asymmetrically labeled (15N,13C/14N,12C) sample. In addition, a 2D IPAP 1H,15N- HSQC experiment47 was used to obtain the one-bond NH scalar coupling (1JN–H) values. RDCs for NH vectors (1DN–H) were calculated from differences in couplings measured in the presence and absence of the ordering agent (15 mg/ml Pf1 phage). Standard pulse sequences48 were used for measurements of the 15N relaxation rates (R1, R2) of HsSen15(36–157). 15N R1 values were determined from spectra recorded with delay values of 100, 300, 700, 1000, 1400, and 1800 ms. 15N R2 values were determined from spectra recorded with delay values of 10, 30, 50, 70, 90, 110, and 130 ms. The rotational correlation time of HsSen15(36–157) was estimated from the 15N R2/R1 ratios by using the program quadric_diffusion.49
Spectra were processed and analyzed, respectively, with the NMRPipe50 and Sparky (http://www.cgl.ucsf.edu/home/sparky) software packages. Sequential backbone assignment was based on the 3D HNCACB, CBCA(CO)NH and HNCO. The GARANT software program51 was used to facilitate the initial backbone assignment. Assignments of 1H, 13C, and 15N signals from non-aromatic side chains were achieved by using data from HBHA(CO)NH, H(CCO)NH, C(CO)NH and HCCH-TOCSY. Assignments of aromatic residue 1Hδ and 1Hε signals were determined from 2D (HB)CB(CGCD)HD and 2D (HB)CB(CGCDCE)HE data sets.
Structure Calculation
For the structure calculation, intramolecular NOEs were derived from 15N-edited 1H,1H-NOESY and 13C-edited 1H,1H-NOESY, and intermolecular NOEs were derived from 3D 13C,15N-filtered/13C-edited 1H,1H-NOESY. Backbone dihedral angles φ and Ψ were predicted from 1Hα, 15N, 13Cα, 13Cβ, and 13C′ secondary chemical shifts by using TALOS52 software. The automated NOE assignment module in CYANA program36, which executed seven cycles of iterative NOE assignment and structure calculation, and one additional round of structure calculation in the end, was used to generate the monomeric fold of HsSen15(36–157) and initial intramolecular NOE assignments. The intramolecular NOEs restraints, combined with intermolecular NOEs and dihedral angle restraints, were then used to calculate 100 dimeric models of HsSen15(36–157), using the standard CYANA torsion angle dynamics protocol36. A number of hydrogen bonds derived from chemical shifts analysis53 and from observed NOEs characteristic for α-helices and β-sheets, were added in the final rounds of structure refinement. On the basis of their lowest target functions, 20 structures were chosen for further refinement using XPLOR program38, in which 1DNH RDC restraints, physical force field terms and explicit solvent term37 were added to the calculation. Due to the fact that HsSen15(36–157) is a symmetric homodimer, non-crystallographic symmetry constraints were also used to enforce the symmetry of the secondary structural elements between the two monomers at the final stage.
Database Depositions
The raw, time-domain NMR data sets and chemical shift assignments have been deposited in the BioMagResBank (BMRB) database under accession number 6860. The three-dimensional coordinates for the structural models have been deposited in the Protein Data Bank (PDB) under the accession number 2GW6.
Acknowledgments
This work was supported by the National Institutes of Health, Protein Structure Initiative through grants P50 GM64598 and U54 GM074901. NMR data were collected at the National Magnetic Resonance Facility at Madison, which is supported in part by NIH grants P41 RR02301 and P41 GM66326. The authors thank the many team members from the Center for Eukaryotic Structural Genomics who provided the infrastructure for this work.
Abbreviations used
- AF
Archeoglobus fulgidus
- AF_endo
endonuclease from AF
- BHB
bulge helix bulge
- HSQC
heteronuclear single-quantum correlation
- HsSen15(36–157)
protein construct consisting of residues 36–157 of the 172-residue full-length HsSen15 plus an N-terminal serine residue (artifact of the TEV protease cleavage site introduced to produce the protein)
- HsSen15
Sen15 from Homo sapiens
- IPAP
in-phase anti-phase
- MBP
maltose binding protein
- MJ
Methanocaldococcus jannaschii
- MJ_endo
endonuclease from MJ
- MmSen15
Sen15 from Mus musculus
- NiNTA
nickel nitrilotriacetic acid
- NOE
nuclear Overhauser enhancement
- NOESY
NOE spectroscopy
- r.m.s.d
root mean standard deviation
- R1
longitudinal relaxation rate
- R2
transverse relaxation rate
- RDC
residual dipolar coupling
- ScSen15
Sen15 from Saccharomyces cerevisiae
- Sen2
Sen15, Sen34, Sen54, subunits of eukaryal endonuclease involved in tRNA splicing
- SpSen15
Sen15 from Schizosaccharomyces pombe
- SS
Sulfolobus solfataricus
- SS_endo
endonuclease from SS
- ST
Sulfolobus tokodaii
- ST_endo
endonuclease from ST
- TEV
tobacco etch virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Coordinates and related data have been deposited at PDB (2GW6) and NMR data at BMRB (6860).
References
- 1.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cech T. In: RNA World. Gesteland RF, Atkins JF, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1993. pp. 239–270. [Google Scholar]
- 3.Peebles CL, Gegenheimer P, Abelson J. Precise excision of intervening sequences from precursor tRNAs by a membrane-associated yeast endonuclease. Cell. 1983;32:525–536. doi: 10.1016/0092-8674(83)90472-5. [DOI] [PubMed] [Google Scholar]
- 4.Thompson LD, Daniels CJ. Recognition of exon-intron boundaries by the Halobacterium volcanii tRNA intron endonuclease. J Biol Chem. 1990;265:18104–18111. [PubMed] [Google Scholar]
- 5.Trotta CR, Miao F, Arn EA, Stevens SW, Ho CK, Rauhut R, et al. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell. 1997;89:849–858. doi: 10.1016/s0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- 6.Englert M, Beier H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res. 2005;33:388–399. doi: 10.1093/nar/gki174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greer CL, Peebles CL, Gegenheimer P, Abelson J. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell. 1983;32:537–546. doi: 10.1016/0092-8674(83)90473-7. [DOI] [PubMed] [Google Scholar]
- 8.Phizicky EM, Schwartz RC, Abelson J. Saccharomyces cerevisiae tRNA ligase. Purification of the protein and isolation of the structural gene. J Biol Chem. 1986;261:2978–2986. [PubMed] [Google Scholar]
- 9.McCraith SM, Phizicky EM. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J Biol Chem. 1991;266:11986–11992. [PubMed] [Google Scholar]
- 10.McCraith SM, Phizicky EM. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol Cell Biol. 1990;10:1049–1055. doi: 10.1128/mcb.10.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trotta CR, Paushkin SV, Patel M, Li H, Peltz SW. Cleavage of pre-tRNAs by the splicing endonuclease requires a composite active site. Nature. 2006;441:375–377. doi: 10.1038/nature04741. [DOI] [PubMed] [Google Scholar]
- 12.Xue S, Calvin K, Li H. RNA recognition and cleavage by a splicing endonuclease. Science. 2006;312:906–910. doi: 10.1126/science.1126629. [DOI] [PubMed] [Google Scholar]
- 13.Abelson J, Trotta CR, Li H. tRNA splicing. J Biol Chem. 1998;273:12685–12688. doi: 10.1074/jbc.273.21.12685. [DOI] [PubMed] [Google Scholar]
- 14.Thompson LD, Daniels CJ. Recognition of exon-intron boundaries by the Halobacterium volcanii tRNA intron endonuclease. J Biol Chem. 1990;265:18104–18111. [PubMed] [Google Scholar]
- 15.Reyes VM, Abelson J. Substrate recognition and splice site determination in yeast tRNA splicing. Cell. 1988;55:719–730. doi: 10.1016/0092-8674(88)90230-9. [DOI] [PubMed] [Google Scholar]
- 16.Tang TH, Rozhdestvensky TS, d'Orval BC, Bortolin ML, Huber H, Charpentier B, et al. RNomics in Archaea reveals a further link between splicing of archaeal introns and rRNA processing. Nucleic Acids Res. 2002;30:921–930. doi: 10.1093/nar/30.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kjems J, Garrett RA. Novel splicing mechanism for the ribosomal RNA intron in the archaebacterium Desulfurococcus mobilis. Cell. 1988;54:693–703. doi: 10.1016/s0092-8674(88)80014-x. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe Y, Yokobori S, Inaba T, Yamagishi A, Oshima T, Kawarabayasi Y, et al. Introns in protein-coding genes in Archaea. FEBS Lett. 2002;510:27–30. doi: 10.1016/s0014-5793(01)03219-7. [DOI] [PubMed] [Google Scholar]
- 19.Diener JL, Moore PB. Solution structure of a substrate for the archaeal pre-tRNA splicing endonucleases: the bulge-helix-bulge motif. Mol Cell. 1998;1:883–894. [PubMed] [Google Scholar]
- 20.Armbruster DW, Daniels CJ. Splicing of intron-containing tRNATrp by the archaeon Haloferax volcanii occurs independent of mature tRNA structure. J Biol Chem. 1997;272:19758–19762. doi: 10.1074/jbc.272.32.19758. [DOI] [PubMed] [Google Scholar]
- 21.Thompson LD, Daniels CJ. A tRNA(Trp) intron endonuclease from Halobacterium volcanii. Unique substrate recognition properties. J Biol Chem. 1988;263:17951–17959. [PubMed] [Google Scholar]
- 22.Reyes VM, Abelson J. Substrate recognition and splice site determination in yeast tRNA splicing. Cell. 1988;55:719–730. doi: 10.1016/0092-8674(88)90230-9. [DOI] [PubMed] [Google Scholar]
- 23.Baldi MI, Mattoccia E, Bufardeci E, Fabbri S, Tocchini-Valentini GP. Participation of the intron in the reaction catalyzed by the Xenopus tRNA splicing endonuclease. Science. 1992;255:1404–1408. doi: 10.1126/science.1542788. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Abelson J. Crystal structure of a dimeric archaeal splicing endonuclease. J Mol Biol. 2000;302:639–648. doi: 10.1006/jmbi.2000.3941. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Trotta CR, Abelson J. Crystal structure and evolution of a transfer RNA splicing enzyme. Science. 1998;280:279–284. doi: 10.1126/science.280.5361.279. [DOI] [PubMed] [Google Scholar]
- 26.Lykke-Andersen J, Garrett RA. RNA-protein interactions of an archaeal homotetrameric splicing endoribonuclease with an exceptional evolutionary history. EMBO J. 1997;16:6290–6300. doi: 10.1093/emboj/16.20.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calvin K, Hall MD, Xu F, Xue S, Li H. Structural characterization of the catalytic subunit of a novel RNA splicing endonuclease. J Mol Biol. 2005;353:952–960. doi: 10.1016/j.jmb.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 28.Tocchini-Valentini GD, Fruscoloni P, Tocchini-Valentini GP. Structure, function, and evolution of the tRNA endonucleases of Archaea: an example of subfunctionalization. Proc Natl Acad Sci USA. 2005;102:8933–8938. doi: 10.1073/pnas.0502350102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randau L, Calvin K, Hall M, Yuan J, Podar M, Li H, et al. The heteromeric Nanoarchaeum equitans splicing endonuclease cleaves noncanonical bulge-helix-bulge motifs of joined tRNA halves. Proc Natl Acad Sci USA. 2005;102:17934–17939. doi: 10.1073/pnas.0509197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paushkin SV, Patel M, Furia BS, Peltz SW, Trotta CR. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell. 2004;117:311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- 31.Ho CK, Rauhut R, Vijayraghavan U, Abelson J. Accumulation of pre-tRNA splicing '2/3' intermediates in a Saccharomyces cerevisiae mutant. EMBO J. 1990;9:1245–1252. doi: 10.1002/j.1460-2075.1990.tb08232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 33.Song J, Tyler RC, Lee MS, Tyler EM, Markley JL. Solution structure of isoform 1 of Roadblock/LC7, a light chain in the dynein complex. J Mol Biol. 2005;354:1043–1051. doi: 10.1016/j.jmb.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Altieri AS, Hinton DP, Byrd RA. Association of Biomolecular Systems via PFG-NMR Diffusion Measurements. J Am Chem Soc. 1995;117:7566–7567. [Google Scholar]
- 35.Güntert P, Mumenthaler C, Wüthrich K. Torsion Angle Dynamics for NMR Structure Calculation with the New Program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 37.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 38.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 39.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 40.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for Checking the Quality of Protein Structures Solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wüthrich K. MOLMOL - A Program for Display and Analysis of Macromolecular Structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 42.Kleman-Leyer K, Armbruster DW, Daniels CJ. Properties of H. volcanii tRNA intron endonuclease reveal a relationship between the archaeal and eucaryal tRNA intron processing systems. Cell. 1997;89:839–847. doi: 10.1016/s0092-8674(00)80269-x. [DOI] [PubMed] [Google Scholar]
- 43.Tocchini-Valentini GD, Fruscoloni P, Tocchini-Valentini GP. Coevolution of tRNA intron motifs and tRNA endonuclease architecture in Archaea. Proc Natl Acad Sci USA. 2005;102:15418–15422. doi: 10.1073/pnas.0506750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Q, Frederick R, Seder K, Thao S, Sreenath H, Peterson F, et al. Production in two-liter beverage bottles of proteins for NMR structure determination labeled with either 15N- or 13C-15N. J Struct Funct Genomics. 2004;5:87–93. doi: 10.1023/B:JSFG.0000029205.65813.42. [DOI] [PubMed] [Google Scholar]
- 45.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 46.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 47.Ottiger M, Delaglio F, Bax A. Measurement of J and Dipolar Couplings from Simplified Two-Dimensional NMR Spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 48.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, et al. Backbone Dynamics of a Free and Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 49.Lee LK, Rance M, Chazin WJ, Palmer AG., III Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J Biomol NMR. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- 50.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPIPE - A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Bartels C, Billeter M, Güntert P, Wüthrich K. Automated Sequence-Specific NMR Assignment of Homologous Proteins Using the Program GARANT. J Biomol NMR. 1996;7:207–213. doi: 10.1007/BF00202037. [DOI] [PubMed] [Google Scholar]
- 52.Cornilescu G, Delaglio F, Bax A. Protein Backbone Angle Restraints from Searching a Database for Chemical Shift and Sequence Homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 53.Wishart DS, Sykes BD. The 13C Chemical Shift Index: A Simple Method for the Identification of Protein Secondary Structure Using 13C Chemical Shifts. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]