

Abstract
Dietary protocols that increase serum levels of ketones, such as calorie restriction and the ketogenic diet, offer robust protection against a multitude of acute and chronic neurological diseases. The underlying mechanisms, however, remain unclear. Previous studies have suggested that the ketogenic diet may reduce free radical levels in the brain. Thus, one possibility is that ketones may mediate neuroprotection through antioxidant activity. In the present study, we examined the effects of the ketones β-hydroxybutyrate and acetoacetate on acutely dissociated rat neocortical neurons subjected to glutamate excitotoxicity using cellular electrophysiological and single-cell fluorescence imaging techniques. Further, we explored the effects of ketones on acutely isolated mitochondria exposed to high levels of calcium. A combination of β-hydroxybutyrate and acetoacetate (1 mM each) decreased neuronal death and prevented changes in neuronal membrane properties induced by 10 μM glutamate. Ketones also significantly decreased mitochondrial production of reactive oxygen species and the associated excitotoxic changes by increasing NADH oxidation in the mitochondrial respiratory chain, but did not affect levels of the endogenous antioxidant glutathione. In conclusion, we demonstrate that ketones reduce glutamate-induced free radical formation by increasing the NAD+/NADH ratio and enhancing mitochondrial respiration in neocortical neurons. This mechanism may, in part, contribute to the neuroprotective activity of ketones by restoring normal bioenergetic function in the face of oxidative stress.
Keywords: glutamate, neurotoxicity, diet, mitochondria, oxidation, stress
Calorie restriction can decrease the risk of neurodegenerative disease and protect the brain against acute insults such as stroke (Mattson et al, 2002). Similarly, the ketogenic diet, a high-fat, low-carbohydrate diet created to mimic the effects of calorie restriction, is an extremely efficacious treatment for medically intractable epilepsy (Freeman et al, 1998; Vining et al, 1998). Several metabolic changes occur during calorie restriction and the ketogenic diet, notably an increase in serum concentrations of the ketones β-hydroxybutyrate (BHB) and acetoacetate (ACA) (Gilbert et al, 2000; Koubova & Guarente, 2003; Denny et al, 2006; Mahoney et al., 2006).
Recent work has shown that ketones exert a protective effect on brain. For example, BHB prevents the death of hippocampal neurons exposed to Aβ1–42, protects cultured mesencephalic dopaminergic neurons from the toxic effects of 1-methyl-4-phenylpyridinium (MPP+, an inhibitor of NADH dehydrogenase that increases oxygen radical formation) and reduces brain injury in rodents subjected to glycolysis inhibition and focal or generalized ischemia (Kashiwaya et al, 2000; Suzuki et al, 2001 & 2002). Furthermore, ACA protects hippocampal neurons against glycolysis inhibition in vivo and in vitro (Massieu et al, 2003). In parallel, clinical data suggest that seizure control in epileptic patients treated with the ketogenic diet correlates with the serum concentration of ketones (Gilbert et al, 2000). The mechanisms underlying the therapeutic effects of ketones remain, however, largely unknown.
Studies in cardiac tissue have suggested that ketones might reduce oxidative stress (Veech et al, 2001), a pathogenic process implicated in many disorders ranging from atherosclerosis and traumatic injuries to diseases more specific to the nervous system (Droge, 2002; Keller et al, 2005). To investigate the antioxidant activity of ketones in neurons, we used a glutamate excitotoxicity model because of its equally important relevance to several neurological diseases (including stroke, epilepsy, trauma and Alzheimer’s disease) and because of the well-known fact that glutamate excitotoxicity is associated with higher cellular levels of reactive oxygen species (ROS) (Nicholls, 2004; Sullivan et al, 2005). The antioxidant effects of BHB and ACA were therefore assessed in acutely dissociated neocortical neurons subjected to glutamate excitotoxicity and in isolated neocortical mitochondria exposed to high concentrations of calcium. Our results show that excitotoxic injury is associated with increased mitochondrial production of ROS and that ketones inhibit these deleterious effects by enhancing NADH oxidation in the mitochondrial respiratory chain.
EXPERIMENTAL PROCEDURES
All protocols were approved by the Barrow Neurological Institute and the University of Kentucky Institutional Animal Care and Use Committees. Chemical products were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Fluorescent indicators were purchased from Molecular Probes (Eugene, OR). Of note, the physiological isomer of β-hydroxybutyrate - i.e., R(-)BHB or D(-)BHB - was used in all experiments.
Tissue Preparation
One to 3 week-old Wistar rats (Charles River Laboratories; Wilmington, MA), deeply anesthetized with halothane, were sacrificed and their brains quickly transferred to ice-cold, oxygenated PIPES buffer (120 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 25 mM glucose, 20 mM PIPES, pH adjusted to 7.0 with Trizma base, 320–330 mOsm). Slices (500 μm) from somatosensory cortex were cut on a vibratome (The Vibratome Company, St. Louis, MO), treated with Pronase (protease, Streptomyces griseus; Calbiochem; San Diego, CA) 0.61 PUK/ml for 20 min at 37°C and then allowed to recover in oxygenated PIPES buffer at room temperature for 1 hour. Small areas (1 mm in diameter) were dissected and mechanically dissociated with fire-polished Pasteur pipettes of progressively decreasing diameter in 35 mm Petri dishes (Becton Dickinson; Franklin Lanes, NJ) containing HEPES buffer (145 mM NaCl, 4.0 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH adjusted to 7.4 with Trizma base, 320–330 mOsm).
Electrophysiology
Borosilicate microelectrodes (1.5 mm external diameter, 0.75 mm internal diameter; WPI; Sarasota, FL) were prepared with a Narishige PP-830 puller (Narishige International USA; East Meadow, NY) and filled with an internal solution (150 mM K-gluconate, 8 mM MgCl2, 10 mM HEPES, pH adjusted to 7.2 with Trizma base) containing 240 μg/ml amphotericin B (from streptomyces, ≈ 80%; Rae et al., 1991). Electrode impedances ranged from 5 to 9 MΩ. Acutely dissociated neurons in Petri dishes were placed under a Zeiss Axiovert 200 microscope (Zeiss, Germany). Electrodes were attached to a Sutter MP-225 micromanipulator (Sutter Instrument Company, Novato, CA) and connected to an Axon Multiclamp 700B controlled by Multiclamp Commander (Axon Instruments; Union City, CA). Electrical signals were digitized with an Axon Digidata 1322A and pClamp 9.2 (Axon Instruments; Union City, CA).
Once a gigaohm seal was achieved, membrane (Rm) and access (Ra) resistances were continuously monitored using the Membrane Test function of pClamp for 10 to 40 min until the access resistance fell below 100 MΩ. Membrane potential (Vm) was recorded for 5 min and if the range did not fluctuate by more than 10 mV, the neuron was selected for further study with glutamate (L-glutamic acid monosodium salt, 99–100%), BHB (R-3-hydroxybutyric acid, ≥ 98%), ACA (lithium acetoacetate, 90–95%) or catalase (from mouse liver). Rm was monitored again at the end of each protocol. In some experiments, cells were imaged under phase contrast to look for morphological changes. Pharmacological substances were applied with an ALA-VM8 pressure-controlled pump (ALA Scientific Instruments; Westbury, NY). Electrophysiological data were analyzed with Clampfit 9.2 (Axon Instruments; Union City, CA).
Fluorescence Imaging
Neurons were dissociated in HEPES buffer containing either 50 μg/ml propidium iodide (PI), 1 μM dihydroethidium (DHE), 50 μM monochlorobimane (MCB) or 5 μM Rhod-2 (Macklis and Madison, 1990; Keelan et al., 2001; Vergun et al., 2001). Intracellular labeling was observed at 64X under the Zeiss Axiovert 200 equipped with an EXFO X-Cite 120 fluorescence system (Photonics Solutions Inc, Canada) and Zeiss filter set 15 (excitation 546 nm, emission 590 nm; Zeiss, Germany) for PI and Rhod-2, Zeiss filter set 10 (excitation 450 to 490 nm, emission 515 to 565 nm; Zeiss, Germany) for DHE or Zeiss filter set 49 (excitation 365 nm, emission 545–550 nm; Zeiss, Germany) for MCB. Images were acquired and analyzed with Axiovision 4.3 (Zeiss, Germany). Neurons exposed to PI and DHE were studied immediately following dissociation whereas those treated with MCB and Rhod-2 were incubated with the dye for 30 and 60 min, respectively, prior to recording. For NAD(P)H fluorescence, neurons were dissociated in customized Petri dishes. The bottoms of the dishes were cut out and replaced with thin cover slides to allow imaging with a 40X oil-immersion objective (Plan-NEOFLUAR 40X / 1.3 oil DIC; Zeiss; Germany). Neuronal fluorescence in all protocols was normalized to background (i.e., cell-free area) fluorescence.
Mitochondrial Isolation
The following procedures were modified from previously described protocols (Sullivan et al., 2000; Sullivan et al., 2003; Brown et al., 2004). Adult male Sprague-Dawley rats (~250g) were anesthetized with carbon dioxide in a sealed chamber and subsequently decapitated. The brains were quickly removed and the cortices dissected off. Cortical tissue was placed in cold isolation buffer with 1 mM EGTA (75 mM sucrose, 215 mM mannitol, 0.1% BSA, 1 mM EGTA, 20 mM HEPES with a pH adjusted to 7.2 using KOH) and all tissues were kept on ice at all times throughout the isolation.
Dissected cortical tissue was put into an all-glass dounce homogenizer with 3 mL of isolation buffer with EGTA, homogenized, split into four 2 mL tubes, topped off with isolation buffer with EGTA, and spun at 1,300 x g for 5 minutes at 4ºC. The supernatant was taken off and saved in separate tubes. The pellet was resuspended in isolation buffer with EGTA and spun at 1,300 x g for 5 min at 4ºC. The supernatant was again taken off and saved in separate tubes. The saved supernatant was topped off with isolation buffer with EGTA and spun at 13,000 x g for 10 min at 4ºC. Supernatant was then discarded (by slinging off) and the pellets were resuspended and combined in 500 μL of isolation buffer with EGTA. The sample was placed in a cold nitrogen cell disruptor for 10 min at 1000 psi to burst synaptoneurosomes formed by homogenization (Brown et al, 2004).
After nitrogen disruption of synaptoneurosomes, the sample was spun on a Percoll gradient, with fresh stocks of 30%, 24%, and 40% Percoll made prior to spin with isolation buffer with EGTA. The gradient was made by first adding 3.5 mL of the 24% stock to the ultracentrifuge tube and then, using a 5 mL syringe, injecting 3.5 mL of the 40% stock to the bottom of the tube. Using equal amounts of 30% Percoll stock and sample a 15% Percoll solution was made and 3 mLs were added to the top of the gradient. The gradient was then spun in a high-speed Sorval centrifuge for 10 min at 30,400 x g. The third fraction containing the purified mitochondria was removed, put into clean tubes, and topped off with isolation buffer without EGTA. The samples were spun in the same centrifuge at 16,700 x g for 15 min. The supernatant was removed and the pellet was resuspended in isolation buffer without EGTA and spun at 13,000 x g in the high-speed Sorval centrifuge for 10 min. After the supernatant was removed and discarded the pellet was resuspended in 1 mM isolation buffer without EGTA and transferred to a microcentrifuge tube, which was then spun at 10,000 x g for 10 min. The supernatant was removed and the pellet was resuspended in enough isolation buffer without EGTA to obtain a concentration of 10–15 μg/μl. A BCA protein assay kit was used to determine protein concentration by measuring absorbance at 560 nm with a BioTek Synergy HT plate reader (Winooskin, VT).
Mitochondrial ROS and NADH Assay
ROS and NADH were measured using a Synergy HTTR com1 plate reader and analyzed using KC4 software program (Bio-Tek Instruments Winooski, VT). A KCl stock respiration buffer (as described above) which also contained 5 mM pyruvate, 2.5 mM malate, 10 μM DCF, and 100 μM horseradish peroxidase (HRP) was used to measure fluorescence. A final concentration of 1 mM calcium was added to one aliquot of stock buffer. Ketones (BHB and ACA, 1 mM each) were added to the stock buffer with and without 1 mM calcium. 25 μg of protein was added to each well containing 50 μL total volume of one of the previously described buffers. ROS production was measured at 37 C at 485nm/528nm and NADH was measured at 360nm/460nm for 15 min at intervals of 1:24 min. Values from blank wells containing only buffer were subtracted from values obtained from sample wells. Values were expressed as % control (mitochondria with calcium-free and ketone-free buffer).
Mitochondrial Respiration
Respiration experiments were performed using a standard Clark-type oxygen electrode that was sealed, constantly stirred, and temperature controlled with Oxygraph software to measure oxygen consumption within the chamber (Hansatech Instruments, Norfolk, England). Based on the BCA protein assay, ~50 μg of protein (mitochondria) was loaded into the oxytherm chamber containing respiration buffer (125 mM KCl, 2 mM MgCl2, 2.5 mM KH2PO4, 0.1% BSA, 20 mM HEPES at pH 7.2) and the oxygen consumption within the chamber in response to substrates was measured. Substrate concentrations were optimized to the following final concentrations: state II respiration was induced by the addition of pyruvate (5 mM), malate (2.5 mM), state III respiration was induced by sequential additions of ADP (150 μM; the second addition of ADP was made when a return to state IV respiration indicated that all the ADP added to the chamber had be phosphorylated to ATP or when after 2 min if no change in respiration was noted), state IV respiration was induced by the addition of oligomycin (1 μM) to inhibit the ATP synthase, complex-I driven, maximum respiration Va was induced by the addition of FCCP (1 μM), complex I respiration was then inhibited completely by the addition of rotenone (0.8 μM), and complex-II driven, maximum respiration Vb was induced by the addition of succinate (10 mM) (Sullivan et al., 2003; Brown et al., 2004) To determine the ability of ketones to recover respiration after calcium addition, KCl respiration buffer was used containing 0.5 mM calcium. Ketones (BHB and ACA, 1 mM each) were added to KCl respiration buffer containing 0.5 mM calcium immediately prior to adding mitochondria and the start of the trace to measure ketone-induced mitochondrial respiration in the presence of calcium. Rates of oxygen consumption during state III respiration (following the second addition of ADP), Va respiration (in the presence of FCCP, prior to the addition of rotenone and succinate), and Vb (following the addition of succinate and in the presence of rotenone) were expressed as % control (calcium-free buffer) run on the same day.
Statistical Analyses
Treatment groups were compared at the beginning and end of experiments with non-parametric tests. Significance was set at p < 0.05. All analyses were performed with SigmaStat (SPSS Inc, Chicago, IL).
RESULTS
Ketones Reduce Neuronal Swelling and Death
The acutely isolated cell preparation was chosen to allow for detailed recording and imaging experiments while obviating the confounding metabolic and technical variables associated with primary cell culture and brain slice paradigms. Neurons were initially incubated with propidium iodide (PI), a fluorescent marker of neuronal death, and then immediately exposed to 10 μM glutamate for 45 min. Out of 12 cells, 6 displayed evidence of injury associated with a sharp increase in PI signal at 20.83 ± 2.91 min (mean ± S.E.M.) following the beginning of glutamate application (Fig. 1). When BHB and ACA (1 mM each) were simultaneously applied, 10 of 10 neurons with similar baseline characteristics survived (Chi-square test, p < 0.01).
Figure 1.
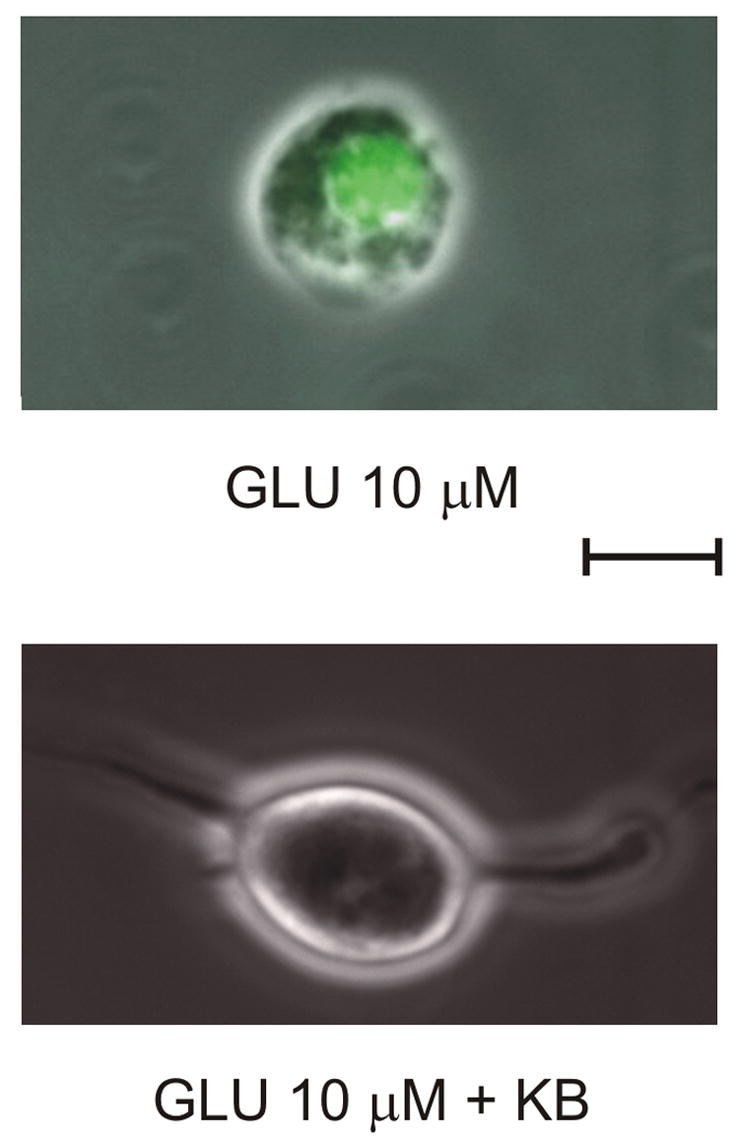
Ketones (KB) prevent neuronal injury induced by prolonged exposure to glutamate (GLU). Continuous exposure to GLU (10 μM) for 45 min resulted in the death of 50% of neurons with significant increase in propidium iodide fluorescence. A combination of 1 mM BHB and 1 mM ACA prevented neuronal death. Calibration bar = 10 μm.
Ketones Prevent Glutamate-induced Changes in Neuronal Membrane Properties
To better understand how ketones protect neurons against glutamate excitotoxicity, we initially tested the hypothesis that BHB and ACA might reduce glutamate-mediated excitability. We chose to investigate the early electrophysiological changes induced by glutamate without the confound of morphological changes that might precede neuronal injury, and as such, glutamate exposure in subsequent experiments was limited to 10 min and PI was removed. Using amphotericin B perforated patch-clamp recordings, we initially demonstrated that individual neurons could be monitored in current-clamp mode with stable membrane potentials (Vm) and membrane resistances (Rm) for over an hour in most cases, provided that a gigaohm seal with an access resistance (Ra) below 100 MΩ was achieved and that Vm remained stable during a 5 min. baseline recording period.
Application of 10 μM glutamate for 10 min resulted in an initial, small depolarizing response (averaging 11.2 ± 0.9 mV) followed, a few minutes later, by a much larger depolarization in 11/13 experiments (Fig. 2A). Vm did not return to baseline after a 10 min washout period and a significant decrease in Rm (Fig. 2B; ANOVA on ranks with Dunn’s post-hoc analyses, p = 0.01) occurred when compared to control, despite a stable morphological appearance under phase contrast (Fig. 2C) (Coulter et al., 1992).
Figure 2.
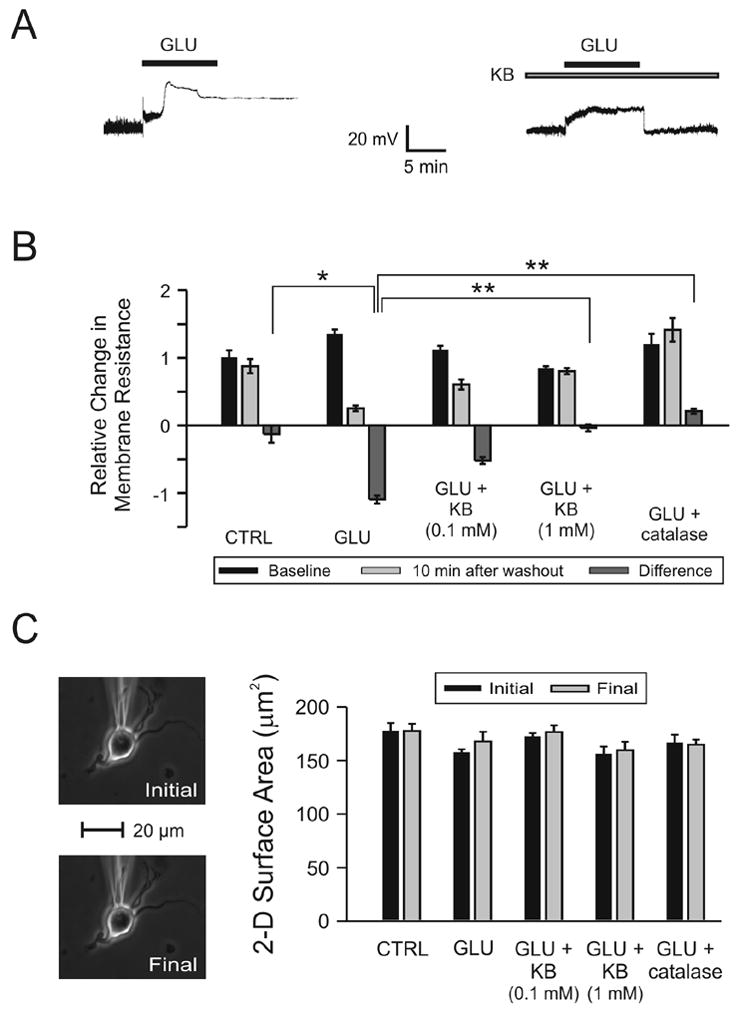
Changes in neuronal membrane properties following glutamate (GLU) excitotoxicity are prevented by ketones (KB). KB were administered as a cocktail in a 1:1 concentration ratio of BHB to ACA (either 0.1 mM or 1.0 mM each). (A) Exposure to GLU (10 μM ) for 10 min (short horizontal bar) alone resulted in an initial, small depolarization followed by a sharp, larger increase of the membrane potential (Vm) without return to baseline despite a 10 min washout period. The same protocol was used in the presence of BHB and ACA (1 mM each; long horizontal bar) but in this case, GLU induced a stable, depolarizing response with return of Vm to baseline after discontinuation of the application. (B) GLU (n = 13) significantly reduced Rm relative to control (CTRL; n = 8; p = 0.02). Following the addition of the 1 mM KB combination 20 (N = 13) or catalase 250 U/ml (n = 6), but not the 0.1 mM KB combination (n = 12), Rm was significantly higher than in cases exposed to GLU alone (p < 0.01). (C) The electrophysiological changes induced by GLU could not be attributed to morphological changes or technical difficulties. The two-dimensional surface area of the neuron exposed to GLU alone (in the absence of any fluorescent dyes) was unchanged by the experimental protocol and the position of the recording electrode was stable throughout the experiment. Neurons in all treatment groups displayed similar surface areas at the beginning and at the end of the experiments.
When both BHB and ACA (1 mM each) were added 5 min prior to glutamate application and maintained throughout the experiment, a stable depolarizing response was elicited during exposure to glutamate (Fig. 2A). The amplitude of the depolarization induced by glutamate (averaging 11.6 ± 0.7 mV) was not affected by pre-treatment with ketones. After a 10 min. washout period, Vm returned to baseline and Rm remained unchanged in 11/13 cells (Fig. 2B). This phenomenon was replicated in 5/6 neurons treated with catalase (250 U/ml), an enzyme that breaks down hydrogen peroxide (H2O2) to oxygen and water, suggesting that ketone-mediated protection was through an antioxidant mechanism.
Ketones Decrease Free Radical Levels
Since ketones did not influence responsiveness to glutamate, we investigated whether their neuroprotective effect might be mediated by a reduction in oxidative stress, as suggested by the catalase experiments. The effects of BHB and ACA on free radical levels were examined with the fluorescent indicator dihydroethidium (DHE), which reflects superoxide (and likely peroxynitrite) levels (Budd et al, 1997; Vergun et al, 2001). Exposure of neurons to DHE over a 25 min period without glutamate or ketones resulted in a slow, modest, and progressive increase in DHE fluorescence over time (Fig. 3A). When neurons were exposed to 10 μM glutamate following a 5 min baseline, a faster and more significant increase in DHE fluorescence occurred and continued during the 10 min washout period. This increase was prevented by the addition of BHB and ACA (1 mM each). Despite similar DHE signals and two-dimensional surface areas at baseline, the group exposed to glutamate alone exhibited significantly higher levels of DHE fluorescence (ANOVA on ranks, p = 0.01). BHB and ACA applied without glutamate also decreased baseline levels of DHE fluorescence. Two-dimensional areas did not change significantly for any experimental condition.
Figure 3.
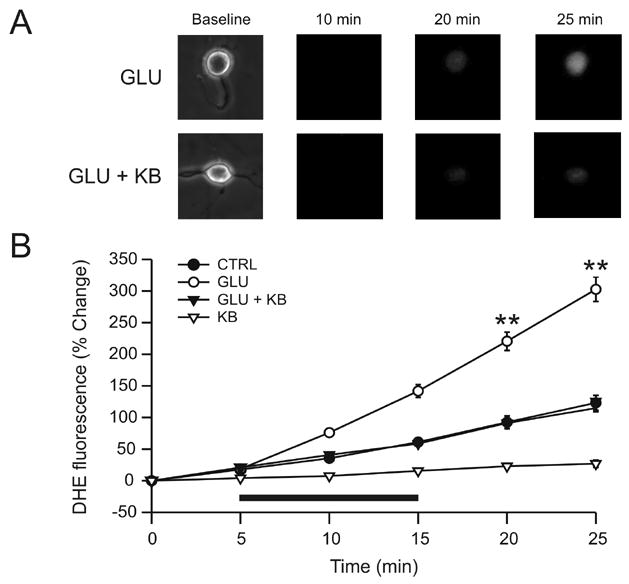
Ketones (KB) block glutamate-induced increases in superoxide radicals. (A) Exposure of neurons to glutamate (GLU; 10 μM) for 10 min led to a time-dependent increase in the fluorescence of DHE, a marker for the free radical superoxide (n = 15). In the presence of both GLU and KB (D-β-hydroxybutyrate and acetoacetate, 1 mM each), the increase in DHE fluorescence was smaller (GLU + KB; n = 16). (B) GLU (horizontal bar) provoked a time-dependent increase in DHE fluorescence that persisted after discontinuation of GLU but that was significantly reduced (p = 0.01) by KB, down to levels similar to the control group (CTRL; n = 11). The application of KB alone (KB; n = 8) significantly decreased the DHE signal below that of control experiments (p = 0.04). ** p < 0.01.
Ketones Decrease Production of Reactive Oxygen Species Rather Than Increase Glutathione
Our data indicated that ketones blocked the increase in DHE signal induced by glutamate. However, a change in DHE fluorescence does not necessarily imply a change in free radical production but rather reflects changes in the steady-state concentrations of ROS (Nicholls, 2004). Consequently, an increase in DHE signal could be secondary to a decrease in endogenous antioxidants such as glutathione. To evaluate this possibility, acutely dissociated neurons were incubated with monochlorobimane (MCB), a fluorescent marker specific for reduced glutathione, the active form of that antioxidant (Keelan et al, 2001). Exposure to 10 μM glutamate for 10 min resulted in a significant decrease of MCB fluorescence that persisted after glutamate washout (Fig. 4A). Ketones reversed the decrease in MCB fluorescence induced by glutamate but not by diamide, a thiol-specific oxidizing agent (Zago et al, 2000; Ault & Lawrence, 2003). Moreover, ketones did not increase MCB fluorescence above baseline. Based on these results, we hypothesized that ketones affected MCB fluorescence in glutamate-treated cells by reducing ROS production rather than by increasing glutathione levels.
Figure 4.
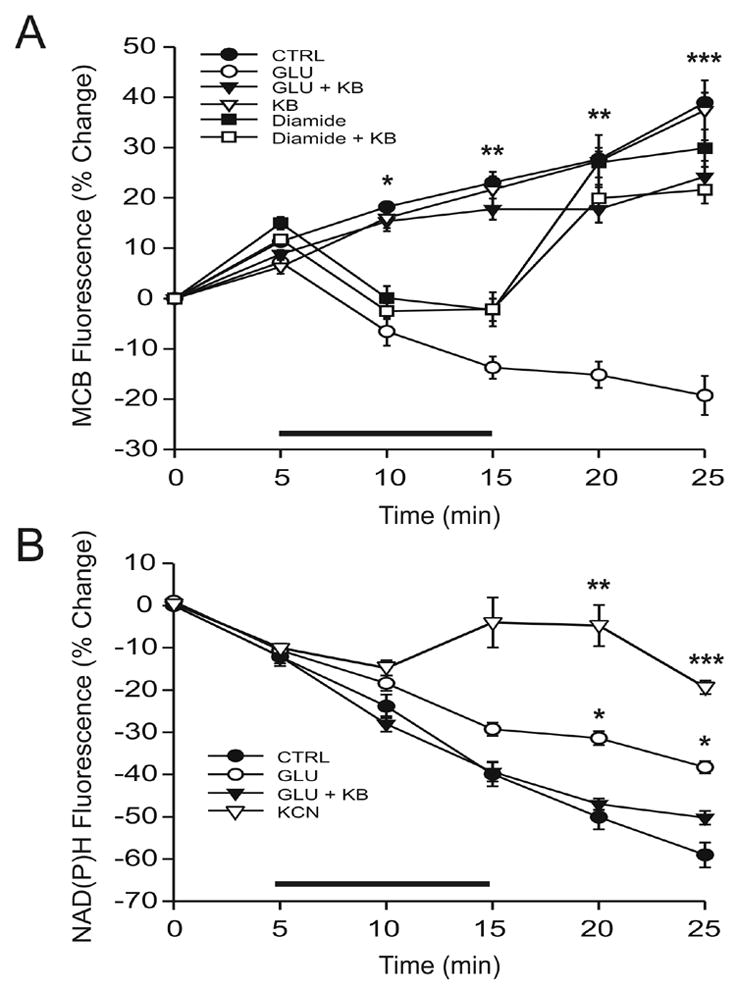
The antioxidant effects of ketones (KB) are mediated by NADH oxidation and not by an increase in reduced glutathione. (A) Acutely dissociated neurons were incubated with monochlorobimane (MCB), a fluorescent marker of reduced glutathione. The decrease in MCB fluorescence induced by 10 μM glutamate alone (GLU; n = 7) for 10 min was prevented by ketones (GLU + KB; n = 8; p < 0.01). The administration of 10 mM diamide, a specific thiol oxidant, for 10 min temporarily decreased MCB fluorescence as well (diamide; n = 10 mM; p = 0.01) but ketones did not have any effect under these conditions (diamide + KB; n = 5). Moreover, ketones did not affect baseline levels of MCB (KB; n = 5). (B) Glutamate (n = 7) increased the NAD(P)H signal relative to controls (n = 6), suggesting that the observed decrease in glutathione levels was secondary to increased production of reactive oxygen species (ROS). Ketones blocked the effect of glutamate on NAD(P)H, confirming that they decrease ROS rather an increase glutathione (n = 6). Differences were statistically significant (p = 0.01 at 20 min and 0.04 at 25 min). The effects of 1 mM potassium cyanide (KCN) are also presented for comparative purposes. Exposure to KCN for 10 min significantly increased NAD(P)H fluorescence almost to baseline levels. Horizontal bars indicate 10 min treatment periods. * p < 0.05; ** p < 0.01; *** p < 0.001.
To extend these findings and determine the mechanism and site of ketone action, NADH autofluorescence was studied in the same preparation. Free radical production is associated with increases in NADH whereas enhanced NADH oxidation results in a reduction of ROS production (Duchen, 1992; Kudin et al, 2004; Sullivan et al, 2004). NADH is produced in mitochondria by the Krebs cycle, and when excited in the ultraviolet range, emits an autofluorescent signal in the blue range. Fluorescence is lost following oxidation by the mitochondrial respiratory chain. NADPH, which is involved in the reduction of glutathione, exhibits a fluorescence profile that is indistinguisable from that of NADH. Fluorescence signals are therefore typically referred to as NAD(P)H transients, although NADH is the predominant source of the autofluorescence due to significantly higher mitochondrial concentrations. Exposing acutely dissociated neurons to 10 μM glutamate for 10 min significantly increased the NAD(P)H signal relative to the control condition of buffer only (Fig. 4B). The addition of ketones reversed the effects of glutamate. This combination of results is in fact the opposite of what would be expected if ketones exerted significant effects on glutathione levels. In order to better assess the magnitude of changes in NAD(P)H levels, we also ran control experiments with the respiratory chain inhibitor potassium cyanide (KCN). Exposure to 1 mM KCN for 10 min led to a significant increase in NAD(P)H fluorescence in isolated neurons to almost pretreatment levels (Fig. 4B).
Ketones Decrease NADH Levels
The effects of ketones on ROS and NAD(P)H levels were further confirmed in isolated mitochondria exposed to high doses of calcium to simulate glutamate excitotoxicity. It is well established that glutamate excitotoxicity increases intracellular calcium concentration and that calcium entry into the mitochondria stimulates ROS production (Hasselbalch et al, 1995; Brookes et al, 2004). Our data clearly demonstrated that exposure to 10 μM glutamate for 10 min led to a significant increase in fluorescence emitted by Rhod-2, an indicator of mitochondrial calcium content. These results suggested that glutamate excitotoxicity increased mitochondrial production of ROS in neocortical neurons (ANOVA on ranks, p < 0.05; Fig. 5A). Importantly, ketones did not reduce calcium entry into the mitochondria.
Figure 5.
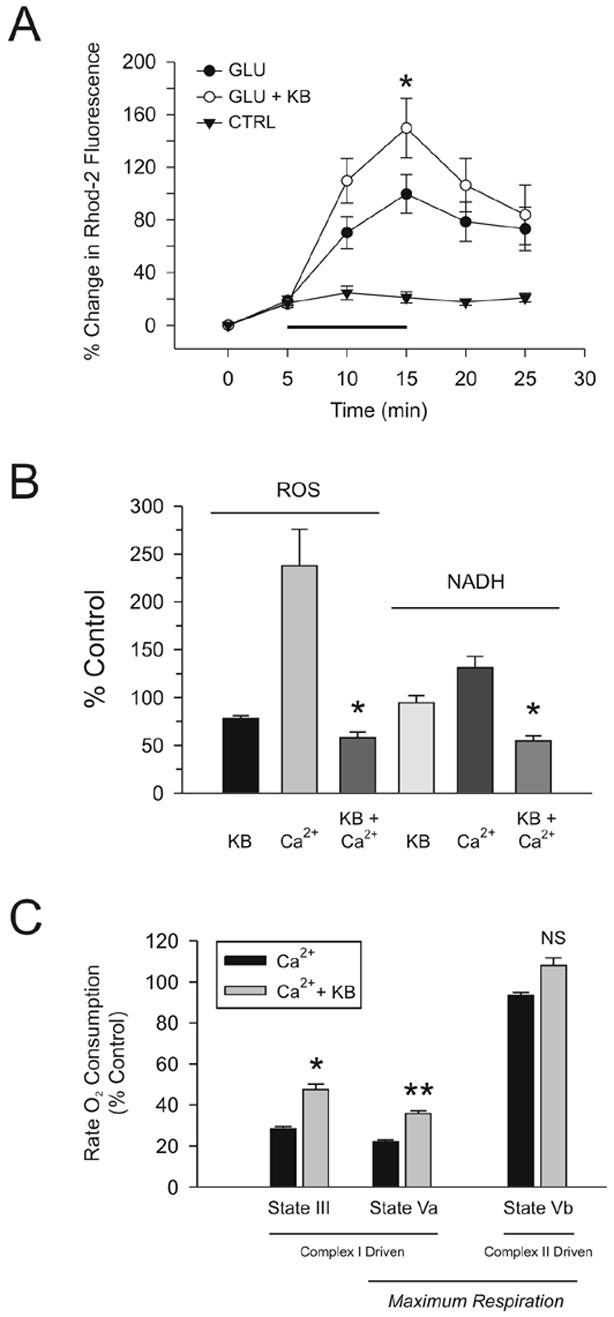
Ketones (KB) reduce calcium-induced alterations in mitochondrial bioenergetics, ROS production and NAD+/NADH cycling. (A) Glutamate-mediated influx of calcium into mitochondria was not affected by ketones. Acutely dissociated neurons were incubated with the fluorescent mitochondrial calcium indicator Rhod-2. Exposure to 10 μM glutamate for 10 min (n = 5) increased mitochondrial calcium levels significantly relative to controls (n = 4). The same findings were observed when 1 mM BHB and 1 mM ACA were added (n = 4; ANOVA on ranks with Dunn’s post-hoc analyses; p < 0.05). (B) 0.5 mM calcium significantly increased mitochondrial ROS production and NADH concentrations. The addition of BHB and ACA (1 mM each) reduced both mitochondrial ROS production and NADH levels to baseline. (C) Mitochondrial respiration was assessed using a Clark-type electrode and demonstrated that calcium significantly impaired complex-I (NADH-linked) driven oxygen consumption both in response to the addition of ADP (state III respiration) or carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone [FCCP] (maximum electron transport system capacity; state Va) but did not significantly alter complex-II (FADH-linked) driven maximum electron transport system capacity (state Vb). The inclusion of 1 mM BHB and 1 mM ACA partially reversed this inhibition of respiration. * p < 0.05; ** p < 0.01.
Following exposure of isolated mitochondria to calcium, both ROS (evaluated with the indicator 2',7'-dichlorofluorescein diacetate) and NAD(P)H levels, measured simultaneously, increased significantly compared to control values (ANOVA on ranks with Dunn’s post-hoc analysis, p < 0.05; Fig. 5B). Moreover, calcium inhibited mitochondrial respiration in the presence of ADP and the uncoupler carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP), but not succinate, thereby suggesting that electrons donated by NADH to complex I of the electron transport chain had an increased probability of slipping from intermediates to form superoxide rather than be utilized for ATP synthesis (Fig. 5C). Calcium effects were completely reversed by the addition of BHB and ACA (1 mM each). ROS and NAD(P)H levels actually decreased below control levels as ketones significantly increased complex I (but not complex II)-driven mitochondrial respiration.
DISCUSSION
The principal finding of our study is that ketones, produced by the liver under conditions of fasting, calorie restriction or treatment with high-fat, low-carbohydrate diets, prevent glutamate excitotoxicity by reducing ROS levels in both acutely dissociated neocortical neurons and in isolated neocortical mitochondria. These results are consistent with previous findings showing that ACA and BHB increase the viability of HT22 hippocampal cell lines and primary hippocampal neurons following glutamate excitotoxicity (Noh et al, 2005). Increased survival of HT22 cells was associated with decreased production of ROS and decreased markers for apoptosis and necrosis. We additionally demonstrate that the reduction in free radical formation by ketones occurs through enhancement of NADH oxidation (i.e., increased NAD+/NADH ratios) and mitochondrial respiration in neocortical neurons without alterations in levels of the endogenous antioxidant glutathione. Our data therefore provide further support for the neuroprotective properties of ketones and offer insights into their antioxidant activity at the mitochondrial level.
Glutamate excitotoxicity is a pathogenic process that can lead to calcium-mediated neuronal injury and death by generating reactive oxygen and nitrogen species, as well as impairing mitochondrial bioenergetic function (Sun et al, 2001; Nagy et al, 2004; Nicholls, 2004). Oxidative stress subsequently damages nucleic acids, proteins and lipids and potentially opens the mitochondrial permeability transition pore which, in turn, can further stimulate ROS production, worsen energy failure and release pro-apoptotic factors such as cytochrome c into the cytoplasm (Nicholls, 2004; Kowaltowski et al, 2001). Although it is generally accepted that calcium influx is associated with increased ROS levels and that mitochondria are the main source of ROS, linking both phenomena in intact neurons has thus far been challenging (Brookes et al, 2004; Hongpaisan et al., 2004; Balaban et al, 2005). Evidence from isolated mitochondria indicates that ROS production requires a hyperpolarized mitochondrial membrane potential, but calcium influx into the mitochondria actually decreases the mitochondrial membrane potential (Nicholls, 2004). Similarly, cerebellar granule cells subjected to glutamate excitotoxicity display increased ROS levels despite a decrease in mitochondrial membrane potential (Ward et al, 2000). These contradictory findings have led some authors to suggest that cytoplasmic enzymes might be the source of ROS during glutamate excitotoxicity, whereas others have hypothesized that oxidative stress results from decreased levels of antioxidants such as glutathione (Atlante et al, 1997; Almeida et al, 1998; Sanganahallli et al, 2005).
In the present study, combining data from acutely dissociated neurons and isolated mitochondria without the use of respiratory inhibitors, glutamate excitotoxicity and calcium resulted in an inhibition of mitochondrial respiration and an increase of mitochondrial ROS production that persisted beyond the period of glutamate administration. At both whole cell and mitochondrial levels, we found increased NAD(P)H fluorescence following glutamate administration (as well as increased DHE and DCF levels), and a reversal of these effects by concomitant ketone administration, suggesting that glutamate excitotoxicity increased the mitochondrial production of ROS and that ketones reduced ROS levels in mitochondria. Moreover, by using monochlorobimane, a fluorescent marker for reduced glutathione, to examine the effects of ketone bodies on glutathione levels following exposure to diamide, a thiol oxidant which is known to inactivate glutathione peroxidase (Armstrong & Jones, 2002), we found that ketones did not significantly affect glutathione depletion or recovery. Finally, our findings pointed specifically to complex I as the site of electron transfer inhibition. Consistent with this result are previous findings suggesting that, in neurons, ROS are generated at complex I by a self-sustaining cycle that maintains ROS production beyond the original injury (Kudin et al, 2004; Turrens, 2003).
Although the neuroprotective properties of ketones have been previously reported, uncertainty surrounds their antioxidant effects. First, ACA might actually stimulate ROS generation in brain mitochondria and endothelial cells (Jain et al, 1998; Tieu et al, 2003). Second, prior work on cardiac myocytes suggested that ketones increased glutathione levels by reducing the NADP/NADPH couple (Veech et al, 2001; Squires et al, 2003). Third, in rat liver mitochondria, although ACA decreased NAD(P)H fluorescence, state IV respiration was not affected and the major consequence was opening of the mitochondrial permeability transition pore, a phenomenon attributed by the authors to impaired glutathione antioxidant function (Zago et al, 2000).
The contradictory findings regarding the effects of ketones most probably reflect technical and tissue-specific differences. First, the source of mitochondrial ROS in neurons (complex I) differs from that in non-neuronal cells such as cardiac myocytes (complex III) (Turrens, 2003). Second, the use of respiratory inhibitors such as rotenone and oligomycin instead of, or in addition to, calcium would undoubtedly influence results (Brookes et al, 2004). Nevertheless, our findings, made in neurons and isolated neuronal mitochondria without added respiratory inhibitors, indicate that a combination of ACA and BHB (which potentially better mirrors brain physiological conditions compared to previous studies employing one ketone body or the other) exert a neuroprotective, antioxidant effect by decreasing ROS production in neuronal tissues.
A separate protective effect has also been proposed for BHB, mainly in cardiac tissue but also in the brain, and involves increased ATP production (Suzuki et al, 2001 & 2002; Veech et al, 2001; Brookes et al, 2004). Our results are consistent with these earlier studies since the improvement of mitochondrial respiration by ketone bodies would help counteract the increased bioenergetic demand that characterizes glutamate excitotoxicity (Nicholls, 2004).
Overall, similarities between the neuroprotective effects of ketones and those of calorie restriction are emerging at the mitochondrial level. Calorie restriction decreases ROS generation, possibly at complex I, and stimulates mitochondrial respiration along with an increase in NAD+ (Merry, 2002 & 2004; Bordone & Guarente 2005; Guarente and Picard, 2005). Although these changes might influence a variety of cellular processes, including modulation of regulatory proteins such as Sirt1 (Chen et al, 2005) or enhancement of ATP production (Nisoli et al, 2005), we propose an additional mechanism whereby the neuroprotective effects of calorie restriction might be mediated by the antioxidant effects of ketones in mitochondria. Ketones may therefore provide a neuroprotective strategy that naturally targets the mitochondria with the potential not only to reduce oxidative injury and death but also to maintain bioenergetic processes and preserve cellular function.
Acknowledgments
This work was supported by NIH grants NS 044846 (JMR), NS048191/NS046426 (PGS) and the Barrow Neurological Foundation. We thank Timothy A. Simeone, Kristina Fenoglio and Heather A. Milligan for critical reading of this manuscript.
The abbreviations used are
- BHB
β-hydroxybutyrate
- ACA
acetoacetate
- MPP+
1-methyl-4-phenylpyridinium
- ROS
reactive oxygen species
- PI
propidium iodine
- DHE
dihydroethidium
- KCN
potassium cyanide
- MCB
monochlorobimane
- HRP
horse radish peroxidase
- H2O2
hydrogen peroxide
- FCCP
carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone
- KB
ketones
- GLU
glutamate
Footnotes
Section Editor: Molecular Neuroscience
W. Sieghart, Brain Research Institute, University of Vienna, Division of Biochemistry and Molecular Biology, Spitalgasse 4, A-1090 Vienna, Austria
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida A, Heales SJ, Bolanos JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res. 1998;790:209–216. doi: 10.1016/s0006-8993(98)00064-x. [DOI] [PubMed] [Google Scholar]
- Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. FASEB J. 2002;16:1263–1265. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Ciotti MT, Marra E, Calissano P. Glutamate neurotoxicity in rat cerebellar granule cells: a major role for xanthine oxidase in oxygen radical formation. J Neurochem. 1997;68:2038–2045. doi: 10.1046/j.1471-4159.1997.68052038.x. [DOI] [PubMed] [Google Scholar]
- Ault JG, Lawrence DA. Glutathione distribution in normal and oxidatively stressed cells. Exp Cell Res. 2003;285:9–14. doi: 10.1016/s0014-4827(03)00012-0. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. J Neurosci Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Sombati S, DeLorenzo RJ. Electrophysiology of glutamate neurotoxicity in vitro: induction of a calcium-dependent extended neuronal depolarization. J Neurophysiol. 1992;68(2):362–373. doi: 10.1152/jn.1992.68.2.362. [DOI] [PubMed] [Google Scholar]
- Denny CA, Kasperzyk JL, Gorham KN, Bronson RT, Seyfried TN. Influence of caloric restriction on motor behavior, longevity, and brain lipid composition in Sandhoff disease mice. J Neurosci Res. 2006;83(6):1028–1038. doi: 10.1002/jnr.20798. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283( Pt 1):41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JM, Vining EPG, Pillas DJ, Pyzik PL, Casey JC, Kelly MT. The efficacy of the ketogenic diet - 1998. A prospective evaluation of intervention in 150 children. Pediatr. 1998;102:1358–1363. doi: 10.1542/peds.102.6.1358. [DOI] [PubMed] [Google Scholar]
- Gilbert DL, Pyzik PL, Freeman JM. The ketogenic diet: seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J Child Neurol. 2000;15:787–90. doi: 10.1177/088307380001501203. [DOI] [PubMed] [Google Scholar]
- Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am J Physiol Endocrinol Metab. 1995;268:E1161–E1166. doi: 10.1152/ajpendo.1995.268.6.E1161. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J Neurosci. 2004;24:10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SK, Kannan K, Lim G. Ketosis (acetoacetate) can generate oxygen radicals and cause increased lipid peroxidation and growth inhibition in human endothelial cells. Free Radic Biol Med. 1998;25:1083–1088. doi: 10.1016/s0891-5849(98)00140-3. [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci U S A. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelan J, Allen NJ, Antcliffe D, Pal S, Duchen MR. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res. 2001;66:873–884. doi: 10.1002/jnr.10085. [DOI] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Koubova J, Guarente L. How does calorie restriction work? Genes Dev. 2003;17:313–321. doi: 10.1101/gad.1052903. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- Macklis JD, Madison RD. Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: a fluorescence scale of membrane integrity. J Neurosci Methods. 1990;31:43–46. doi: 10.1016/0165-0270(90)90007-3. [DOI] [PubMed] [Google Scholar]
- Mahoney LB, Denny CA, Seyfried TN. Caloric restriction in C57BL/6J mice mimics therapeutic fasting in humans. Lipids Health Dis. 2006;5:13. doi: 10.1186/1476-511X-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massieu L, Haces ML, Montiel T, Hernandez-Fonseca K. Acetoacetate protects hippocampal neurons against glutamate-mediated neuronal damage during glycolysis inhibition. Neuroscience. 2003;120:365–378. doi: 10.1016/s0306-4522(03)00266-5. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL, Duan W. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol Rev. 2002;82:637–672. doi: 10.1152/physrev.00004.2002. [DOI] [PubMed] [Google Scholar]
- Merry BJ. Molecular mechanisms linking calorie restriction and longevity. Int J Biochem Cell Biol. 2002;34:1340–1354. doi: 10.1016/s1357-2725(02)00038-9. [DOI] [PubMed] [Google Scholar]
- Merry BJ. Oxidative stress and mitochondrial function with aging--the effects of calorie restriction. Aging Cell. 2004;3:7–12. doi: 10.1046/j.1474-9728.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- Nagy K, Kis B, Rajapakse NC, Bari F, Busija DW. Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J Neurosci Res. 2004;76:697–704. doi: 10.1002/jnr.20120. [DOI] [PubMed] [Google Scholar]
- Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res. 2005;83:702–709. doi: 10.1002/jnr.20736. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Sanganahalli BG, Joshi PG, Joshi NB. Xanthine oxidase, nitric oxide synthase and phospholipase A(2) produce reactive oxygen species via mitochondria. Brain Res. 2005;1037:200–203. doi: 10.1016/j.brainres.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Squires JE, Sun J, Caffrey JL, Yoshishige D, Mallet RT. Acetoacetate augments beta-adrenergic inotropism of stunned myocardium by an antioxidant mechanism. Am J Physiol Heart Circ Physiol. 2003;284:H1340–1347. doi: 10.1152/ajpheart.00473.2002. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol. 2000;48:723–729. [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–717. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J Comp Neurol. 2004;474:524–534. doi: 10.1002/cne.20130. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sun DA, Sombati S, DeLorenzo RJ. Glutamate injury-induced epileptogenesis in hippocampal neurons: an in vitro model of stroke-induced "epilepsy". Stroke. 2001;32:2344–2350. doi: 10.1161/hs1001.097242. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A. Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn J Pharmacol. 2001;87:143–150. doi: 10.1254/jjp.87.143. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol. 2002;89:36–43. doi: 10.1254/jjp.89.36. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF., Jr Ketone bodies, potential therapeutic uses. IUBMB Life. 2001;51:241–247. doi: 10.1080/152165401753311780. [DOI] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR. Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol. 2001;531:147–163. doi: 10.1111/j.1469-7793.2001.0147j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vining EPG, Freeman JM, Ballaban-Gil K, Camfield CS, Camfield PR, Holmes GL, Shinnar S, Shuman R, Trevathan E, Wheless JW. A multicenter study of the efficacy of the ketogenic diet. Arch Neurol. 1998;55:1433–1437. doi: 10.1001/archneur.55.11.1433. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago EB, Castilho RF, Vercesi AE. The redox state of endogenous pyridine nucleotides can determine both the degree of mitochondrial oxidative stress and the solute selectivity of the permeability transition pore. FEBS Lett. 2000;478:29–33. doi: 10.1016/s0014-5793(00)01815-9. [DOI] [PubMed] [Google Scholar]