

Abstract
Anger is an independent predictor of coronary heart disease (CHD) events, although the mechanisms for this relation are unclear. We examined the effects of an anger-provoking interview vs. a neutral interview on endothelium-dependent and endothelium-independent vasodilation (EDV, EIV) assessed by brachial artery ultrasound in 14 healthy subjects without CHD risk factors. The anger provocation condition, but not the neutral condition, caused a significant impairment in EDV at 90 minutes compared to baseline (p=.004) and 30 minutes (p=.013). Similarly, EIV was significantly impaired at 90 minutes following the angry interview, compared to baseline (p=.003) and 30 minutes (p=.001). The decrease in EDV and EIV was greater following the anger-provoking interview than the neutral interview, especially between 30 and 90 minutes. In conclusion, our preliminary results suggest that an episode of anger is associated with a dysregulation in both endothelium-dependent and endothelium-independent pathways, suggesting these mechanisms might contribute to the link between anger and CHD events.
Although anger is associated with an increased short-term and long-term risk of coronary heart disease (CHD) events, independent of traditional CHD risk factors,1–6 the mechanisms that underlie this relation are unknown. Endothelial dysfunction plays a major role in the development of atherosclerosis.7 Traditional CHD risk factors are associated with endothelial dysfunction8 and may also impair arterial vasodilation in response to exogenous nitric oxide (NO), suggesting concomitant vascular smooth muscle dysfunction.8,9 Thus, the higher risk of CHD events associated with anger may be similarly mediated through an impairment in endothelium-dependent vasodilation (EDV) with or without an impairment in endothelium-independent vasodilation (EIV). To our knowledge, the effects of anger on EDV and EIV have never previously been examined. To test this hypothesis, we examined the effects of anger induction in humans on EDV and EIV assessed by brachial artery ultrasonography.
Methods and Results
Fourteen healthy adult participants (mean age 35 ± 8 years; range 22–52 years, 8 women) were screened from the community through advertisements. Participants were enrolled if they did not report a history of cardiovascular disease; CHD risk factors including hypertension, diabetes, hypercholesterolemia, smoking history, and family history of premature CHD; or concurrent medication use, including over-the-counter drugs. Written informed consent was obtained from all subjects, and the study was approved by the Institutional Review Board of Columbia University Medical Center.
Participants were exposed to an anger-provoking and a neutral interview, one-month apart, in randomized order. Anger provocation was accomplished by administering the Extended Type A Structured Interview (ESI),10 a 12-minute structured interview that elicits anger by asking participants about their characteristic responses to a variety of different provocative situations, posed in a challenging manner. The 12-minute neutral version of the interview contains the same questions, but the interviewer does not challenge the respondent. Subjects were given a general outline of the nature of the study, but were not provided specific details of the protocol including the content of the interviews.
Blood pressure (BP) and heart rate were recorded at 6-minute intervals throughout the study period using an ambulatory BP monitor (Spacelabs model 90207, Redmond, WA). Brachial artery EDV was measured by high-resolution ultrasonography as the response to reactive hyperemia induced by 5-minute BP cuff occlusion of the forearm.11 End-diastolic images were digitized at baseline and 1 minute after cuff deflation. EIV was then assessed by measuring brachial artery response to 0.4 mg sublingual nitroglycerin. A blinded reader analyzed brachial artery diameters off-line using analysis software. EDV and EIV were calculated as the percentage increase in brachial artery diameter from rest, during reactive hyperemia and after nitroglycerin administration. 12,13 Intra-observer variability for EDV measurements was 1.3%. 11 To assess the level of anger experienced, subjects answered the following question prior to the ultrasound assessments: “How angry do you feel at this moment?”, on a 5-point Likert-type scale (1=“not at all” and 5=“extremely”).14 To avoid distraction, no measurement was taken during the interview. Thus, brachial artery EDV and EIV, and the severity of anger experienced were assessed at baseline (pre-interview), and at 30 and 90 minutes after the end of each interview.
Data are expressed as mean value ± SD unless stated otherwise. Nonparametric tests, which do not require assumptions of normality, were used for statistical comparisons. The Friedman test was used to evaluate the effects of the interview (angry vs. neutral) on EDV and EIV, hemodynamic parameters, and angry mood ratings. Comparisons of measures at baseline and at each of the three time points between the neutral and anger-provocation conditions were analyzed by the Wilcoxon test. A p value <.05 was considered significant.
Baseline systolic BP and diastolic BP, heart rate, brachial artery diameter, EDV, EIV, and anger rating scores did not differ significantly between the neutral and anger-provoking interviews. Systolic BP and diastolic BP significantly increased after anger provocation, but no change was observed after the neutral interview (Figure 1). Feelings of anger also significantly increased following anger provocation, but no change in anger was observed in the neutral condition.
Figure 1.
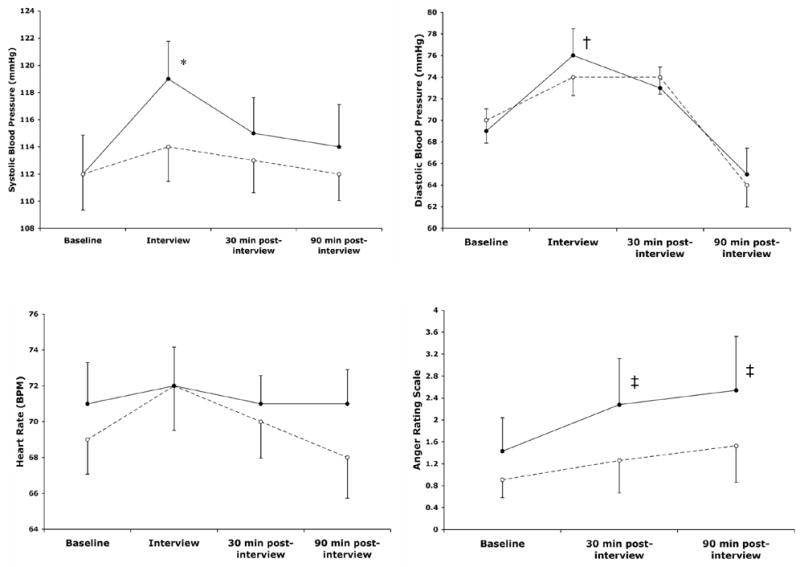
Mean changes (± SEM) in systolic and diastolic blood pressure, heart rate, and self-reported anger before and after the stimulus interviews.
● anger provocation condition, ○ neutral condition
*p=.006 vs. baseline, †p=.001 vs. baseline, ‡p=.02 vs. baseline.
Following the angry interview (Figure 2A), EDV significantly decreased over the three intervals (7.5±4.3% at baseline, 6.9±7.6% after 30 minutes, 0.6±5.6% after 90 minutes; χ2 Friedman = 12.00, p=.002). Similar findings (Figure 2B) were seen with EIV after anger provocation (25.6±9.7% at baseline, 27.3±8.9% after 30 minutes, and 14.6±10.8% after 90 minutes; χ2 Friedman = 16.00, p=.001). In the neutral condition, none of the individual changes in EDV at any of the intervals were significant (6.3±4.2% at baseline, 3.4±4.5% after 30 minutes, and 3.8±4.8% after 90 minutes, ps>.05 for all comparisons), although significant changes were observed in EIV (26.0±9.4% at baseline, 21.9±9.1% after 30 minutes, and 17.5±11.9% after 90 minutes; χ2 Friedman = 9.57, p=.008). EIV at 30 and 90 minutes were both significantly lower than at baseline (p=.03, 30 minutes vs. baseline; p=.008, 90 minutes vs. baseline). However, unlike in the anger condition, the change between 30 and 90 minutes in the neutral condition was not significant.
Figure 2.
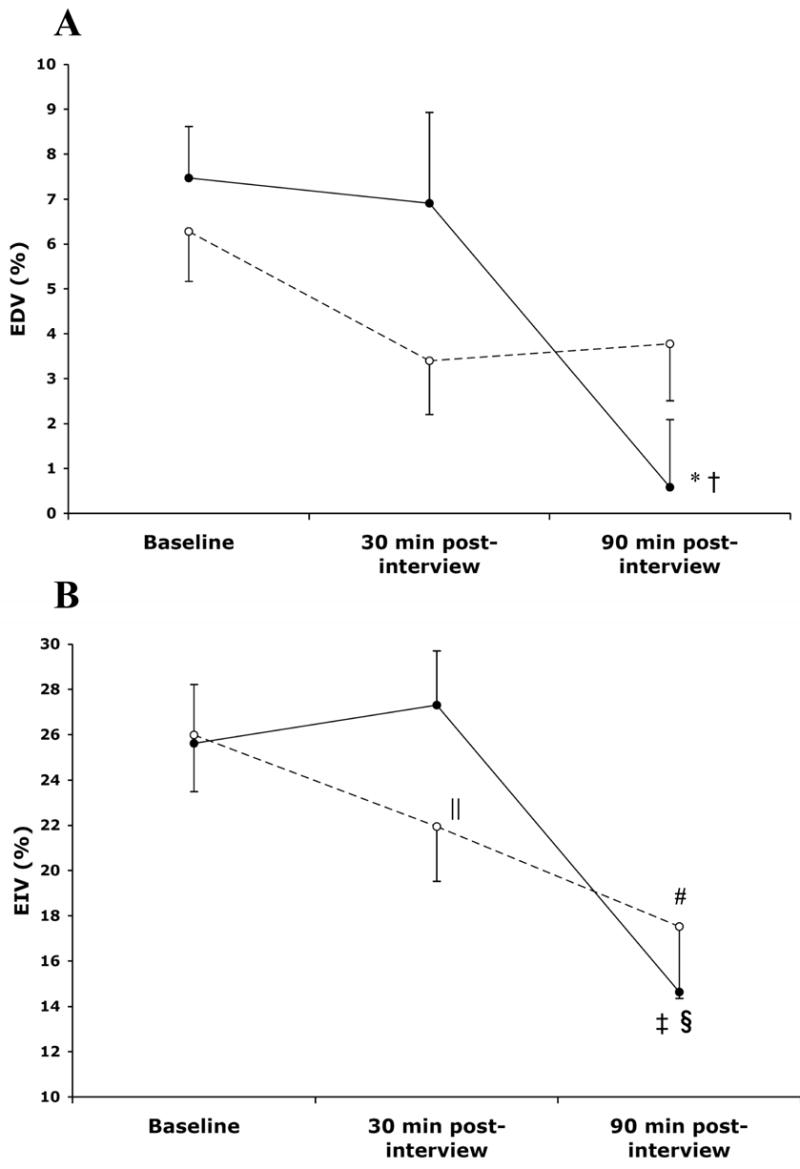
Mean changes (± SEM) in EDV (A) and EIV (B) before and after the stimulus interviews.
*p=.004 vs. baseline, †p=.013 vs. 30 minutes.
‡p=.003 vs. baseline, §p=.001 vs. 30 minutes.
||p=0.03 vs. baseline, #p=0.008 vs. baseline.
● anger provocation condition, ○ neutral condition.
EDV = endothelium-dependent vasodilation, EIV = endothelium-independent vasodilation
Resting brachial artery diameters measured prior to brachial artery occlusions at 30 and 90 minutes were not significantly different after anger provocation compared to the neutral condition. Compared to the neutral condition, anger was associated with a greater reduction in EDV, especially between 30 and 90 minutes (p=.07, 90 vs. 30 minutes; p=.06, 90 minutes vs. baseline). Between baseline and 30 minutes, a significantly greater reduction in EIV was seen in the neutral condition than after anger provocation. However, as the session progressed from 30 to 90 minutes, EIV after anger provocation declined substantially and significantly more than in the neutral condition (p=.016). Thus, similar to EDV, the decrease in EIV appeared to be greater following the anger-provoking interview than after the neutral interview, especially between 30 and 90 minutes.
Discussion
We demonstrated an acute dysregulation of both flow-mediated and nitroglycerin-induced vasodilation, two pathways of the NO-cyclic guanosine monophosphate vasodilator system, following anger induction. Possible mechanisms that could account for these findings include decreased NO bioavailability or smooth muscle dysfunction. If decreased bioavailability of NO plays a role, it is unlikely that a reduction in NO synthesis is the cause, as we observed a reduction in vasodilation in response to the administration of exogenous nitrates after anger-provocation. Alternatively, increased production of oxygen-derived free radicals may have led to decreased bioavailability of NO to the smooth muscle cell, impairing the ability of the brachial artery to dilate to endogenous and exogenous NO. However, this mechanism is also unlikely because nitroglycerin is converted to NO inside smooth muscle cells,15 and oxidative stress impairs vasodilation in response to nitroglycerin to a lesser extent than to endogenous NO.16,17
Therefore, our findings suggest that smooth muscle dysfunction plays a primary role in the effect of anger on vascular reactivity. It is possible that structural abnormalities, such as fibrosis or atrophy, could have prevented the artery from dilating in response to either reactive hyperemia or nitroglycerin. However, anger would not have acutely altered EDV and EIV in this scenario. Rather, decreased responsiveness or activity of the NO-cyclic guanosine monophosphate pathway at the level of the vascular smooth muscle most likely accounted for the study findings. To our knowledge, an acute impairment of the smooth muscle NO- cyclic guanosine monophosphate vasodilator system after induction of a specific negative emotion such as anger has never previously been described. Several studies have demonstrated that a standardized mental stress task, as opposed to anger, reduces EDV but not EIV. 12, 13, 18, 19 Proposed mechanisms to explain the impairment in EDV include a dysregulation in the sympathetic nervous system,20,21 increased cortisol production,19 and the release of other non-sympathomimetic vasoconstrictors such as endothelin.13 However, these are unlikely to play a major role in the impairment in EDV and EIV observed after anger provocation. Sympathetic nervous system activation21 and increased cortisol production only affect EDV and not EIV.19 Further, anger was not associated with a change in resting brachial artery diameters, suggesting that vasoconstrictors did not play a substantial role. Therefore, our findings suggest a novel pathway by which anger may affect vascular reactivity and the subsequent risk of CHD events.
An interesting aspect of the present study is that the effect of anger on EDV and EIV was delayed, as a significant effect was not seen until 30 to 90 minutes. Reasons for this delayed effect are unknown, however, it is worth noting that the time course of anger’s effect on brachial artery reactivity is consistent with the time course of myocardial infarction risk, which reaches its peak in apparently healthy subjects within a 2-hour period after an angry episode.1,2
Strengths of the study include the random assignment to either angry or neutral condition first, the use of an active neutral condition, and the within-subjects design in which each subject served as their own control. A potential limitation of our study is the relatively small sample size. However, differences consistent with our hypotheses were observed, and the use of a within-subjects design, which minimizes the error variance associated with individual differences, provided adequate power to detect effects. Further research should be conducted to replicate our preliminary findings, and to examine whether impaired EDV and EIV mediate the relation between anger and incident CHD events.
Footnotes
The authors have no financial conflicts of interest. This work was supported by grants RR00645, HL072866, and HL076857 from the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mittleman MA, Maclure M, Sherwood JB, Mulry RP, Tofler GH, Jacobs SC, Friedman R, Benson H, Muller JE. Triggering of acute myocardial infarction onset by episodes of anger. Determinants of Myocardial Infarction Onset Study Investigators. Circulation. 1995;92:1720–1725. doi: 10.1161/01.cir.92.7.1720. [DOI] [PubMed] [Google Scholar]
- 2.Moller J, Hallqvist J, Diderichsen F, Theorell T, Reuterwall C, Ahlbom A. Do episodes of anger trigger myocardial infarction? A case-crossover analysis in the Stockholm Heart Epidemiology Program (SHEEP) Psychosom Med. 1999;61:842–849. doi: 10.1097/00006842-199911000-00019. [DOI] [PubMed] [Google Scholar]
- 3.Strike PC, Perkins-Porras L, Whitehead DL, McEwan JR, Steptoe A. Triggering of acute coronary syndromes by physical exertion and anger: clinical and sociodemographic characteristics. Heart. 2006;92:1035–1040. doi: 10.1136/hrt.2005.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang PP, Ford DE, Meoni LA, Wang NY, Klag MJ. Anger in young men and subsequent premature cardiovascular disease: the precursors study. Arch Intern Med. 2002;162:901–906. doi: 10.1001/archinte.162.8.901. [DOI] [PubMed] [Google Scholar]
- 5.Williams JE, Paton CC, Siegler IC, Eigenbrodt ML, Nieto FJ, Tyroler HA. Anger proneness predicts coronary heart disease risk: prospective analysis from the atherosclerosis risk in communities (ARIC) study. Circulation. 2000;101:2034–2039. doi: 10.1161/01.cir.101.17.2034. [DOI] [PubMed] [Google Scholar]
- 6.Kawachi I, Sparrow D, Spiro A, 3rd, Vokonas P, Weiss ST. A prospective study of anger and coronary heart disease. The Normative Aging Study. Circulation. 1996;94:2090–2095. doi: 10.1161/01.cir.94.9.2090. [DOI] [PubMed] [Google Scholar]
- 7.Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 8.Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF, Jr, Lehman BT, Fan S, Osypiuk E, Vita JA. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation. 2004;109:613–619. doi: 10.1161/01.CIR.0000112565.60887.1E. [DOI] [PubMed] [Google Scholar]
- 9.Adams MR, Robinson J, McCredie R, Seale JP, Sorensen KE, Deanfield JE, Celermajer DS. Smooth muscle dysfunction occurs independently of impaired endothelium-dependent dilation in adults at risk of atherosclerosis. J Am Coll Cardiol. 1998;32:123–127. doi: 10.1016/s0735-1097(98)00206-x. [DOI] [PubMed] [Google Scholar]
- 10.Hall P, Davidson KW, MacGregor MW, MacLean D. Expanded structured interview administration training manual. Vol. 1998 Halifax: Heart Health of Nova Scotia; 1998. [Google Scholar]
- 11.Shimbo D, Grahame-Clarke C, Miyake Y, Rodriguez C, Sciacca R, Di Tullio M, Boden-Albala B, Sacco R, Homma S. The association between endothelial dysfunction and cardiovascular outcomes in a population-based multi-ethnic cohort. Atherosclerosis (Epub ahead of print) 2006 doi: 10.1016/j.atherosclerosis.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O’Connor G, Betteridge J, Klein N, Steptoe A, Deanfield JE. Mental stress induces transient endothelial dysfunction in humans. Circulation. 2000;102:2473–2478. doi: 10.1161/01.cir.102.20.2473. [DOI] [PubMed] [Google Scholar]
- 13.Spieker LE, Hurlimann D, Ruschitzka F, Corti R, Enseleit F, Shaw S, Hayoz D, Deanfield JE, Luscher TF, Noll G. Mental stress induces prolonged endothelial dysfunction via endothelin-A receptors. Circulation. 2002;105:2817–2820. doi: 10.1161/01.cir.0000021598.15895.34. [DOI] [PubMed] [Google Scholar]
- 14.Lampert R, Joska T, Burg MM, Batsford WP, McPherson CA, Jain D. Emotional and physical precipitants of ventricular arrhythmia. Circulation. 2002;106:1800–1805. doi: 10.1161/01.cir.0000031733.51374.c1. [DOI] [PubMed] [Google Scholar]
- 15.Mehta JL. Endothelium, coronary vasodilation, and organic nitrates. Am Heart J. 1995;129:382–391. doi: 10.1016/0002-8703(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 16.Hanspal IS, Magid KS, Webb DJ, Megson IL. The effect of oxidative stress on endothelium-dependent and nitric oxide donor-induced relaxation: implications for nitrate tolerance. Nitric Oxide. 2002;6:263–270. doi: 10.1006/niox.2001.0412. [DOI] [PubMed] [Google Scholar]
- 17.Mugge A, Elwell JH, Peterson TE, Hofmeyer TG, Heistad DD, Harrison DG. Chronic treatment with polyethylene-glycolated superoxide dismutase partially restores endothelium-dependent vascular relaxations in cholesterol-fed rabbits. Circ Res. 1991;69:1293–1300. doi: 10.1161/01.res.69.5.1293. [DOI] [PubMed] [Google Scholar]
- 18.Gottdiener JS, Kop WJ, Hausner E, McCeney MK, Herrington D, Krantz DS. Effects of mental stress on flow-mediated brachial arterial dilation and influence of behavioral factors and hypercholesterolemia in subjects without cardiovascular disease. Am J Cardiol. 2003;92:687–691. doi: 10.1016/s0002-9149(03)00823-3. [DOI] [PubMed] [Google Scholar]
- 19.Broadley AJ, Korszun A, Abdelaal E, Moskvina V, Jones CJ, Nash GB, Ray C, Deanfield J, Frenneaux MP. Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. J Am Coll Cardiol. 2005;46:344–350. doi: 10.1016/j.jacc.2005.03.068. [DOI] [PubMed] [Google Scholar]
- 20.Harris KF, Matthews KA. Interactions between autonomic nervous system activity and endothelial function: a model for the development of cardiovascular disease. Psychosom Med. 2004;66:153–164. doi: 10.1097/01.psy.0000116719.95524.e2. [DOI] [PubMed] [Google Scholar]
- 21.Hijmering ML, Stroes ES, Olijhoek J, Hutten BA, Blankestijn PJ, Rabelink TJ. Sympathetic activation markedly reduces endothelium-dependent, flow-mediated vasodilation. J Am Coll Cardiol. 2002;39:683–688. doi: 10.1016/s0735-1097(01)01786-7. [DOI] [PubMed] [Google Scholar]