

Abstract
Marinobufagenin (MBG) is a cardiotonic steroid of the bufadienolide class of compounds which has the ability to inhibit the ubiquitous enzyme, Na+/K+-ATPase, resulting in natriuresis. The involvement of MBG in the pathogenesis of volume expansion-mediated forms of hypertension has been suggested for some time, and we have proposed that MBG participates in the hypertension noted in preeclampsia. We examined the hypothesis that MBG might contribute to these forms of hypertension by promoting the activity of the mineralocorticoid receptor (MR). However, our data demonstrate that instead, MBG interferes with the functioning of the MR by inhibiting the transcriptional activity of the receptor, and this is reflected in a reduced interaction between the SRC-3 coactivator and the MR. Thus, the ability of MBG to cause a natriuresis may be due, not only to inhibition of Na+/K+-ATPase activity, but also its ability to interfere with MR-dependent expression of the Na/K/H exchanger in the late distal nephron.
Keywords: Cardiac glycosides, Marinobufagenin, Mineralocorticoid receptor, Preeclampsia, Volume expansion-mediated hypertension, Coactivator, Transcription
INTRODUCTION
The renal mechanisms that regulate sodium excretion in the urine are influenced by a number of factors. These include hormones that influence the glomerular filtration rate and the tubular readsorption of electrolytes. Mineralocorticoids such as aldosterone promote sodium retention by stimulating its readsorption from the renal tubular contents. Consistent with the relatively high expression of mineralocorticoid receptors (MRs) in the distal nephron, the most significant effects of aldosterone on sodium reabsorption are observed in this region of the nephron. This is especially the case with respect to the cortical collecting duct [1]. In this latter nephron segment, mineralocorticoids increase the expression of the epithelial sodium channel (ENaC) on the apical surface of renal tubular cells, enabling the entrance of sodium ions [2]. Aldosterone also increases the expression of Na+/K+-ATPase molecules on the basolateral surface of these cells [3]. Consequently, mineralocorticoids stimulate sodium uptake into the tubular cells. The sodium ions are then transported into the peritubular capillary blood in an ATP-dependent process.
Marinobufagenin (MBG) is a cardiac glycoside which belongs to the bufadienolide class of compounds [4]. These agents are natriuretic, vasoconstrictive (resulting in hypertension) and are cardiac inotropes [5]. Furthermore, they circulate in the blood and are excreted in the urine [6; 7]. Their natriuretic activity is thought to relate to the fact that the bufadienolides, like other cardiotonic steroids such as ouabain and its congeners, inhibit the activity of the enzyme Na+/K+-ATPase resulting in decreased sodium transport of the renal tubular contents into the peritubular blood [8]. MBG has a predilection for the alpha-1 isoform of the enzyme [9]. Retention of excessive water (volume expansion) and volume expansion-mediated hypertension are associated with vasoconstriction, increased mineralocorticoid action and sodium retention [10; 11]. Elevated blood and urine levels of MBG have been measured in volume expansion and volume expansion-mediated hypertension [7; 12-14]. This raises the possibility that MBG may contribute to the pathogenesis of these conditions. Since the MR plays an important role in volume expansion-mediated hypertensive states [15], and MBG is a cardiotonic steroid [4] we wondered if this compound could positively influence MR functions, either directly or through other pathways. The studies described in this communication provide evidence for an effect of MBG to interfere with the function of the MR.
MATERIALS AND METHODS
Chemicals and Plasmid DNAs
Aldosterone was purchased from Steraloids, Inc (Newport, RI), and 17β-estradiol (E2) was obtained from Sigma Chemical Company (St. Louis, MO). The marinobufagenin (MBG) was a gift from Dr. G. Robert Pettit, Arizona State University, Department of Biological Sciences. The mammalian expression plasmid for human mineralocorticoid receptor (pRShMR [16]) was a gift from Dr. Ronald Evans (Salk Institute, San Diego, CA), and vectors for ERα [pCR3.1-hERα [17]] and Flag-epitope-tagged SRC-3 [pCMV-Flag-SRC-3 [18]] were described previously as were the synthetic target genes, PRE-E1b-Luc (a PR and MR responsive reporter [19]) and EREE1b-Luc [17].
Cell Culture and Trans-activation assays
Cos-1 (African green monkey kidney) cells were obtained from ATCC and routinely maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Twenty four hours prior to transfections, cells were plated in six-well culture dishes at a density of 2 × 105 cells per well in phenol red-free DMEM with 5% dextran-coated charcoal stripped fetal bovine serum (sFBS). Plasmid DNA was introduced into cells in the indicated amounts using Lipofectamine (Invitrogen, Carlsbad, CA), according to the manufacturer's guidelines. Four hours later, serum-free media was replaced with phenol red-free DMEM supplemented with 5% sFBS, and cells were treated with the indicated amounts of various hormones for 24 h. Thereafter cells were harvested and extracts were assayed for luciferase activity using the Luciferase Assay Systems kit (Promega) and a Luminoskan Ascent luminometer (Thermo Fisher Scientific Inc., Waltham, MA). Relative luciferase units were normalized to total cellular protein, as determined by Bio-Rad Protein Assay. Experiments were done in duplicate and values represent the mean ± SEM of at least three individual experiments.
Western Blot Analyses
To assess MR expression 1.2 × 105 cells/well were transfected as described above and cultured in media containing 10% sFBS, treated the next morning with the indicated compounds and harvested for Western blot analysis 24 h thereafter. Cell pellets were resuspended in 50 mM Tris buffer (pH 8.0) containing 400 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.2% Sarkosyl and a Complete Protease Inhibitor Cocktail tablet (Roche Diagnostics, Indianapolis, IN), incubated on ice for 30 minutes, and centrifuged at 20,000 × g for 10 minutes at 4°C. The resulting supernatant was resolved by a NuPAGE Novex Bis-Tris 4-12% Gel (Invitrogen) and electrotransferred to nitrocellulose. Filters were blocked in 5% skim milk powder in PBS containing 0.1% Tween-20 (PBS-T), and incubated sequentially with primary antibodies against MR (N-17; Santa Cruz Biotechnology, Santa Cruz, CA) or actin (Chemicon International Inc., Temecula, CA) and horseradish peroxidase (HRP)-conjugated anti-goat (Santa Cruz) or anti-mouse IgG (ICN, Aurora, OH), respectively. Immunodetection was performed with ECL Plus Western Blotting Detection System reagents as recommended by the manufacturer (Amersham, Piscataway, NJ).
Aldosterone Binding Assay
The ability of MBG to bind to MR was determined in vivo essentially as previously described [20]. Briefly, 1 μg of the MR expression plasmid was transfected into Cos-1 cells using Lipofectamine and 24 h thereafter media was aspirated from wells and replaced with DMEM containing 5% sFBS and ∼1.0 nM [3H]aldosterone (39.0 Ci/mmol) (Perkin Elmer Life Sciences, Shelton, CT) in the presence of either vehicle, up to 200 nM aldosterone or increasing concentrations (from 10−8 to 10−6 M) of unlabeled MBG. After incubation for 2 hours at 37°C media was aspirated, cells were washed 3 times with ice-cold PBS and incubated in 100% ethanol for 10 minutes at room temperature to extract bound steroid. The amount of MR-bound [3H]aldosterone in the ethanol extract was quantified with a Beckman LS 6500 scintillation counter (Beckman Instrument, Fullerton, CA) and Biodegradable Counting Scintillant (Amersham).
Coimmunoprecipitation
Cos-1 cells were plated at a density of 1 × 106 cells/100 mm Petri dish in phenol-red free DMEM containing 10% sFBS, and transfected with 500 ng each of pCMV-Flag-SRC-3 and pRShMR using Lipofectamine. After 24 h, cells were treated with either 0.1% ethanolic vehicle, 10−8 M aldosterone or 10−8 M aldosterone with 10−6 MBG for 30 min and then harvested and incubated in lysis buffer [20 mM Tris (pH 7.5) containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 5% glycerol, 1mM Na3VO4 and 1mM NaF) supplemented with a complete Mini-Tablets protease inhibitor tablet (Roche Diagnostics) at 4°C with rotation for 30-60 min. The cell lysate was centrifuged for 5 min at 20,000 × g, and 1.5 mg cell lysate protein was pre-cleared with 25 μl of a 50% slurry of prewashed protein G agarose beads (Santa Cruz) in a total volume of 1 ml lysis buffer. The resulting lysate was incubated with rotation with 20 μl EZview™ Red ANTI-FLAG® M2 Affinity Gel (Sigma) or 3 μg normal mouse IgG (Santa Cruz) for 2 hours at 4°C, prior to the addition of 25 μl of a 50% slurry of prewashed protein G agarose beads and an additional 2 h incubation with rotation. The immunocomplex was washed 4 times with lyses buffer at 4°C and subsequently heated to 95°C for 10 min in 25 μl of 1x Laemmli buffer and resolved by a NuPAGE Novex 3-8% Tris-Acetate gel (Invitrogen). The Western blot was probed with anti-AIB1 (BD Biosciences, San Jose, CA), anti-MR or anti-actin antibodies in PBS-T containing 5% skim milk powder, followed by anti-mouse or anti-goat antibodies conjugated to horseradish peroxidase. Protein bands were detected using ECL Plus reagents as described above.
RESULTS
In order to ascertain whether MBG altered the transcriptional activity of the mineralocorticoid receptor, expression plasmids for this receptor along with the PRE-E1b-Luc reporter gene were transfected into Cos-1 kidney cells. The PRE-E1b-Luc synthetic target gene possesses two copies of a DNA sequence, termed the progesterone response element, which is identical to a response element for the MR [21], linked upstream to TATA box and luciferase reporter gene. As expected, treatment of Cos-1 cells with aldosterone led to a strong induction of luciferase gene expression (Fig. 1A). Although exposure of these cells to MBG alone did not affect the very low basal activity of the MR, it was observed that treatment of cells with 10−6 M MBG reduced MR transcriptional activity induced by 10−9 M aldosterone by ∼65% and activity induced by 10−8 M aldosterone was inhibited by ∼50%. Western blot analysis (Fig. 1B) revealed the anticipated reduction in MR expression in cells treated with aldosterone [22]. MBG alone did not affect MR expression, and MR levels in cells treated with aldosterone and MBG were similar to those treated with aldosterone alone indicating that MBG did not interfere with MR transcriptional activity via alterations in MR expression.
Figure 1.
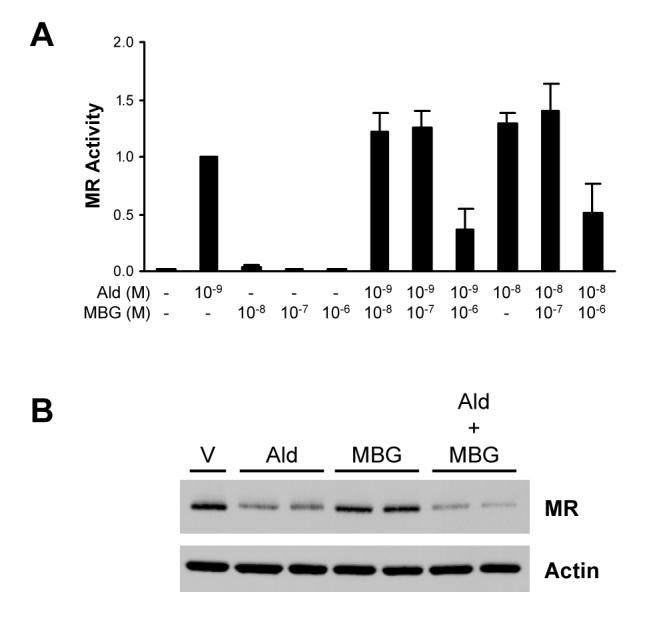
Inhibition of MR transcriptional activity by MBG. A) Cos-1 cells were transfected with 100 ng pRShMR and 1 μg of PRE-E1b-Luc and treated with vehicle (0.1% ethanol) or the indicated concentrations of aldosterone (Ald) or marinobufagenin (MBG). B) Cos-1 cells were transfected with 500 ng pRShMR and 1 μg PRE-E1b-Luc and treated with either vehicle (V), 10−9 M Ald and/or 10−6 M MBG.
To determine whether MBG inhibition of MR activity was a reflection of altered ligand binding capacity by the receptor, in vivo competitive ligand binding assays were performed. Cos-1 cells transfected with MR expression plasmid were incubated with [3H]aldosterone in the absence or presence of unlabeled aldosterone or MBG. As shown in Fig. 2, incubation of cells with increasing concentrations of cold aldosterone competed for binding to MR, such that an ∼200-fold excess of radio-inert aldosterone effectively competed with [3H]aldosterone for binding to the receptor. In contrast, incubation of the cells with 10−9 to 10−6 M MBG was unable to reduce MR binding to tritiated aldosterone, and the lowest levels appeared to modestly stimulate ligand binding to the receptor. An in vitro competitive ligand binding assay conducted with cell extracts prepared from MR-transfected Cos-1 cells yielded consistent results (data not shown) in which MBG was unable to inhibit MR binding to [3H]aldosterone. Thus, MBG does not inhibit MR transcriptional activity by inhibiting receptor binding to its cognate ligand.
Figure 2.
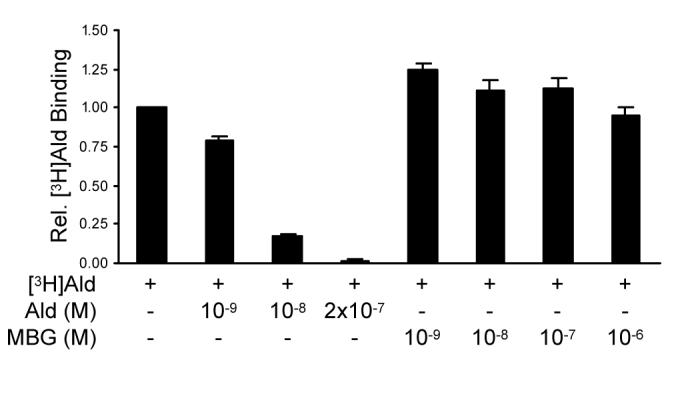
MBG does not bind to MR. Cos-1 cells were transfected with MR expression plasmid and incubated with [3H]aldosterone in the absence or presence of increasing concentrations of unlabeled aldosterone or MBG.
To determine whether MBG inhibition of MR transcriptional activity was specific to this transcription factor, we tested whether it would inhibit the activity of another member of the nuclear receptor superfamily, estrogen receptor-α (ERα). Treatment of Cos-1 cells with MBG reduced the basal transcriptional activity of this receptor in a dose-dependent fashion, suggesting that it reduced transcriptional activity independent of any effects on ligand binding (Fig. 3). ERα transcriptional activity induced by the receptor's cognate ligand, estradiol, was also reduced by MBG.
Figure 3.
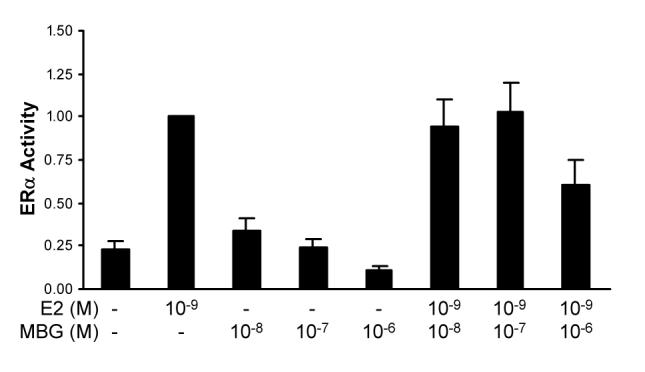
Inhibition of ERα transcriptional activity by MBG. Cos-1 cells were transfected with 10 ng pCR3.1-hERα and 1 μg ERE-E1b-Luc, and treated with vehicle (0.1% ethanol), 10−9 M E2 and/or the indicated concentrations of marinobufagenin (MBG).
The ability of MBG to inhibit the transcriptional activity of two members of the nuclear receptor family suggests that these effects may be achieved by negative regulation of an event common to both transcription factors. Nuclear receptors such as ERα and MR require coactivator proteins to stimulate the expression of their respective target genes [23; 24], and experiments were therefore conducted to determine if MBG treatment influenced the expression of a major family of nuclear receptor coactivators, the steroid receptor coactivator family [25]. Experiments were also performed to determine whether exposure of cells to MBG affected the ability of aldosterone to promote interactions between MR and coactivator. Of the three members of the SRC family of coactivators, SRC-3 is exquisitely sensitive to the intracellular signaling environment [26], and it was therefore chosen as the representative coactivator for these studies. Using a co-immunoprecipitation approach, it was observed in multiple experiments that MBG treatment did not affect the expression of SRC-3 or MR, but rather reduced the interaction between these two molecules (Fig. 4).
Figure 4.

MBG reduces interaction between MR and SRC-3. Cos1 cells were transfected with expression vectors for Flag-SRC-3 and MR and 24h later treated with either 0.1% ethanol (V), 10−8 M aldosterone (Ald) or 10−8 M aldosterone with 10−6 marinobufagenin (Ald + MBG) for 30 min. Cells were lysed and aliquots of input extracts (lanes 1-3) as well as material immunoprecipitated with EZview™ Red ANTI-FLAG® M2 Affinity Gel (lanes 4-6) or IgG (lane 7) were subjected to Western blot analyses for SRC-3 (top), MR (middle) and actin (bottom). The blot is representative of four experiments.
DISCUSSION AND CONCLUSIONS
The administration of bufadienolides results in an increase in sodium excretion [27]. This effect of MBG and other similar cardiac glycosides on sodium transport has been attributed to their ability to inhibit the action of the ubiquitous enzyme Na+/K+-ATPase located on the basolateral surface of the renal tubular cell [28]. The elevated levels of MBG in volume expansion-mediated hypertension suggested that MBG may exert effects in addition to those mediated by Na+/K+ exchange. Based on the data described in this communication it appears that MBG (and, perhaps, other of the bufadienolides) inhibits the transcriptional activity of the MR, not via direct effects on this steroid receptor, but more likely via targeting the receptor's functional interactions with a coactivator. Taken together, our data would suggest that there may be a role for increased MBG during volume expansion to limit MR-mediated effects such as sodium retention.
The expression of Na+/K+-ATPase in the late distal nephron is positively regulated by the MR [3] and the ability of MBG to inhibit MR function suggests that MBG reduces Na+/K+-ATPase activity by reducing the expression of this enzyme as well as its activity. Moreover, the gene for the epithelial sodium channel, ENaC, in the collecting ducts is also an MR target [2]. It is therefore possible that MBG also reduces the expression of this transporter. Both of these possibilities will need to be tested in in vivo experiments. However, if this thesis is correct, then sodium ions leaving the proximal nephron would be less likely to be reabsorbed in the more distal nephron. Accordingly, blockade of more than one nephron sodium transport site would result in a more potent natriuretic effect of these agents [29; 30].
The transcriptional activity of the MR, as for other members of the nuclear receptor superfamily, is positively influenced by coactivator molecules which are necessary for receptor interactions with the general transcriptional machinery [25]. The coactivator SRC-3 (also known as AIB1 or RAC3) interacts with the ligand binding domain of agonist-bound steroid receptors and helps to recruit additional factors such as CBP and PCAF which ultimately results in chromatin remodeling, recruitment of RNA polymerase II and activation of gene expression. Our data indicate that treatment of cells with MBG reduces MR transcriptional activity, and that this occurs, at least in part, via a reduction in the interaction between MR and SRC-3. The mechanism(s) by which MBG treatment reduces MR binding to SRC-3 is unknown at the present time. However, the lack of any effect of MBG on MR expression or ligand binding suggests the possibility that MBG may alter intracellular signaling pathways leading to post-translational modifications that can alter receptor-coactivator interactions. The MR is a phosphoprotein whose transcriptional activity can be regulated by signaling pathways [31], and recent work demonstrates that the activity of coactivators such as SRC-3 is modulated by post-translational modifications [26]. Indeed, loss of SRC-3 phosphorylation results in poor stimulation of receptor-dependent gene expression, and reduced interaction with the CBP coactivator [18]. Thus, intracellular signaling pathways originating from distinct classes of molecules can have profound influences on MR and SRC-3 function and consequently steroid receptor-dependent gene expression. MBG increases the activities of Src kinase, ERK1/2 and GSK3 in muscle cells [32], and several of these signaling molecules (e.g. ERK1/2 and GSK3) can affect the activity of SRC-3 [18].
We have proposed that MBG plays a pathogenetic role in at least some patients with the preeclamptic syndrome [33], related to excessive volume expansion [34]. These data demonstrate that MBG does not stimulate MR transcriptional activity and indicates that the in vivo effects of MBG are unlikely to be mediated via the enhanced expression of MR target genes involved in sodium transport in the kidney. A search for the proximate cause of the excessive elaboration of MBG in this syndrome is underway.
ACKNOWLEDGMENTS
These studies were supported by a grant-in-aid from Dialysis Clinic, Incorporated, Nashville, Tennessee (to JBP) and NIH DK64038 (to CLS). The authors thank Dr. Bert O'Malley for his advice and support. We thank Mrs. Dolores Todaro for the production of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Marver D, Kokko JP. Renal target sites and the mechanism of action of aldosterone. Miner Electrolyte Metab. 1983;9:1–18. [PubMed] [Google Scholar]
- 2.Mick VE, Itani OA, Loftus RW, Husted RF, Schmidt TJ, Thomas CP. The alpha-subunit of the epithelial sodium channel is an aldosterone-induced transcript in mammalian collecting ducts, and this transcriptional response is mediated via distinct cis-elements in the 5'-flanking region of the gene. Mol Endocrinol. 2001;15:575–588. doi: 10.1210/mend.15.4.0620. [DOI] [PubMed] [Google Scholar]
- 3.Kolla V, Litwack G. Transcriptional regulation of the human Na/K ATPase via the human mineralocorticoid receptor. Mol Cell Biochem. 2000;204:35–40. doi: 10.1023/a:1007009700377. [DOI] [PubMed] [Google Scholar]
- 4.Schoner W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur J Biochem. 2002;269:2440–2448. doi: 10.1046/j.1432-1033.2002.02911.x. [DOI] [PubMed] [Google Scholar]
- 5.Schoner W, Scheiner-Bobis G. Endogenous cardiac glycosides: hormones using the sodium pump as signal transducer. Semin Nephrol. 2005;25:343–351. doi: 10.1016/j.semnephrol.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 6.Bagrov AY, Dmitrieva RI, Fedorova OV, Kazakov GP, Roukoyatkina NI, Shpen VM. Endogenous marinobufagenin-like immunoreactive substance. Am J Hypertens. 1996;9:982–990. doi: 10.1016/0895-7061(96)00148-3. [DOI] [PubMed] [Google Scholar]
- 7.Vu HV, Ianosi-Irimie MR, Pridjian CA, Whitbred JM, Durst JM, Bagrov AY, Fedorova OV, Pridjian G, Puschett JB. Involvement of marinobufagenin in a rat model of human preeclampsia. Am J Nephrol. 2005;25:520–528. doi: 10.1159/000088461. [DOI] [PubMed] [Google Scholar]
- 8.Bagrov AY, Roukoyatkina NI, Pinaev AG, Dmitrieva RI, Fedorova OV. Effects of two endogenous Na+, K+-ATPase inhibitors, marinobufagenin and ouabain, on isolated rat aorta. Eur J Pharm. 1995;274:151–158. doi: 10.1016/0014-2999(94)00735-p. [DOI] [PubMed] [Google Scholar]
- 9.Fedorova OV, Kolodkin NI, Agalakova NI, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous α-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension. 2001;37:462–466. doi: 10.1161/01.hyp.37.2.462. [DOI] [PubMed] [Google Scholar]
- 10.Raymond KH, Stein JH. Efferent limb of volume homeostasis. In: Brenner BM, Stein JH, editors. Body Fluid Homeostasis. Churchill; Livingstone, NY: 1987. pp. 33–68. [Google Scholar]
- 11.Hall JH. Role of angiotensin and aldosterone in volume homeostasis. In: Brenner BM, Stein JH, editors. Body Fluid Homeostasis. Churchill; Livingstone, NY: 1987. pp. 69–100. [Google Scholar]
- 12.Fedorova OV, Doris PA, Bagrov AY. Endogenous marinobufagenin-like factor in acute plasma volume expansion. Clin Exp Hypertens. 1998;20:581–591. doi: 10.3109/10641969809053236. [DOI] [PubMed] [Google Scholar]
- 13.Bagrov AY, Fedorova OV, Dmitrieva RI, French AW, Anderson DE. Plasma marinobufagenin-like and ouabain-like immunoreactivity during saline volume expansion in anesthetized dogs. Cardiovasc Res. 1996;31:296–305. [PubMed] [Google Scholar]
- 14.Gonick HC, Ding Y, Vaziri ND, Bagrov AY, Fedorova OV. Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin Exp Hypertens. 1998;20:617–627. doi: 10.3109/10641969809053240. [DOI] [PubMed] [Google Scholar]
- 15.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schutz G. Mineralocorticoid receptor knock-out mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- 17.Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu R-C, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Selective phosphorylations of the SRC-3/AIBI coactivator integrate genomic responses to multiple cellular signaling pathways. Mol.Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 19.Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai M-J, O'Malley BW. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol. 1999;19:1182–1189. doi: 10.1128/mcb.19.2.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walters MR, Dutertre M, Smith CL. SKF-82958 is a subtype-selective estrogen receptor-α (ERα) agonist that induces functional interactions between ERα and AP-1. J.Biol.Chem. 2002;277:1669–1679. doi: 10.1074/jbc.M109320200. [DOI] [PubMed] [Google Scholar]
- 21.Ribeiro RCJ, Kushner PJ, Baxter JD. The nuclear Hormone receptor gene superfamily. Ann.Rev.Med. 1995;46:443–453. doi: 10.1146/annurev.med.46.1.443. [DOI] [PubMed] [Google Scholar]
- 22.Yokota K, Shibata H, Kobayashi S, Suda N, Murai A, Kurihara I, Saito I, Saruta T. Proteasome-medeiated mineralocorticoid receptor degradation attenuates transcriptional response to aldosterone. Endocrine Res. 2004;30:611–616. doi: 10.1081/erc-200043783. [DOI] [PubMed] [Google Scholar]
- 23.Baxter JD, Funder JW, Apriletti JW, Webb P. Towards selectively modulating mineralocorticoid receptor function: lessons from other systems. Mol Cell Endocrinol. 2004;217:151–165. doi: 10.1016/j.mce.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson J-Å. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 25.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 26.Wu R-C, Smith CL, O'Malley BW. Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr Rev. 2005;26:393–399. doi: 10.1210/er.2004-0018. [DOI] [PubMed] [Google Scholar]
- 27.Pamanani MB, Shanwan C, Bryant HJ, Schooley JF, Eliades DC, Yuan CM, Haddy FJ. Effects of three sodium-potassium adenosine triphosphatase inhibitors. Hypertension. 1991;18:316–324. doi: 10.1161/01.hyp.18.3.316. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz A, Lindenmayer GE, Allen JC. The sodium-potassium adenosine triphosphatase: pharmacological, physiological and biochemical aspects. Pharmacol Rev. 1975;27:3–134. [PubMed] [Google Scholar]
- 29.Puschett JB, Winaver J. Section 8: Renal Physiology. In: Windhager EE, editor. Handbook of Physiology,The American Society of Physiology. Oxford University Press; New York: 1992. pp. 2335–2406. [Google Scholar]
- 30.Puschett JB. Sites and mechanisms of the renal actions of diuretics in man. In: Kruck F, Mutschler E, Knauf H, editors. Progress in Pharmacology and Clinical Pharmacology, Clinical Pharmacology of Diuretics. Gustav Fischer Verlag; Stuttgart: 1992. pp. 117–130. [Google Scholar]
- 31.Lim-Tio SS, Fuller PJ. Intracellular signaling pathways confer specificity of transactivation by mineralocorticoid and glucocorticoid receptors. Endocrinology. 1998;139:1653–1661. doi: 10.1210/endo.139.4.5928. [DOI] [PubMed] [Google Scholar]
- 32.Kotova O, Al-Khalili L, Talia S, Hooke C, Fedorova OV, Bagrov AY, Chibalin AV. Cardiotonic steroids stimulate glycogen synthesis in ERK1/2-dependent mechanism. J Biol Chem. 2006;281:20085–20094. doi: 10.1074/jbc.M601577200. [DOI] [PubMed] [Google Scholar]
- 33.Alper AA, Outland J, Finigan K, Pridjian G, Puschett JB. Phenotypic characteristics shared by preeclamptic patients and an animal model of preeclampsia: a pilot study. Am J Hypertens. 2006;19:947–950. doi: 10.1016/j.amjhyper.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Puschett JB. The role of excessive volume expansion in the pathogenesis of preeclampsia. Medical Hypotheses. 2006;67:1125–1132. doi: 10.1016/j.mehy.2006.04.059. [DOI] [PubMed] [Google Scholar]