

Abstract
Phylogenetic footprinting is a method for the discovery of regulatory elements in a set of orthologous regulatory regions from multiple species. It does so by identifying the best conserved motifs in those orthologous regions. We describe a computer algorithm designed specifically for this purpose, making use of the phylogenetic relationships among the sequences under study to make more accurate predictions. The program is guaranteed to report all sets of motifs with the lowest parsimony scores, calculated with respect to the phylogenetic tree relating the input species. We report the results of this algorithm on several data sets of interest. A large number of known functional binding sites are identified by our method, but we also find several highly conserved motifs for which no function is yet known.
One of the great challenges currently facing biologists is to understand the varied and complex mechanisms that regulate gene expression. We focus on one important aspect of this challenge, the identification of binding sites for the factors involved in such regulation.
A number of computer algorithms have been proposed for the discovery of novel regulatory elements in nucleotide sequences. Most of these try to deduce the regulatory elements by considering the regulatory regions of several (putatively) coregulated genes from a single genome. Such algorithms search for overrepresented motifs in this collection of regulatory regions, these motifs being good candidates for regulatory elements. Examples of this approach include van Helden et al. (1998), Hertz and Stormo (1999), Hughes et al. (2000), Sinha and Tompa (2000), and Workman and Stormo (2000).
We adopt an orthogonal approach of deducing regulatory elements by considering orthologous regulatory regions of a single gene from several species. This approach is called “phylogenetic footprinting” (Tagle et al. 1988). The simple premise underlying phylogenetic footprinting is that selective pressure causes functional elements to evolve at a slower rate than that of nonfunctional sequences. This means that unusually well conserved sites among a set of orthologous regulatory regions are excellent candidates for functional regulatory elements. This approach has proved successful for the discovery of regulatory elements for many genes, including ɛ-globin (Tagle et al. 1988; Gumucio et al. 1993), γ-globin (Tagle et al. 1988), rbcL (Manen et al. 1994), cystic fibrosis transmembrane conductance regulator (Vuillaumier et al. 1997), tumor necrosis factor-α (Leung et al. 2000), and interleukin (IL)-4, IL-13, and IL-5 (Loots et al. 2000). See the review by Duret and Bucher (1997) for more details. The same idea of using comparative analysis to identify conserved elements, but among only two or three species (particularly human and mouse), has recently become popular (Hardison et al. 1997; Jareborg et al. 1999; Dubchak et al. 2000; Wasserman et al. 2000; Mouchel et al. 2001; Wu et al. 2001).
The major advantage of phylogenetic footprinting over the single genome, multigene approach mentioned earlier is that the latter requires a reliable method for assembling the requisite collection of coregulated genes. In contrast, phylogenetic footprinting is capable of identifying regulatory elements specific even to a single gene, as long as they are sufficiently conserved across many of the species considered. Genome projects are quickly producing sequences from a wide variety of organisms, so the data necessary for phylogenetic footprinting are becoming increasingly available.
The standard method that has been used for phylogenetic footprinting is to construct a global multiple alignment of the orthologous regulatory sequences and then to identify conserved regions in the alignment. A tool such as CLUSTALW (Thompson et al. 1994) is appropriate for this purpose, as it can take advantage of knowledge of the phylogeny relating the species. To see why this approach to phylogenetic footprinting does not always work, consider typical lengths of the sequences involved. Regulatory elements tend to be quite short (5 to 20 nucleotides long) relative to the entire regulatory region in which we search for them (a 1000-bp promoter region would be typical). Given these relative lengths, if the species are somewhat diverged, it is likely that the noise of the diverged nonfunctional background will overcome the short conserved signal. The result is that the alignment may not align the short regulatory elements together. In that case, the regulatory elements would not appear to belong to conserved regions and would go undetected. Thus, when the entire regulatory regions considered are moderately to highly diverged, global multiple alignment is likely to miss significant signals.
Cliften et al. (2001) made similar observations in conjunction with their comparative analysis of several Saccharomyces species. They discovered that if the species are too closely related, the sequence alignment is obvious but uninformative, because the functional elements are not sufficiently better conserved than the surrounding nonfunctional sequence. On the other hand, if the species are too distantly related, it is difficult or impossible to find an accurate alignment (for discussion of these issues, see Tompa 2001).
Rather than relying on multiple alignment, a more successful recent approach to phylogenetic footprinting is to use one of the existing motif discovery programs—such as MEME (Bailey and Elkan 1995), Projection (Buhler and Tompa 2001), Consensus (Hertz and Stormo 1999), AlignAce (Roth et al. 1998), or ANN-Spec (Workman and Stormo 2000)—or the segment-based multiple alignment program DIALIGN (Morgenstern et al. 1998, Morgenstern 1999). Cliften et al. (2001), for instance, reported some successes using AlignAce when global multiple alignment tools failed. Another example of this approach is the work of McCue et al. (2001), who used a Gibbs sampler to perform phylogenetic footprinting in bacterial sequences. Such general motif discovery algorithms were designed for a different purpose, however, and have their own drawback: None take into account the phylogenetic relationship of the given sequences; that is, these methods assume the input sequences to be independent. This can be problematic, for example, in data sets containing a mixture of some closely related species and some distant ones. If the phylogeny underlying the data is ignored, similar sequences from the set of closely related species will have an unduly high weight in the choice of motifs reported. Even if these methods were modified to weight the input sequences unequally, this would still not capture the information in an arbitrary phylogenetic tree. The method we present does capture this information.
In this paper, we describe an algorithmic method designed specifically for phylogenetic footprinting in multiple species. Because it is tailored to this purpose, it avoids the drawbacks described above of both multiple alignment and general motif discovery algorithms. Given a set of unaligned orthologous sequences, our approach identifies all DNA motifs that appear to have evolved unusually slowly compared with the surrounding sequence. More precisely, given n orthologous input sequences and the phylogenetic tree T relating them, our algorithm is guaranteed to produce every set of k-mers, one from each input sequence, that have parsimony score at most d with respect to T, where k and d are parameters that can be specified by the user.
As orthologous sequences from more and more species are included in the input, the distinction between conserved motif and diverged background generally becomes clearer. However, when including many orthologous sequences, particularly distantly related ones, there is increased chance that some of them may have lost or completely altered some regulatory elements over the course of evolution. For example, a species may not need the regulatory mechanism in which some regulatory element was involved, in which case selective pressure would no longer operate. As an example of an altered regulatory element, LexA has an entirely different binding motif in gram-positive bacteria than in gram-negative bacteria (McGuire et al. 2000). For these reasons, we developed a variant of our phylogenetic footprinting algorithm that identifies motifs that occur in many, but not necessarily all, of the input sequences. This variant requires some way of comparing the levels of conservation among motifs that occur in different subsets of the input species and with different parsimony scores. For example, should one prefer a motif that occurs in all the species with parsimony score 2 or a motif that occurs in most of the species but with parsimony score 0? To address this, for each parsimony score s, we allow the user to set a minimum threshold on the fraction of the phylogeny that must be spanned by any reported motif with score s. For example, the user can ask to see all motifs with parsimony score 0 that span at least 200 Myrs of the phylogenetic tree (i.e., the sum of all branch lengths induced by the leaves containing the motif is at least 200 Myrs), plus those with score 1 that span at least 350 Myrs, plus those with score 2 that span at least 500 Myrs. Thresholds are to be set in such a way that the motifs reported are conserved at a statistically significant level (see Methods).
The focus of the present paper is an explanation of what the new algorithms do and a discussion of the results obtained on several interesting data sets available in the public databases. Although a high-level description of how the algorithm works is given in Methods, the details of the algorithm, and in particular several algorithmic optimizations that render it practical on realistic problems, are beyond the scope of the present paper. These algorithmic details are described in a companion paper devoted to that purpose (Blanchette et al. 2002).
The algorithms are implemented in a program called FootPrinter that has been used to obtain the results presented here. FootPrinter is available at http://bio.cs.washington.edu/software.html.
RESULTS
In this section, we report the highly conserved motifs found by FootPrinter in nine sets of orthologous or paralogous sequences. We identified many previously known regulatory elements, as well as many highly conserved motifs with unknown function. The data sets considered in this study were chosen according to two main criteria: (1) the availability of several orthologous promoter sequences in GenBank and (2) the availability of information about the regulation of the genes considered (to validate our results). Some sets of orthologous sequences come from the ACUTS database (Duret and Bucher 1997), which lists a number of genes for which regulatory regions have been sequenced in several vertebrates. Other data sets were built by the authors directly from GenBank. The sequences, accession numbers, phylogenetic trees, and detailed results from FootPrinter can be found at http://bio.cs.washington.edu/GR/. The phylogenetic relationships among the sequences considered were derived from Murphy et al. (2001) and Maddisson (2002), unless mentioned otherwise (see Discussion). All results are summarized in Table 1, but there is more detail available at the web site mentioned above.
Table 1.
Motifs Found by Phylogenetic Footprinting
| DNA region(a) | Species(b) | Motif (length) (position)(c) | Score (species)(d) | Ref.(e) |
|---|---|---|---|---|
| Metallothionein family 5′ UTR + promoter (590 bp) | Human (Ia, Ih, II, IV), rat (I, II, III), mouse (III), hamster (I, II), sheep (Ia, II), rabbit (I), cow (I), frog (a), trout (a), pike, icefish (I, II), carp, loach, urchin (I, II), mussel, C. elegans (I, II) | 1. GCTATAAAc (8) (Human II,−103) 2. CATGCGCAGg (9) (Rat III, −143) 3. cCGTGTGCAg (8) (Human II, −239) CGTGTGCAggc (8) (Human II, −156) 4. TTTGCACACG (10) (Pike, −142) 5. tGCGCCCGG (8) (Human II, −222) TGCACTCG (8) (Human II, −126) 6. TAACTGATAAA (10) (C. ele. I, −324) 7. TACACTCAG (9) (Rat III, −207) 8. TCCCACCAA (9) (Rat III, −497) 9. CAGGCACCT (9) (Rat III, −284) 10. TGCACACGG (9) (Human II, −374) 11. tGTACATTGTga (9) (C. elegans I, −129) 12. GCTTTAAAA (9) (Pike, −114) | 2 (see Figure 1) 2 9 (*) 9 (*) 4 5 4 0 1 1 1 1 2 0 | 1.1 1.2 1.3 1.4 1.5 1.6 1.7 |
| Insulin family 5′ promoter (500 bp) | Human, chimp, aotus, pig, rat (I, II), mouse (I, II) | 1. gttAAGACTCTAAtgacc (10) (−223) 2. tcagcccccaGCCATCTGCC (10) (−122) 3. CTATAAAGcc (8) (−32) 4. GGGAAATG (8) (−145) | 0 (Mutated in rodents (I)) 1 0 0 (Absent from rodents) | 2.1 2.2 2.3 2.4 |
| c-myc 5′ promoter (1000 bp) | Goldfish, frog, chicken, rat, pig, marmoset, human | 1. aGTTTATTC (8) (−611) 2. TTGCTGGG (8) (−570) 3. GGCGCGCAGT (10) (−359) 4. CAGCTGTTCCgc (10) (−325) 5. TGTTTACATCc (10) (−173) 6. ccaCCCTCCCC (8) (−105) 7. AGCAGAGGGCG (10) (−69) 8. GGCGTGGG (8) (−62) 9. ATCTCCGCCCAcc (8) (−26) | 1 (Absent from goldfish) 3 (Absent from chicken) 2 (Chicken + mammals) 2 (Chicken + mammals) 2 (Chicken + mammals) 4 2 (Chicken + mammals) 2 (Absent from goldfish) 2 (Chicken + mammals) | 3.1 3.2 3.3 3.4 |
| c-myc second intron (971 to 1376 bp) | Chicken, pig, rat, marmoset, gibbon, human | 1. CATTTTAATT (10) (303) 2. TGAATGAATT (10) (375) 3. tTTTGAACACT (10) (542) 4. TAGGGAGTTG (10) (670) 5. ATTTGCAGCTat (10) (698) 6. GAAGTGTTCT (10) (725) 7. TTGGTAAAGT (10) (733) 8. GCTTTGCTTTGGGTGTGT (10) (780) 9. GCCTCATTAAGTCTTAGGTAAG (10) (795) 10. TTCCTTTCTT (10) (1362) | 0 (Mammals) 0 (Mammals) 0 (Mammals) 2 2 2 0 (Mammals) 0 (Mammals) 0 (Mammals) 2 | 4.1 |
| c-fos 5′ UTR + promoter (800 bp) | Tetraodon, chicken, mouse, hamster, pig, human | 1. CAGGTGCGAATGTTC (10) (−615) 2. TTCCCGCCTCCCCTCCCC (10) (−583) 3. GAGTTGGCTGcagcc (10) (−527) 4. GTTCCCGTCAATCcct (10) (−504) 5. CACAGGATGTcc (10) (−479) 6. AGGACATCTG (10) (−462) 7. GTCAGCAGGTTTCCACG (10) (−439) 8. TACTCCAACCGC (10) (−159) | 0 (Mammals) 0 (Mammals) 3 (Tetraodon + mammals) 1 (Chicken + mammals) 4 1 (Chicken + mammals) 0 (Mammals) 0 (Mammals) | 5.5 5.1 5.2 5.3 5.4 |
| c-fos first intron (376 to 758 bp) | Fugu, tetraodon, chicken, pig, mouse, hamster, human | 1. GGGTGTGTAAgg (10) (404) 2. GTTTCATTGATAAAAAGCGAGTTCATTCT GGAGACTCCGGAGCGGCG (10) (417) 3. agcgcagacgtcAGGGATATTTA (10) (472) | 3 (Absent from fugu) 1 (Absent from fishes) 1 | 6.1 6.1 6.1 |
| Growth-hormone 5′ UTR + promoter (380 bp) | Salmon, trout, white fish, seriola, lates, tilapia, fugu, grass carp, catfish, chicken, rat, mouse, dog, sheep, goat, human | 1. GGGAGGAG (8) (−198) 2. ATTATCCAT (9) (−183) 3. TTAGCACAA (9) (−174) 4. GTCAGTGG (8) (−162) 5. gcATAAATGTA (9) (−146) 6. GAAACAGGT (9) (−131) 7. cagggTATAAAAAGggc (9) (−97) 8. TCATGTTTt (9) (Salmon, −138) | 3 (Chicken + mammals) 1 (Mammals) 3 (Human, rodents, chicken) 3 (Chicken + mammals) 2 (Chicken + mammals) 1 (Human, rodents, salmonida) 6 (Absent from catfish) 2 (Fishes, except catfish, trout) | 7.1 7.2 7.3 7.4 7.5 |
| Interleukin-3 5′ UTR + promoter (490 bp) | Rat, mouse, cow, sheep, human, macaca | 1. TTGAGTACTagaaagt (8) (−228) 2. GATGAATAATt (8) (−208) 3. GTCTGTGGTTTtCTATGGAGGTTCCATGT CAGATAAAG (8) (−195) 4. TCTTCAGAGc (8) (−56) 5. AGGACCAG (8) (−40) | 1 1 0 1 1 | 8.1 8.2 |
| Histone H1 5′ UTR + promoter (650 bp) | Chicken, duck, frog, mouse | 1. CAATCACCAC (10) (Mouse, −107) 2. gAAACAAAAGTtt (10) (Mouse, −427) | 3 1 | 9.1 |
DNA regions considered.
Species (and isoforms) considered.
Highly conserved motifs found by FootPrinter. Overlapping motifs reported by FootPrinter have been merged, but all nucleotides of the motifs in this Table belong to at least one solution of the given length and with a parsimony score matching our statistical significance threshold. (See Methods.) Capitalization is only relevant with respect to column d. The sequences and positions reported are those for the human sequences, except where otherwise noted. Negative positions are measured in the 5′ direction from the start codon, and positive positions in the 3′ direction from the 5′ end of the intron. A few conserved regions found by FootPrinter that are of low complexity or otherwise uninteresting are not reported.
Parsimony score of the capitalized motif in the subset of species listed. The capitalized region is that with the lowest parsimony score. When no subset is mentioned, the motif is found in all sequences. FootPrinter identified motifs marked by an asterisk in several subsets of the species where shown in Fig. 1, but not in the whole set of species. These subsets were merged by hand to produce Fig. 1. The parsimony score given is that for the whole set of species.
Known functional information about the motif. Unless otherwise noted, the information comes from TRANSFAC (Wingender et al. 1996), with accession number in brackets. Metallothionein: 1.1 TATA-box [R03173], 1.2 MREe [R08295], 1.3 MREb [R08294], 1.4 MREa [R01816], 1.5 MREa [R08293], 1.6 MREd [R08298], and 1.7 MREg [R08296]. Insulin: 2.1 CT-II [R02709], 2.2 IEB1 [R04457], 2.3 TATA-box [GenBank annotation], and 2.4 GG-II [R02711]. C-myc: 3.1 Near NRE [R02571], 3.2 NHE [R01804], 3.3 P1 promoter [R04076], and 3.4 TCE [R04076]. C-myc second intron: 4.1 Part of 3′ splice site. C-fos: 5.1 SIF-E, SIE [R00458, R08485], 5.2 [many factors bind in this region; R00466, R00465, R00464, R01889], 5.3 [many factors bind in this region; R00467, R00463, R04047, R04046, R00462, R00461], 5.4 [part of DSE in SRE; R00467], 5.5 MatInspector (Quandt et al. 1995) hit: MTZ1 (Myeloid zinc factor 1). C-fos first intron: 6.1 (Transcription elongation signals; Mechti et al. 1991), Motif 3 contains a CREB binding site (Lange and Bading 2001). Growth hormone: 7.1 GHF-2 [R02050 in rat], 7.2 dGHF-1 [R00611], 7.3 [R04639 in rat], 7.4 pGHF-1 [R00612], and 7.5 nT3RE [R03959 in rat]. IL-3: 8.1 [R02736], 8.2 [R02682, R05026, R05027]. Histone H1: 9.1 CAAT signal [GenBank annotation].
Metallothionein Gene Family
The metallothionein gene family is particularly well suited to show the merits of our approach, as a large number of promoter sequences are available from a wide variety of species, the phylogenetic relationships among these sequence have been studied, and a large number of regulatory elements have been experimentally determined in several species. Notice that although we described phylogenetic footprinting as applied to orthologous sequences, the approach applies equally well to paralogous sequences, in which two sequences diverged because of duplication rather than speciation, as long as the gene family tree is known.
The primary function of proteins in the metallothionein family is to bind to heavy metal ions and to mediate cellular detoxification of metals. They have also been shown to act as antioxidant agents, protecting DNA from free radicals (for review on the function of metallothionein, see Ghoshal and Jacob 2001).
The metallothionein gene family appears to have evolved through a series of gene tandem duplications and losses. Most mammals have four major isoforms (MT-I, -II, -III, and -IV). Humans actually have 13 copies of the MT-I gene. Some nonmammals (Caenorhabditis elegans, sea urchin, icefish, and trout) also have several copies of the metallothionein gene, but the duplication events that led to this situation most likely took place quite recently, in such a way that, for example, the MT-I C. elegans gene is not more closely related to the MT-I mouse gene than to the MT-II mouse gene. The phylogenetic relationships among the various members of the gene family have been studied by Binz and Kägi (1997), and the phylogenetic tree used here (see Fig. 1) is derived from theirs.
Figure 1.

FootPrinter identified 12 highly conserved motifs in the metallothionein gene family. Each input sequence is the 590 bp located upstream of the start codon. For more details on each motif, see Table 1. The phylogenetic tree is derived from Binz and Kägi (1997; branch lengths not to scale). Numbers along branches indicate when each motif was created (unboxed) or lost (boxed), ignoring any less conserved occurrences of the motif not reported by FootPrinter.
Genes from the metallothionein family are known to be regulated by a number of transcription factors. The most important of them, MTF-1, required for basal expression, binds to cis-acting elements known as metal response elements (MREs). A metallothionein promoter typically contains several MREs. In addition to MREs, the mouse MT-I promoter contains one or more GC boxes bound by Sp1, and major late transcription factor/antioxident response element (MLTF/ARE) binding site. The human MT-II promoter contains three basal level enhancer (BLE) elements known to bind transcription factors from the AP-2 family, and a glucocorticoid-responsive element (GRE), bound by the glucocorticoid receptor. Some of these bindings sites have also been identified in other metallothionein promoters. Most binding sites known in any of the species we consider occur within 300 bp of the start codon (for more on the regulation of genes in the metallothionein family, see Ghoshal and Jacob 2001).
We ran FootPrinter on the 590 bp of sequence located upstream of the start codon of each of the metallothionein genes listed in Figure 1. The 5′-UTR is usually short (between 50 and 100 bp for species for which the transcription start site is known), and was included in the sequences considered. We searched for conserved elements of the lengths 7, 8, 9, and 10, in consecutive runs, each time adjusting the parameters to ensure that the motifs reported are well conserved at a statistically significant level (see Methods).
Because the family contains both orthologous and paralogous genes, the ability of FootPrinter to allow for losses of regulatory elements is particularly crucial. Indeed, duplicated copies of a gene may evolve to have slightly different functions, and it is likely that the same holds for their promoter regions.
Our analysis identified 12 motifs, plotted in Figure 1 and listed in Table 1. Motifs labeled 3, 4, 5, and 10 all correspond to different variants of MREs. That is, they are all experimentally verified binding sites of the same transcription factor MTF-1 in at least one of the sequences. The most common motif, labeled 3, corresponds to MREs located on the reverse strand, whereas the others are on the forward strand. Motif 3 is present in all isoforms of all deuterostomes (echinoderms and vertebrates) studied. It is often present in multiple copies, with up to four copies in human MT-IV and rat and hamster MT-II. Note that other promoter regions in Figure 1 may also contain more copies of this motif than shown, but not sufficiently conserved to be reported (see Discussion). Motif 5 was only found in mammalian MT-I and MT-II, and motif 10 was only found in mammalian MT-II, whereas motif 4 appears to have been lost on the branch leading to mammalian MT-I, -II, and -IV. In all, FootPrinter identified five of the seven MREs documented in the TRANSFAC database for human MT-II, but only two of the six known in rat MT-I (see Discussion for an explanation of why these were missed).
The other motif known to have regulatory function is motif 1, a TATA-box, which was found in all isoforms of all mammals, except in MT-I of nonrodents. Had we insisted that the motif be present in all genes of the MT-I/MT-II mammal phyla, its parsimony score would have increased greatly, as it is so poorly conserved in nonrodent MT-I, to a point at which it would no longer have been significant.
The other seven conserved motifs identified are not documented in TRANSFAC (Wingender et al. 1996). None of them were found in the mammalian MT-I and MT-II families, which may partly explain our lack of knowledge about them, as genes in these families have received much more attention than the other isoforms. Motifs 7, 8, and 9, found only in frog and in the MT-III gene family, may be of particular interest. (Motif 2 also occurs in this set of sequences, but its order with respect to that of the other motifs is not as well conserved.) Another interesting point is that most of the motifs found are present in more than one isoform family, which indicates that we gained accuracy by considering the gene family as a whole, instead of running FootPrinter separately on each of the four mammal isoforms.
Insulin Gene Family
The upstream sequences of insulin genes are currently known only for three primates, two rodents (with two gene copies in each), and pig. This set of species is much less diverged than the metallothionein gene family. Because of this, we searched only for motifs with 0 mutations (for motif length 8) or 1 mutation (for motif lengths 9 and 10) at most, as motifs with more mutations would be likely to happen by chance in nonfunctional sequences. Our search identified four conserved motifs, all of them corresponding to known binding sites for that gene. (See Table 1 for this gene family and those that follow in the remainder of this section.) Several other binding sites were missed by FootPrinter because they contained a few too many mutations (see Discussion). Had we had a few more diverged species (or many other mammalian sequences), we may have been able to identify these sites as well. Another way to counteract the lack of diversity is to search for longer motifs, as we would then be able to accept motifs with more mutations. However, in this case, searches for motifs of the lengths 12 and 15 did not yield any new motifs.
IL-3
The data set for IL-3 is quite similar to that of insulin, as only six mammalian sequences are available. We were thus limited to reporting only motifs with at most one mutation. Two of the five motifs identified are known binding sites. In fact, motif 3 actually contains binding sites for a number of factors.
C-myc Promoter
The upstream sequences of c-myc are known for only 7 species, but these contain members from diverse animal phyla (fishes, batrachians, birds, and mammals). This allowed us to do a much more sensitive motif search. Four of our nine predictions are known binding sites. Notice that some of these were found only in mammals and chicken, others in all terrestrial animals, and one in all species considered, which again illustrates the necessity to search for conservation in subsets of the species. All these known binding sites are located in a 120-bp promoter region known to be very rich in binding sites. We also identified motifs with unknown function but which are as well conserved as those corresponding to known sites. Most of these novel motifs are located well upstream of the known regulatory-rich region, in an area where very few binding sites were previously known.
C-myc Second Intron
Although regulatory elements are often located in the 5′-promoter region, some genes also have regulatory elements in their introns, and this is believed to be the case for c-myc. The second intron of c-myc varies in size between 971 bp (in chicken) and 1376 bp (in human). This intron was shown by Spicer and Sonenshein (1992) to contain an antisense promoter that results in the transcription of the reverse strand of part of the c-myc gene. Our analysis indeed identified 10 highly conserved motifs. Many of these are located just downstream of the transcription start site for the reverse strand (located at position 832 in rat, corresponding to position 860 in human). All but one of these are novel motifs.
C-fos Promoter
Our set of sequences for c-fos covers a set of species that is similar to that for c-myc. The results are also quite similar. Five of our eight predictions are known binding sites, four of them concentrated in an 80-bp area located about 500 bp upstream of the start codon.
C-fos First Intron
The first intron of the c-fos gene is known to contain a long sequence that acts as a transcription terminator by blocking elongation (Mechti et al. 1991). The presence of Ca2+ has been shown to prevent this premature termination of transcription. The mechanism that underlies this interesting regulation mode is still unclear. FootPrinter very clearly identified that long sequence, as it is almost perfectly conserved in all tetrapoda considered. However, we also observed that this 103-bp-long sequence is broken into three segments. The first segment is conserved in all species considered, except fugu. The second one was found only in tetrapoda. The third segment is conserved in all species and contains a cAMP response element in humans (Lange and Bading 2001). This division into three segments indicates that each segment has a different function. Notice that in the absence of the two fish species, we would not have seen this division and would not have learned anything new about the region.
Growth Hormone 1
The growth hormone data set is our second largest; it contains sequences from nine fishes, one bird, and six mammals. Again, the subsets of species containing the motifs found vary greatly, from mammals only or fishes only to 15 of the 16 species. Five of the eight motifs identified are known binding sites in either rat or human.
DISCUSSION
Known Versus Predicted Binding Sites
In the Results section, we showed that FootPrinter identifies a large number of binding sites with a function that has been established experimentally. However, there are also many known binding sites that were not found by our approach. It is illuminating to analyze why FootPrinter did not detect those motifs. Of course, in the end, the reason must be that these motifs were not sufficiently well conserved to be reported, but a more detailed study is instructive.
The known binding sites missed by FootPrinter can be divided into five categories. First, some binding sites appear to have no significant matches in most other species. For example, the thyroid hormone receptor T3R binding site upstream of the rat growth hormone 1, and the Pur-1 binding site upstream of the rat insulin gene are both conserved only in rodents. There is very little hope of detecting these sites by phylogenetic footprinting, unless a large number of closely related species (in this case, rodents) are available.
Second, some binding sites show very good conservation, but only over a region that is shorter than the ones we looked for. This is the case for the GC-box of metallothionein, the sequence GGGGCGG of which is perfectly conserved in the four MT-II sequences and has only one mutation in the MT-III sequences. The substring GGGG is actually conserved in almost all mammalian isoforms. We may have been able to detect these kinds of motifs, had we searched for motifs of that length. (We did search for motifs of length 7, but the GC-box was not reported because it did not span a large enough part of the tree.) However, such short motifs are often likely to occur simply by chance in nonfunctional sequences. We could have allowed such short motifs, but our results would have been more likely to contain false positives.
Third, a small number of binding sites appear to be relatively well conserved but have had insertions or deletions (although it is not clear if the sequences with insertions or deletions are still functional). FootPrinter can allow for insertions and deletions in the motifs found, but we chose not to use this option, as it is believed that insertions and deletions are rare in binding sites. Allowing for insertions and deletions would thus have produced a few more true positives, but most likely at the price of many more false positives.
Fourth, some motifs are quite well conserved, but they barely fail to meet our statistical significance thresholds. This is the case for the CREB binding sites and CT-I regulatory element of insulin, both of which have parsimony score 3 over a motif of length 8. Again, allowing for that many mutations would have produced a number of false positives. However, if sequences from more organisms had been available, these two motifs might have been detected without increasing the false positive rate.
Fifth, some transcription factors bind as dimers, in which case the binding site may consist of two conserved regions, separated by a few variable nucleotides. For example, in metallothionein MT-II, transcription factors from the AP-2 dimer family are known to bind the BLE element. Visual inspection reveals that the pattern TGACnnnnnGCGG (where n is a variable nucleotide) is perfectly conserved in all four MT-II genes. Because of the variable internal sequence, FootPrinter did not discover this motif. However, a future version of the program will allow one to search for motifs containing a variable sequence in the middle, where mutations should not be counted. More generally, it is well known that some transcription factors can tolerate more than one type of nucleotide at a given position of the binding site. For example, the MRE binding sites of metallothionein can be described by the consensus string CTCTGCRCNCSGCCC, in which bold characters are absolutely required for metal response, R is A or G, and S is C or G (Ghoshal and Jacob 2001). In this case, one would want to assign a smaller penalty to purine-purine transitions at position 7 of the motif than to other substitutions. The current implementation of FootPrinter assigns equal cost to each type of substitution, but we are investigating ways for the program to learn different mutation cost matrices for each position in the sequence.
When reading the results presented in Figure 1 or Table 1, the reader should be aware that there may be more occurrences of regulatory elements than shown, if they are not sufficiently well conserved.
Comparison to Other Computational Methods
A number of existing computational techniques have been used or could be used to identify conserved motifs in orthologous sequences, although none has been designed precisely for that purpose (see Introduction). By far the tool most commonly used for phylogenetic footprinting is CLUSTALW (Thompson et al. 1994), a tree-based global multiple alignment program. We also consider DIALIGN (Morgenstern 1999), a segment-based multiple alignment program, and MEME (Bailey and Elkan 1995), a motif-finding technique based on expectation maximization.
The output of both CLUSTALW and DIALIGN is a global multiple alignment of the input sequences. Given a correct multiple alignment, one can easily identify conserved motifs, for example, by computing the parsimony score of each column of the alignment and outputting motifs with low overall parsimony score. One could also allow for motif losses and compare the score of a motif to its evolutionary span, as we propose in this paper. (Note that neither CLUSTALW nor DIALIGN currently uses either of these approaches.) However, correctly aligning a set of diverged sequences is a difficult task. For example, CLUSTALW produces very good alignments for closely related sequences (e.g., those from the insulin family and from IL-3, which all come from mammals), but most often incorrectly aligns more highly diverged sequences, thus failing to show the conservation of some motifs. DIALIGN produces better alignments for the purpose of phylogenetic footprinting, because it starts by identifying short conserved regions and then incorporates them into a multiple alignment. In fact, for most data sets, DIALIGN correctly aligned most of the conserved motifs found by FootPrinter (and vice versa: Most conserved regions present in the alignment of DIALIGN were reported by FootPrinter). However, in the metallothionein data set, several conserved sites were misaligned by DIALIGN. In general, we believe that for large data sets containing weakly conserved motifs, or motifs present in a small subset of the input sequences, the advantage of FootPrinter over DIALIGN will become clearer.
MEME is a motif-finding program that searches for motifs with high information content, but makes no use of phylogenetic information. Moreover, MEME does not consider the position at which motifs are found in each sequence, so that the motifs reported may occur in a different order in each input sequence (see Methods). Nonetheless, the majority of the motifs reported in this paper are also found by MEME. This is probably because these motifs are very highly conserved, which makes them relatively easy to identify. A notable exception is again the large metallothionein gene family, for which MEME fails to find many of the motifs that occur in small subsets of the input sequences.
From the point of view of running time, DIALIGN is about 10 times slower than FootPrinter on large data sets, with motif lengths and scores as in Table 1, whereas CLUSTALW and MEME run roughly as fast as FootPrinter.
A more quantitative analysis of the accuracy of each method on biological data such as that considered here is problematic, as there is no definitive classification of false positives and false negatives. We are currently conducting such comparative experiments on simulated data. Please refer to Blanchette et al. (2002) for more details on how the methods compare on biological sequences.
Phylogenetic Information
Throughout this paper, we assume that we are given the correct phylogenetic relationship among the sequences under study. It is the use of this phylogenetic information that allows FootPrinter to accurately identify regions of interest. The phylogenetic tree should represent the evolutionary history of the sequences considered, which may be different from that of the species they come from, because of lateral gene transfers. In vertebrates, such events appear relatively rare, and we thus used the species tree as an estimate of the sequence tree. When such a trusted tree is unavailable, one could infer the phylogenetic tree directly from the sequences considered or from their neighboring coding regions. (This is what Binz and Kägi [1997] did in the case of the metallothionein gene family, and this is what we did for the insulin gene family.) In cases in which the correct topology of the phylogenetic tree remains unclear, an unresolved multifurcating tree can be used.
The correctness of the parsimony scores computed obviously depends on that of the tree. Using a completely incorrect tree may greatly affect the accuracy of FootPrinter. However, using a tree with a small number of topological errors should still yield better results not than using a tree at all.
Improving Accuracy
The predictions of FootPrinter could be made more accurate by injecting more prior knowledge as to what interesting solutions ought to look like. For example, the order and orientation in which regulatory elements occur in a sequence should be the same in all species, unless large-scale genome rearrangements occurred. Using this information may allow us to reject spurious motifs with order that is not consistent across species. Regulatory elements often occur several times in the same promoter (e.g., some metallothionein promoters contain up to 15 imperfect copies of MREs). Incorporating this type of information may allow us to detect regulatory element that are not sufficiently conserved to be reported by FootPrinter but that occur in several copies in each input sequence, thus boosting the statistical significance of the motif. Finally, if one had some idea about the transcription factors potentially regulating a given gene, one may want to allow motifs that look like potential binding sites for those factors to have slightly larger parsimony scores.
METHODS
Algorithm
For the sake of clarity, we present here the simplest (but least efficient) version of the algorithm of FootPrinter, and we also assume that the only mutations allowed are point substitutions. The interested reader can find the extension to handle more general mutations, and the details of optimizations that make the algorithm truly practical, in a companion paper (Blanchette et al. 2002). The basic method is a dynamic programming algorithm similar to one presented by Sankoff and Rousseau (1975) for the computation of the parsimony score of a fixed set of aligned sequences (whereas what we seek is the most parsimonious choice of k-mer from each of the input sequences).
The inputs to the algorithm are n homologous sequences S1, S2, …, Sn; the phylogenetic tree T relating them; the length k of the motifs sought; and the maximum parsimony score d allowed. The algorithm proceeds from the leaves of T to its root. At each node u of T, it computes a table Wu containing 4k entries, one for each possible k-mer. For each such k-mer s, let Wu[s] be the best parsimony score that can be achieved for the subtree of T rooted at u, if the ancestral sequence at u was forced to be s. Let the set of children of u be denoted C(u); let h(s,t) be the number of positions at which k-mers s and t differ; and let Σ = {A,C,G,T}. The table Wu is computed according to the following recurrence:
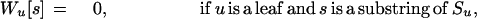 |
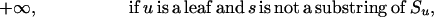 |
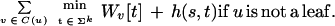 |
A straightforward implementation of this recurrence computes all W tables in time O(nk(42k + l)), where l is the average length of the input sequences S1, S2, …, Sn. The main term nk · 42k in this expression comes from the fact that, for each of the O(n) edges (u,v) of T, for each of the 4k possible values of s labeling u, and for each of the 4k values of t labeling v, the recurrence calls for the computation of h(s,t).
If r is the root of T, each entry of Wr that is at most d gives rise to one or more solutions to be reported. For each such entry, the corresponding k-mers of the n input sequences can be recovered by retracing the recurrence from the root back to the leaves. By maintaining appropriate pointers that reflect the computation of the W tables, the set of solutions can be recovered in time linear in its size. In nonrepetitive biological sequences, the number of solutions is usually small (when d is small), and the time to enumerate them is negligible compared to the time to compute the W tables.
The 42k factor in the complexity of the algorithm as described makes it impractical to use for most interesting values of k. In the companion paper (Blanchette et al. 2002), we show how various algorithmic optimizations can reduce the running time to O(nk min (l(3k)d/2, 4k + l)), which makes it quite practical for the type of data sets given in Results. Notice that the running time is proportional to nl, which is the total length of all the input sequences. This means that the performance of the algorithm's scales, as well as the number of species or length of regulatory region provided, is increased.
Although the running time is exponential in either d/2 or k (depending on which of l(3k)d/2 or 4k + l is the lesser), in practice both of these parameters are quite small: Typical values in our experiments were k = 10 and d = 3. Using a desktop workstation, a typical run of the algorithm on a data set of n = 10 sequences of length l = 700 each might take 30 seconds if only substitutions are allowed or a few minutes if insertions and deletions are allowed as well.
Handling Motif Losses
Here we discuss the generalization of the phylogenetic footprinting algorithm to identify motifs that may be missing (or highly mutated) in some of the input sequences. For this problem to make sense, there must be a way to compare two solutions containing motifs from different subsets of species. To do so, consider the total amount of evolution (measured, e.g., in millions of years) that the motif has survived. Motifs that have resisted a large amount of evolution are more likely to be interesting than those that span a short time.
To be able to estimate the amount of evolution spanned by a set of species, the algorithm must be given not only the phylogenetic tree T that relates the species, but also the length of each of its branches. We estimated branch lengths by computing pairwise alignment scores for the input sequences and using the Fitch-Margoliash algorithm (Fitch and Margoliash 1967) from the PHYLIP package (Felsenstein 1989) to find the branch lengths that make the tree distances match the pairwise distances as closely as possible. Estimating branch lengths is a notoriously difficult problem, and our estimates may be inaccurate. However, our experience indicates that the quality of the results obtained by the method does not depend very strongly on the accuracy of these estimates.
The algorithm identifies motifs that have small parsimony score but span a large part of T. More precisely, the algorithm solves the following problem. In addition to the inputs provided to the basic phylogenetic footprinting algorithm (described above), the user also provides thresholds δ0, δ1, …, δd. The problem is to find all sets of k-mers, one from each of the leaves i1, i2, …, im, where {i1, i2, …, im} is any subset of the n leaves of T, such that the parsimony score P of this set of k-mers on the subtree induced by the leaves i1, i2, …, im is at most d, and such that the subtree induced by the leaves i1, i2, …, im has total branch length at least δP. For example, the user can ask to see all motifs with parsimony score 0 that span at least 200 Myrs of T (i.e., δ0 = 200 Myrs), plus those with score 1 that span at least 350 Myrs (i.e., δ1 = 350 Myrs), plus those with score 2 that span at least 500 Myrs (i.e., δ2 = 500 Myrs).
The algorithm that solves this generalized problem is very similar in spirit to the dynamic programming algorithm described above. It is a few times slower than the algorithm that does not allow losses, but it produces much more accurate results. This is the algorithm that was used to identify the motifs reported in Table 1. The interested reader can find further details of the algorithm in Blanchette et al. (2002).
Other Useful Parameters
FootPrinter has a number of options that help to find more actual binding sites, although leaving out spurious hits. We briefly discuss some of them here. First, notice that in our formulation of the phylogenetic footprinting problem, the position at which a motif is found in each sequence is ignored. This is a typical feature of local alignment methods, to which our approach belongs. However, in some circumstances, it is desirable to penalize motifs with positions in the set of homologous sequences that vary too much and are thus unlikely to be instances of a single conserved binding site. There is a natural way to incorporate the notion of position into a parsimony score: We simply augment the definition of motif (which until now was just a k-mer) with a number that indicates the approximate position of the k-mer in the sequence. For this study, we usually divided each input sequence into 10 equal-sized regions and assigned a cost of one mutation for a motif to move to an adjacent region. In fact, to avoid inaccuracies when a motif occurs near a region boundary, we view each motif as also occurring in the two adjacent regions. This approach of dividing the sequences into regions only makes sense if we believe that corresponding regions of each input sequence are approximately homologous. For upstream sequences, this may be a reasonable assumption. However, for introns we did not use this option, as the variation in intron size makes it unclear which portions are homologous.
In our original definition of the phylogenetic footprinting problem allowing for regulatory element losses, there is no cost associated with losing a regulatory element, except that the motif spans a smaller part of the tree. This sometimes leads to undesirable situations, in which FootPrinter finds a motif that seems well conserved in two very distantly related species X and Y (thus spanning a large part of the tree) but that appears to have been lost independently in all phyla branching between X and Y. These multiple independent losses are quite unlikely in evolution, and one would like to penalize motifs that have been lost along too many branches. Once again this fits very nicely into our parsimony framework. We do so by assigning a cost to losing a motif along a given branch. For the results reported in this paper, we equate this cost to one substitution. However, one could also assign different loss costs along different branches, so that losses along long branches cost less than those along short branches. In the case of gene families, one may want to assign a smaller cost to motifs lost on branches that follow duplication events, as regulatory elements may be likely to be lost at these times.
Finally, it is often useful to restrict the number of mutations along any given branch of the phylogenetic tree. For example, in cases in which a motif is very well conserved in some subset of the sequences, this avoids finding spurious poorly conserved instances of the motif in sequences that actually do not contain the true binding site. Limiting the number of mutations per branch also has a very positive effect on the running time of the algorithm.
Statistical Significance
Any set of sequences contains some best conserved motif, but that does not mean that this motif was actually under selective pressure. To make sure that the motifs reported have a mutation rate significantly less than that of the surrounding nonfunctional sequence, we generated a set of random sequences with approximately the same evolutionary history as the input sequences. This set of sequences was generated by simulating evolution over the given phylogenetic tree with the inferred branch lengths. These simulated sequences thus mimic the real input sequences, except that the mutation rate is the same at all sites, and thus we should not find any unusually well conserved motifs in them. In this paper, a motif M with parsimony score s over a tree of size δs was reported only if the probability of finding such a motif (or one better conserved) in simulated sequences is <5% (for more details on measuring statistical significance, see Blanchette et al. 2002).
WEB SITE REFERENCES
http://bio.cs.washington.edu/GR/; Sequences, accession numbers, phylogenetic trees, and detailed results from FootPrinter.
http://bio.cs.washington.edu/software.html; FootPrinter, the computer algorithm described in this paper.
http://tolweb.org; The tree of life web project.
http://transfac.gbf.de/TRANSFAC/; A database on transcription factors and their DNA binding sites.
http://www.unizh.ch/∼mtpage; University of Zurich web site.
Acknowledgments
We thank Benno Schwikowski for helpful discussions. This material is based on work supported in part by a Natural Sciences and Engineering Research Council of Canada (NSERC) fellowship, by the National Science Foundation and DARPA under grant DBI-9601046, and by the National Science Foundation under grant DBI-9974498.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tompa@cs.washington.edu; FAX (206) 543-8331.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.6902.
REFERENCES
- Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning. 1995;21:51–80. [Google Scholar]
- Binz P-A, Kägi JHR. Molecular evolution of metallothioneins: Contributions from coding and non-coding regions. Poster at the Second European Meeting of the Protein Society, Cambridge. 1997. [Google Scholar]
- Blanchette, M., Schwikowski, B., and Tompa, M. 2002. Algorithms for phylogenetic footprinting. J. Comput. Biol. (in press). [DOI] [PubMed]
- Buhler J, Tompa M. Finding motifs using random projections. In: Lengauer T, Sankoff D, Istrail S, Pevzner P, Waterman M, editors. Proceedings of the Fifth Annual International Conference on Computational Biology. 2001. pp. 69–76. . Montreal, Canada. [Google Scholar]
- Cliften P, Hillier L, Fulton L, Graves T, Miner T, Gish W, Waterston R, Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. doi: 10.1101/gr.182901. [DOI] [PubMed] [Google Scholar]
- Dubchak I, Brudon M, Loots GG, Pachter L, Mayor C, Rubin EM, Frazer KA. Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Res. 2000;10:1304–1306. doi: 10.1101/gr.142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Bucher P. Searching for regulatory elements in human noncoding sequences. Curr Opin Struct Biol. 1997;7:399–405. doi: 10.1016/s0959-440x(97)80058-9. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP: Phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Fitch WM, Margoliash E. Construction of phylogenetic trees. Science. 1967;155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- Ghoshal K, Jacob SM. Regulation of metallothionein gene expression. Prog Nucleic Acids Res Mol Biol. 2001;66:357–384. doi: 10.1016/s0079-6603(00)66034-8. [DOI] [PubMed] [Google Scholar]
- Gumucio D, Shelton D, Bailey W, Slightom J, Goodman M. Phylogenetic footprinting reveals unexpected complexity in trans factor binding upstream from the ɛ-globin gene. Proc Natl Acad Sci. 1993;90:6018–6022. doi: 10.1073/pnas.90.13.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;8:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Hughes J, Estep P, Tavazoie S, Church G. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Jareborg N, Birney E, Durbin R. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 1999;9:815–824. doi: 10.1101/gr.9.9.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Bading H. The role of putative intragenic control element in c-fos regulation by calcium and growth factor signaling pathways. J Neurochem. 2001;77:1293–1300. doi: 10.1046/j.1471-4159.2001.00342.x. [DOI] [PubMed] [Google Scholar]
- Leung J, McKenzie E, Uglialoro A, Florez-Villanueva P, Sorkin B, Yunis E, Hartl D, Goldfeld A. Identification of phylogenetic footprints in primate tumor necrosis factor-α promoters. Proc Natl Acad Sci. 2000;97:6614–6618. doi: 10.1073/pnas.97.12.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor C M, Wang ZE, Miller W, Rubin EM, Frazer KA. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- Maddisson, D.R. 2002. The tree of life web project. http://tolweb.org.
- Manen JF, Savolainen V, Simon P. The atpB and rbcL promoters in plastid DNAs of a wide dicot range. J Mol Evol. 1994;38:577–582. doi: 10.1007/BF00175877. [DOI] [PubMed] [Google Scholar]
- McCue L, Thompson W, Carmack C, Ryan M, Liu J, Derbyshire V, Lawrence C. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire AM, Hughes JD, Church GM. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 2000;10:744–757. doi: 10.1101/gr.10.6.744. [DOI] [PubMed] [Google Scholar]
- Mechti N, Piechaczyk M, Blanchard JM, Jeanteur P, Lebleu B. Sequence requirements for premature transcriptional arrest within the first intron of the mouse c-fos gene. Mol Cell Biol. 1991;11:2832–2840. doi: 10.1128/mcb.11.5.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern B. DIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999;15:211–218. doi: 10.1093/bioinformatics/15.3.211. [DOI] [PubMed] [Google Scholar]
- Morgenstern B, Frech K, Dress A, Werner T. DIALIGN: Finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14:290–294. doi: 10.1093/bioinformatics/14.3.290. [DOI] [PubMed] [Google Scholar]
- Mouchel N, Tebbutt SJ, Broackes-Carter FC, Sahota V, Summerfield T, Gregory DJ, Harris A. The sheep genome contributes to localization of control elements in a human gene with complex regulatory mechanisms. Genomics. 2001;76:9–13. doi: 10.1006/geno.2001.6603. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, Johnson W, Zhang YP, Ryder OA, O'Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth F, Hughes J, Estep P, Church G. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Sankoff D, Rousseau P. Locating the vertices of a Steiner tree in arbitrary metric space. Math Prog. 1975;9:240–246. [Google Scholar]
- Sinha S, Tompa M. A statistical method for finding transcription factor binding sites. In: Altman R, Bailey TL, Bourne P, Gribskov W, Lengauer T, Shindgalov IN, Ten Eyck LF, Weissig H, editors. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. San Diego, CA: AAAI Press; 2000. pp. 344–354. [PubMed] [Google Scholar]
- Spicer DB, Sonenshein GE. An antisense promoter of the murine c-myc gene is localized within intron 2. Mol Cell Biol. 1992;12:1324–1329. doi: 10.1128/mcb.12.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagle D, Koop B, Goodman M, Slightom J, Hess D, Jones R. Embryonic ɛ and γ globin genes of a prosimian primate (Galago crassicaudatus): Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988;203:439–455. doi: 10.1016/0022-2836(88)90011-3. [DOI] [PubMed] [Google Scholar]
- Thompson J, Higgins D, Gibson T. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M. Identifying functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1143–1144. doi: 10.1101/gr.197101. [DOI] [PubMed] [Google Scholar]
- van Helden J, André B, Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol. 1998;281:827–842. doi: 10.1006/jmbi.1998.1947. [DOI] [PubMed] [Google Scholar]
- Vuillaumier S, Dixmeras I, Messai H, Lapoumeroulie C, Lallemand D, Gekas J, Chebab F, Perret C, Elion J, Denamur E. Cross-species characterization of the promoter region of the cystic fibrosis transmembrane conductance regulator gene reveals multiple levels of regulation. Biochem J. 1997;327:652–662. doi: 10.1042/bj3270651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nature Genet. 2000;26:225–228. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, C.T. and Stormo, G.D. 2000. ANN-Spec: A method for discovering transcription factor binding sites with improved specificity. Pac Symp Biocomput. 467–478. [DOI] [PubMed]
- Wu Q, Zhang T, Cheng J-F, Kim Y, Grimwood J, Schmutz J, Dickson M, Noonan JP, Zhang MQ, Myers R M, et al. Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res. 2001;11:389–404. doi: 10.1101/gr.167301. [DOI] [PMC free article] [PubMed] [Google Scholar]