

Abstract
Microsporidians are a group of emerging pathogens typically associated with chronic diarrhea in immunocompromised individuals. The number of reports of infections with these organisms and the disseminated pathology is growing as diagnostic tools become more readily available. However, little is known about the innate immune response induced by and generated against these parasites. Using a coculture chemotaxis system, primary human macrophages were infected with Encephalitozoon cuniculi or Encephalitozoon intestinalis, and the recruitment of naïve monocytes was monitored. Encephalitozoon spp. induced an average threefold increase in migration of naïve cells 48 h postinfection, which corresponded to optimal infection of monocyte-derived-macrophages. A limited microarray analysis of infected macrophages revealed several chemokines involved in the inflammatory responses whose expression was upregulated, including CCL1, CCL2, CCL3, CCL4, CCL7, CCL15, CCL20, CXCL1, CXCL2, CXCL3, CXCL5, and CXCL8. The levels of 6 of 11 chemokines also present in the microarray were confirmed to be elevated by protein profiling. Kinetic studies confirmed that secreted CCL2, CCL3, and CCL4 were expressed as early as 6 h postinfection, with peak expression at 12 to 24 h and expression remaining until 48 h postinfection. Neutralization of these chemokines, specifically CCL4, significantly reduced the number of migrating cells in vitro, indicating their role in the induction of monocyte migration. This mechanism of recruitment not only supports the evidence that in vivo cellular infiltration occurs but also provides new hosts for the parasites, which escape macrophages by rupturing the host cell. To our knowledge, this is the first documentation that chemokine production is induced by microsporidian infections in human macrophages.
Microsporidiosis is a disease that is caused by an obligate intracellular eukaryotic parasite and has gained recognition as an opportunistic infection in AIDS patients, commonly causing chronic diarrhea leading to malabsorption of nutrients and wasting (23, 32). The true extent of microsporidiosis is difficult to determine, because it is often underreported due to a lack of proper facilities to diagnose this disease (13). Epidemiological reports indicate that anywhere from 5 to 50% (19) of patients with chronic diarrhea are positive for microsporidians, most notably Enterocytozoon bieneusi and Encephalitozoon spp. (7). Based on projections that in the course of human immunodeficiency virus (HIV) AIDS about 93% of individuals have at least one bout of chronic diarrhea (10), these pathogens could conservatively account for chronic diarrhea in approximately 1.3 to 13.5 million people suffering from a life-threatening loss of nutrients and liquids in the sub-Saharan region alone. (The number of cases of microsporidiosis was estimated using previously published reports [19] of the incidence [5 to 50%] of infection in individuals who have chronic diarrhea and are HIV positive. An estimated 93% of AIDS patients develop chronic diarrhea [10] in regions, such as sub-Saharan Africa, where water treatment and highly active antiretroviral therapy are limited. According to the 2005 UNAIDS/WHO AIDS report, 29 million individuals are infected with HIV in this region.)
More recently, cases of microsporidiosis in patients who received immunosuppressive therapy (2, 12, 14) or in immunocompetent individuals (26) who presented with symptoms of chronic diarrhea have been described. Although the most common symptoms of the disease are enteric, there have been many reports of more severe disseminated diseases, such as keratoconjunctivitis, sinusitis, tracheobronchitis, encephalitis, interstitial nephritis, hepatitis, cholecystitis, osteomyelitis, and myositis (7, 22).
Microsporidians are ubiquitous in nature and are known to infect a variety of vertebrate and invertebrate organisms (4, 6, 38, 40). Although these organisms were once classified as protozoans, new evidence based on phylogenetic analysis suggests that they are more closely related to fungi (4, 11, 39). Of the more than 1,200 species of microsporidians, only 14 have been reported to infect humans (4). Microsporidian infections are believed to occur when a spore in contaminated water or food is ingested (4). Infections of epithelial and endothelial cells and macrophages are common (19). Classically, cellular invasion occurs when a spore encounters a host cell and everts a polar tube, which penetrates the cell membrane of the host cell and injects the sporoplasm. Alternatively, spores can be internalized through phagocytosis or endocytosis (8). Further proliferation and spore production occur through merogony and sporogony within a parasitophorous vacuole, followed by lysis of the host cell and release of mature spores (9).
While most studies of microsporidians have focused on their genomes and life cycles (7), there are limited data on the host responses to these opportunistic pathogens and especially their roles in human macrophage infection and disseminated disease (8, 30). Some reports indicate that macrophages/monocytes are the source of the disseminated pathogen (33). In individuals with multifocal organ involvement, infiltrates of infected macrophages are evident in lesions, microabscesses, and granulomas (37). In animal models, injection of infectious spores results in a peritoneal infiltrate with predominately monocytes/macrophages, which is followed by disappearance of these cells and presumably spread to the lymph nodes and other tissues (28, 36). Determining which chemokines are present is critical in the development of therapeutic strategies that prevent dissemination of the pathogen and the resulting disease. Based on previous reports, we investigated two species of microsporidians that are known to cause disseminated diseases, Encephalitozoon cuniculi and Encephalitozoon intestinalis, and defined the production of a chemotactic gradient that is induced by infection with these intracellular parasites. This paper provides the first description of the innate immune response in disseminating infections with regard to chemokines and provides a foundation for describing the initial host reaction to these pathogens involving human macrophages.
MATERIALS AND METHODS
Reagents and antibodies.
Unless indicated otherwise, all tissue culture media and plastic ware were purchased from VWR (West Chester, PA), all neutralizing antibodies and protein array kits were purchased from R&D Systems (Minneapolis, MN), all enzyme-linked immunosorbent assay (ELISA) reagents were purchased from Biosource (Invitrogen, Carlsbad, CA), and all gene microarray materials were purchased from SuperArray Bioscience Corp. (Frederick, MD).
Cell culture.
Peripheral blood mononuclear cells were isolated from buffy coats from healthy donors (Our Lady of the Lake Regional Medical Center, Baton Rouge, LA) by gradient centrifugation on lymphocyte separation media (Cambrex). Monocyte-derived macrophages (MDM) were obtained by adherence assays. Briefly, monocytes were plated in 75-cm2 flasks (1 × 107 monocytes), six-well culture plates with cover glasses, or 24-well culture dishes (Greiner Bio-One; Cellstar) (1 × 106 monocytes/ml) for 3 h in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 μg/ml gentamicin, and 0.25 μg/ml amphotericin B. Cells were stringently washed with phosphate-buffered saline to remove nonadherent mononuclear cells and then allowed to differentiate for 7 days in complete medium with 10% fetal calf serum (FCS). Monocytes used as target cells for recruitment in chemotaxis assays were isolated from mononuclear cells by magnetic bead separation using a Dynal monocyte negative isolation kit (Invitrogen) and resuspended in complete medium with 10% FCS.
Microsporidians.
E. cuniculi III and E. intestinalis (a generous gift from Elizabeth Didier, Tulane National Primate Research Center, Covington, LA) were grown in a rabbit kidney cell line (ATCC CCL-37) and harvested from tissue culture supernatants. Spores were washed once in phosphate-buffered saline containing 0.2% Tween 20, resuspended in supplemented DMEM, and counted with a hemacytometer (5). Some spores were inactivated by treatment with a 10% bleach solution for 30 min and washed once with water. All spores were used at a spore-to-MDM infection ratio of 5:1 (5 × 106 spores and 1 × 106 macrophages), unless indicated otherwise.
Microscopy.
MDM adhering to cover glasses were monitored for spore uptake by challenging them with either E. cuniculi or E. intestinalis spores labeled with PKH26 fluorescent cell linker used according to the manufacturer's instructions (Sigma, St. Louis, MO) for various times at 37°C in 5% CO2. Cells were fixed in 10% formalin (Sigma) and labeled with PKH67 fluorescent cell linker (Sigma), a general cell membrane label. Cover glasses were mounted in Prolong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen) to detect nuclear staining of MDM.
Microsporidian proliferation and infection were monitored using the bromodeoxyuridine (BrdU) assay system (Invitrogen). MDM were challenged with spores for 3 days and fixed with 4% paraformaldehyde. Twenty-four hours prior to fixation, 100 μM BrdU was added to each well. Cells were washed with 1% Triton X-100 and treated with 1 N HCl for 10 min on ice, with 2 N HCl for 10 min at room temperature, and with 2 N HCl for 20 min at 37°C. Cells were neutralized in 0.1 M borate buffer and washed with 1% Triton X-100. Coverslips were incubated on ice with a 1/50 dilution of anti-BrdU (Invitrogen) in 0.5% bovine serum albumin and 1% Triton X-100 for 90 min. The coverslips were washed with 1% Triton X-100 and mounted in Prolong Gold antifade reagent (Invitrogen).
Cells were viewed using a Leica DMI 6000 B inverted fluorescence microscope, and images were captured and analyzed with a Leica DFX300 FX charge-coupled device camera and the Image Pro Plus v5.1 software (MediaCybernetics, Silver Spring, MD).
Chemotaxis assay.
Coculture chemotaxis assays were performed as previously described (15). Briefly, 7-day-old MDM were infected with E. cuniculi or E. intestinalis for 24 h at 37°C in 5% CO2. Sterile 3-μm polycarbonate transwell inserts (Corning Costar) were placed in 24-well plates that contained infected MDM. Target monocytes were labeled with a red fluorescent cell linker (Sigma) by following the manufacturer's instructions, added to the top of the transwell inserts (1 × 106 cells), and cocultured with infected MDM for 24 h at 37°C in 5% CO2. After this coculture step, supernatants were removed, and cells were fixed with 10% formalin. For some of the cultures, supernatants were removed and collected after 24 h of infection and then replaced with complete DMEM with 10% FCS containing one of the following neutralization antibodies: anti-CCL2 (0.9 μg/ml), anti-CCL3 (0.5 μg/ml), anti-CCL4 (0.3 μg/ml), or mouse immunoglobulin G1/immunoglobulin G2b (1.0 μg/ml). The migration of fluorescently labeled target cells was observed and quantified using an inverted fluorescence microscope (Leica DMI 6000 B) and the Image Pro Plus v5.1 software (Mediacybernetics). Each experiment was performed in duplicate, and 10 fields of view were counted for each condition. Ten donors were analyzed for infection with E. cuniculi, and three donors were analyzed for infection with E. intestinalis. Fold changes in monocyte recruitment were calculated by dividing the number of cells migrating under the experimental conditions by the number of cells in the untreated wells.
Focused microarray.
All focused microarray analyses were performed according to the manufacturer's instructions (R&D Systems). Total RNA was isolated from MDM that were infected for 6 h with E. cuniculi and were grown in 75-cm2 flasks using a QIAGEN RNeasy mini kit (Valencia, CA); then the RNA was quantified with a NanoDrop ND-1000 spectrophotometer, and the quality was assessed with an Experion automated electrophoresis system (Bio-Rad). Total RNA was amplified and labeled with biotin-16-UTP (Roche Applied Science) using a TrueLabeling-AMP 2.0 kit (SuperArray). Amplified cRNA was quantified again and equal amounts of cRNA from three donors was pooled. Pools of cRNAs were hybridized to a chemokine and receptor Oligo GEArray (OHS-022) overnight in a hybridization oven at 60°C. Expressed genes were detected using a CDP-Star chemiluminescence kit (SuperArray), and images were obtained using the Bio-Rad Gel Doc 2000 system and Quantity One software (Bio-Rad). The data were analyzed with the GEArray expression analysis suite (SuperArray). All data were normalized to data for the glyceraldehyde-3-phosphate dehydrogenase and β-actin housekeeping genes and background corrected using blank values. Fold changes in gene expression are expressed as the means of two experiments.
Focused protein array.
All focused protein array analyses were performed according to the manufacturer's instructions. Supernatants from overnight infections with E. cuniculi were grouped by using pools from six donors and incubated overnight with human cytokine array panel A (R&D Systems). Horseradish peroxidase substrate (Bio-Rad) was used to detect protein expression, and data were captured by exposure to Kodak BioMax Light film. Arrays were scanned into a computer, and optical density measurements were obtained with the Image Pro Plus v 5.1 software (Mediacybernetics). The data are expressed below as the mean fold changes for two experiments.
ELISA.
Supernatants were collected from MDM cultures at different times after infection with E. cuniculi or E. intestinalis, and CCL2, CCL3, and CCL4 chemokine production was analyzed by ELISA. Samples were assayed in duplicate.
Statistical analysis.
Student's paired two-tailed t test was used. P values of ≤0.05 were considered significant. Analyses were performed using the InStat software (GraphPad).
RESULTS
Infection of primary human macrophages with Encephalitozoon spp. recruits monocytes in vitro.
To evaluate the host response to the initial spore entry, experiments were conducted to determine the proper kinetics for obtaining cultures of primary macrophages in which the majority of cells had visible vacuoles containing microsporidians. MDM grown on cover glasses were challenged with labeled spores (red) and fixed in 10% buffered formalin at various times. Host cells were labeled with a green membrane stain and a blue nuclear stain. Using fluorescence microscopy, the cells positive for spores in 10 consecutive fields were counted. In agreement with the kinetics reported previously (9), it was established that more than 50% of cells had perinuclear vacuoles containing spores between 24 and 48 h postinfection (Fig. 1A). Distinct vacuoles containing labeled spores could be readily observed by 48 h postinfection (Fig. 1B). To determine if spore uptake resulted in microsporidian proliferation and productive infection, a BrdU assay was performed. Since MDM are terminally differentiated, only replicating meronts incorporated the BrdU nucleotide. At 72 h postinfection, approximately 27% of the cells contained a perinuclear vacuole that stained positive for BrdU (Fig. 1C).
FIG. 1.

Encephalitozoon spp. uptake and replication in macrophages. (A) MDM were challenged with labeled E. cuniculi spores (red), and the number of cells having vacuoles containing spores was determined in 10 fields at several times. By 48 h postchallenge a majority of MDM were positive for parasitophorous vacuoles. (B) Labeled spores in vacuoles that are perinuclear (blue) are indicated by arrows (n = 3). (C) By 72 h postinfection, approximately 27% of MDM were positive for vacuoles containing replicating microsporidians, as indicated by the incorporation of BrdU (green) (n = 3). Replication was not observed in MDM challenged with chlorine-treated spores. DIC, Differential interference contrast microscopy.
To determine whether infection of macrophages by Encephalitozoon spp. induces recruitment of monocytes, a previously described (15) coculture chemotaxis system was used to detect a functional chemotactic gradient. Initial experiments were conducted to determine the ratio of spores to MDM (2:1, 5:1, and 10:1) needed to provide reproducible infection rates and immune responses. An infection ratio of 5:1 gave consistent responses for all donors and was used in all experiments described here (Fig. 2A). The fluorescently labeled target cells in the transwell system allowed the times of optimal cell migration to be monitored. MDM were infected for 24 h before the analysis was performed. After 24 h, the supernatants containing free spores were removed, and new media were added. Monocytes were labeled and placed into the upper chamber of a coculture plate. The cells were analyzed for an additional 24 h to determine the prime chemotaxis response. In the initial experiments, the incubated cocultures were monitored at 30-min intervals for up to 2 h, and subsequently they were monitored at 1-h intervals up to 8 h and at 24 h (data not shown). Incubation for 24 h yielded the optimal response. Infection of macrophages with either E. cuniculi or E. intestinalis induced a 2.9-fold increase in monocyte migration after infection for 48 h (Fig. 2B). To ensure that the observed monocyte migration was induced by an MDM-generated gradient, only E. cuniculi spores were added to some wells. Spores by themselves did not induce migration (Fig. 2B).
FIG. 2.
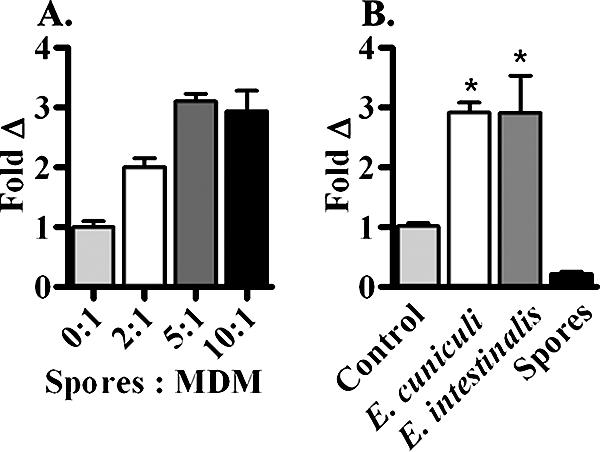
Encephalitozoon spp. infection of human macrophages induces monocyte migration. (A) MDM were infected with E. cuniculi spores for 24 h at spore-to-MDM ratios of 2:1, 5:1, and 10:1 to determine the optimal spore infection ratio for monocyte recruitment. At 24 h, labeled monocytes were added to transwell inserts and cocultured with infected MDM for an additional 24 h. A spore-to-MDM ratio of 5:1 resulted in consistent infection rates with human donors. (B) In coculture chemotaxis assays, infection of MDM with either E. cuniculi or E. intestinalis resulted in 2.9-fold recruitment of naïve monocytes. There were no significant differences in the chemotactic response between the Encephalitozoon spp. Wells that contained only spores and no MDM did not induce a chemotactic response. The error bars indicate standard errors of the means. An asterisk indicates that the P value is ≤0.05.
Focused gene array reveals upregulation of chemokines and receptors by E. cuniculi infection.
To establish which chemokines and receptors may be responsible for initiating monocytic infiltration, a limited, pathway-focused gene array analysis of chemokine signaling was performed to screen for possible candidates. Total RNA was isolated from MDM in medium or from MDM infected with E. cuniculi for 6 h, converted to biotinylated cRNA, and amplified. The amplified cRNA was hybridized overnight and detected using a chemiluminescent substrate.
Stimulation with E. cuniculi spores induced a twofold or greater increase in expression of 40 of 128 genes. Among the 40 genes whose expression was increased were the genes encoding several key chemokines and chemokine receptors, as shown in Tables 1. E. cuniculi infection of MDM elicited expression of several important chemokines which are capable of recruiting the granulocytes (CXCL1, CXCL2, CXCL3, CXCL5, and CXCL8), supporting the hypothesis that other cell types are present at sites of infection (29, 37). However, the vast majority of chemokines whose expression was increased by microsporidian infection were found to recruit monocytes, including CCL1, CCL2, CCL3, CCL4, CCL4L1, CCL5, and CCL7. It is important to note that these chemokines can also act on lymphocytes, such as TH1 cells, and dendritic cells to influence their migration. Additionally, increases in the levels of two vital monocyte receptors, CCR1 and CCR5, were also observed.
TABLE 1.
Microsporidian infection increases chemokine and receptor gene expression
| Chemokine or receptor class | Gene | Common name of chemokine | Accession no. | Avg fold increase in expressiona |
|---|---|---|---|---|
| C-C chemokines | Ccl1 | I-309 | NM_002981 | 11.2 |
| Ccl2 | Monocyte chemoattractant protein 1 | NM_002982 | 2.3 | |
| Ccl3 | Macrophage inflammatory protein 1α | NM_002983 | 2.3 | |
| Ccl4 | Macrophage inflammatory protein 1β | NM_002984 | 12.3 | |
| Ccl4l1 | LAG-1 | NM_207007 | 22.7 | |
| Ccl5 | RANTES | NM_002985 | 25.6 | |
| Ccl7 | Monocyte chemoattractant protein 3 | NM_006273 | 2.9 | |
| Ccl15 | Macrophage inflammatory protein 1δ | NM_004167 | 8.4 | |
| Ccl20 | Macrophage inflammatory protein 3α | NM_004591 | 6.2 | |
| C-X-C chemokines | Cxcl1 | Growth-regulated oncogene α | NM_001511 | 20.7 |
| Cxcl2 | Growth-regulated oncogene β | NM_002089 | 21.4 | |
| Cxcl3 | Growth-regulated oncogene γ | NM_002090 | 5.2 | |
| Cxcl5 | Epithelial cell-derived neutrophil-activating protein 78 | NM_002994 | 5.7 | |
| Cxcl8 | Interleukin-8 | NM_000584 | 9.9 | |
| Cxcl16 | NM_022059 | 6.4 | ||
| C-C receptors | Ccr1 | NM_001295 | 2.9 | |
| Ccr5 | NM_000579 | 4.5 | ||
| Ccr7 | NM_001838 | 10.9 | ||
| Ccr12 | NM_003965 | 3.6 | ||
| C-X-C receptor | Cxcr4 | NM_003467 | 2.2 |
Average fold increases in MDM gene expression were determined at 6 h after infection with E. cuniculi. Each array experiment was performed with pooled cRNA from three donors (n = 2).
Proteomic profiling confirmed that levels of chemokines increased after E. cuniculi infection.
To verify the results obtained with the DNA arrays, an analysis of a focused protein array which included several, but not all, of the chemokines of interest was performed. Equal amounts of supernatants from MDM cultures from six donors infected for 6 h with E. cuniculi were pooled and incubated overnight with human cytokine array panel A. Detection using chemiluminescence revealed significant increases (≥2-fold) in the levels of only a few of the proteins screened for by the limited array (Table 2). For both CCL3 and CCL4, which are monocyte chemoattractants, there was a >15.0-fold increase, and for CCL5, a potent chemokine for lymphocytes, there was an approximately 25.0-fold increase. Moderate increases (≥1.5-fold) were observed for CCL1, CCL2, and CXCL1. The modest increase in the level of CXCL1 can be attributed to high levels in both the control and treated arrays, and this was also true for CXCL8.
TABLE 2.
Proteome profiler array
| Chemokine | Common name | Avg fold increase in expression
|
|
|---|---|---|---|
| Protein arraya | Gene arrayb | ||
| CCL1 | I-309 | 1.6 | 11.2 |
| CCL2 | Monocyte chemoattractant protein 1 | 1.9 | 2.3 |
| CCL3 | Macrophage inflammatory protein 1α | 16.4 | 2.3 |
| CCL4 | Macrophage inflammatory protein 1β | 23.3 | 35.0e |
| CCL5 | RANTES | 25.2 | 25.6 |
| CXCL1 | Growth-regulated oncogene α | 1.5 | 20.7 |
| CXCL8c | Interleukin-8 | 1.1 | 9.9 |
| CXCL10c | Interferon-inducible protein 10 | 0.9 | 1.0 |
| CXCL11d | I-TAC | 1.0 | 1.0 |
| CXCL12d | Stromal cell-derived factor 1 | 1.0 | 1.0 |
| G-CSFd | Colony-stimulating factor 3 | 1.0 | 6.9 |
The average fold increase was calculated by dividing the mean optical density of infected MDM by the mean optical density of uninfected MDM (n = 2).
Average fold increases in gene expression from Table 1.
Equivalent levels of protein were detected under both experimental conditions.
Protein was not detected under either condition.
Sum of the average fold increases for both CCL4 and CCL4L1 gene expression. These two protein products are represented by macrophage inflammatory protein 1β on the protein array.
Sustained chemotactic levels are different among Encephalitozoon spp.
Because microsporidian dissemination is thought to be associated with moncytic infiltrates, the data presented here focus on the chemokines that primarily recruit monocytes, namely, CCL2, CCL3, and CCL4. To establish the time at which the levels of these selected chemokines are elevated during Encephalitozoon infections, kinetic data were obtained at 1, 3, 6, 12, 24, 48, and 72 h, and protein levels were determined by ELISA (Fig. 3). Significant differences in the levels of all three chemokines were detected at 6 h postinfection for infections with both E. cuniculi and E. intestinalis. Infections with E. cuniculi resulted in peak levels of CCL3 and CCL4 at 24 and 12 h, respectively, followed by declines. The levels of these cytokines appeared to undulate but remained moderately high after the initial peak. CCL2 production continued to increase until 48 h and then appeared to remain at the elevated level. Similarly, infection with E. intestinalis resulted in peak levels of CCL3 and CCL4 at 24 h, followed by decreases. The amount of CCL2 that was produced in response to E. intestinalis peaked by 12 h, and the level remained elevated up to 72 h. The levels of all the chemokines produced were similar for the Encephalitozoon spp. throughout the kinetics of infection.
FIG. 3.
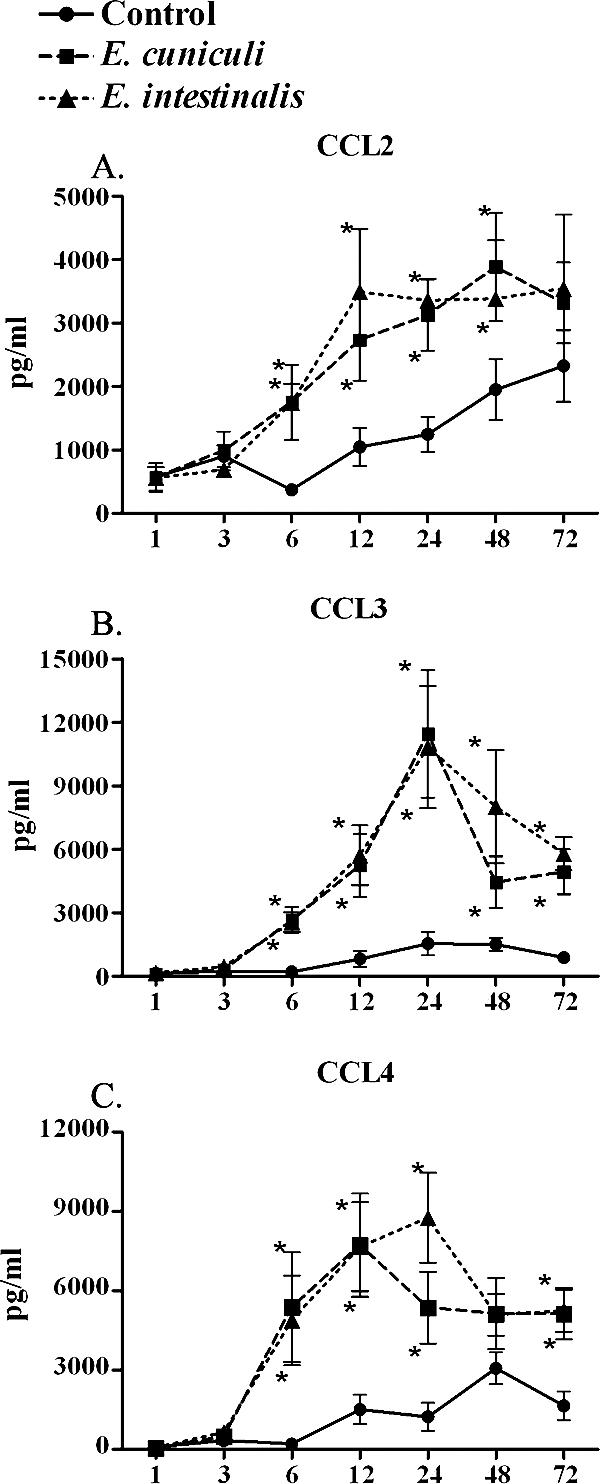
Levels of CCL2, CCL3, and CCL4 increase over time. ELISA confirmed the results of the gene array and protein array analyses for CCL2, CCL3, and CCL4. Supernatants were collected from cocultures of MDM and E. cuniculi or E. intestinalis at multiple times during infection. Both Encephalitozoon spp. induced a strong chemotactic profile after approximately 24 h of infection. After this, the expression of both CCL3 (n = 6) and CCL4 (n = 6) began to decline, whereas the CCL2 levels (n = 5) remained the same after 24 h. The error bars indicate standard errors of the means. An asterisk indicates that the P value is ≤0.05.
Inhibiting expression of CCL4 significantly reduces the number of cells responding to Encephalitozoon infection.
To evaluate whether any or all of the chemokines evaluated by ELISA contributed significantly to the infiltration of monocytes in the coculture system, MDM were infected for 24 h with E. cuniculi or E. intestinalis. The spent media were removed, and fresh complete media containing antibodies against CCL2, CCL3, or CCL4, a combination of the three antibodies, or an isotype-matched control were added prior to introduction of the responding monocytes into the coculture chemotaxis system.
In E. cuniculi assays, neutralizing antibodies against CCL2 or CCL3 resulted in modest but significant decreases in the levels of recruited monocytes (25% and 31%, respectively). In contrast, inhibition of CCL4 markedly decreased the number of migrating cells (49% decrease) (Fig. 4A). Similar results were obtained in cultures infected with E. intestinalis (Fig. 4B), in which antibodies against CCL2, CCL3, and CCL4 decreased migration by 28%, 36%, and 50%, respectively. Furthermore, E. cuniculi cultures inhibited with combinations of two antibodies resulted in modest decreases in the levels of infiltrating monocytes (38% with CCL2 and CCL3 and 45% with CCL2 and CCL4), while antibodies against CCL3 and CCL4 further reduced recruitment by one-half. Cultures containing a mixture of all three antibodies reduced the size of the population of migrating cells by 58%.
FIG. 4.
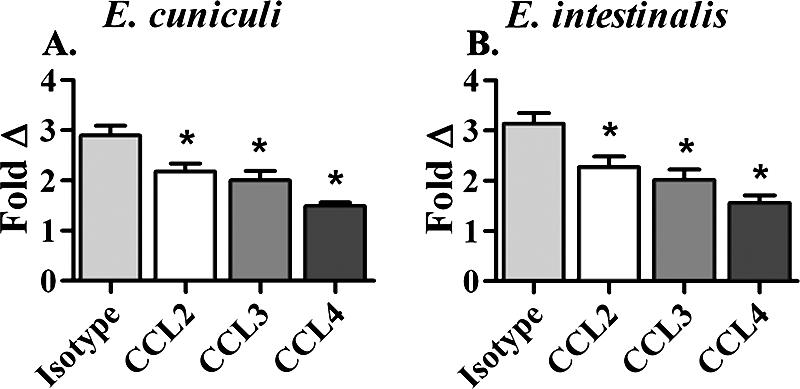
Neutralization of chemokines results in decreased monocyte recruitment. Neutralizing CCL2 or CCL3 resulted in small but significant decreases in monocyte recruitment, whereas blocking CCL4 resulted in a >50% decline in monocytic infiltration. Significant differences in cellular recruitment due to infection with either E. cuniculi (n = 10) or E. intestinalis (n = 3) were not observed. The error bars indicate standard errors of the means. An asterisk indicates that the P value is ≤0.05.
DISCUSSION
Limited data concerning the innate immune response against microsporidians are available. The number of reports of microsporidian infections not only in immunocompromised individuals but also in immunocompetent individuals is increasing, indicating that the incidence of infection may be higher than the incidence that is typically reported (1, 2, 12, 14, 26). Histological studies of microsporidiosis show that infiltrates are often composed of several cell types, including monocytes, granulocytes, and lymphocytes (1, 3, 31, 41); however, infected macrophages have been observed in disseminated disease (30) and are thought to be the vehicle for multiorgan infections.
The production of chemokines to recruit various immune cells during parasitic infections is important in innate immunity (24). Our data indicate that MDM infected with Encephalitozoon spp. induce a functional chemokine gradient that supports the recruitment of monocytes to sites of infection. In our study, E. cuniculi- and E. intestinalis-infected primary human macrophages induced an approximately threefold increase in migration of naïve monocytes after 24 h of incubation in a chemotaxis system. The delay in migration may reflect parasitic evasion of the immune response, providing time to invade and replicate (Fig. 1C).
Focused microarray analyses of MDM infected with E. cuniculi revealed increases in expression of several critical chemokines and receptors needed to recruit a variety of effector cells in order to mount a successful immune response. In a case study, Boldorini et al. found that in a patient with AIDS, an Encephalitozoon sp. infection of kidney epithelium was surrounded by lymphocytes, plasma cells, and macrophages (1), suggesting that these cells have a role in clearing the infection. We found that several genes involved in neutrophil recruitment (namely, the genes encoding C-X-C chemokines) were strongly induced, as were genes involved in the chemotaxis of dendritic and TH1 cells (namely, the genes encoding CCL1 and CCL5) (24). Khan and Moretto reported that in a murine model a TH1 response against microsporidian infections was critical (18), and later Khan et al. further defined a role for CD8+ and γδ T cells in resolving these infections in mice (20, 25). CCL5 is a potent chemoattractant for memory T cells, interleukin-2-activated T cells, and eosinophils (24). Among the upregulated genes that are important in monocyte recruitment (24) are the genes encoding CCL2, CCL3, CCL4, and CCL4L1. These four chemokines have been identified as agonists for the key monocyte receptors CCR1 and CCR5 (27), both of which exhibited increased expression in our model of infection. A similar response has also been demonstrated for Candida albicans infection of human monocytes, where increased expression of CCL2, CCL3, and CCL4 chemokine genes and CCR1 and CCR5 receptor genes has been observed (21). Interestingly, as observed in our model, C. albicans induces CCL4 expression that is many-fold greater than expression of either CCL2 or CCL3 at 6 h postinfection (21).
To determine which of the genes give rise to protein products, a focused protein analysis was performed using proteome profiler arrays. Data obtained from the limited array revealed that there were >15-fold increases for the chemokines CCL3, CCL4, and CCL5 (Table 2). Both CCL3 and CCL4 are known to be strong inducers of movement of monocytes to areas of infection and are also ligands for CCR5, an HIV coreceptor, whereas CCL5 predominately recruits T cells. In addition, the levels of expression CCL2, which is another potent monocyte chemoattractant, were moderate but significant.
The secretion kinetics of the monocyte chemokines most likely to participate in the spread of infection were further analyzed by ELISA to determine whether their expression coincided with the delayed recruitment observed. The increases in levels began around 6 h, and the levels peaked between 12 and 24 h. These times corresponded to the observed chemotactic response. It has yet to be determined whether microsporidians can dampen immediate immune responses during host uptake until they have established the meront stage within the macrophage. Delayed responses to other fungal pathogens, including Aspergillus fumigatus, have been attributed to altered host responses based on the recognition of conidia verses hyphal forms or to C. albicans yeast and hyphal forms which are thought to contribute to the pathology observed in opportunistic infections (35).
To determine the individual roles of these chemokines in the recruitment of potential new hosts for microsporidians, neutralizing antibodies were employed in the coculture system. This analysis revealed that CCL2 and CCL3 contributed to the migration of monocytes, but inhibiting CCL4 in the cultures resulted in the most dramatic reduction (Fig. 4). Neutralization of all three major chemokines reduced the levels of migrating monocytes to levels near the level of uninfected MDM, suggesting that these three chemokines were the chemoattractants responsible for the monocytic infiltration and, therefore, are potential targets of chemotherapeutic agents for controlling microsporidiosis (34). In comparison, using a murine model, Huffnagle et al. showed that Cryptococcus neoformans, a yeast-forming fungus known to cause disseminated disease, could evoke a monocyte chemoattractant protein 1 and macrophage inflammatory protein 1α response; in neutralization studies, the cryptococcal burden in the lungs of mice increased, while a decrease in macrophage and CD4+ T-cell recruitment was observed, resulting in inhibited clearance of the infection (16, 17).
While the immune response generated against Encephalitozoon spp. can result in recruitment of monocytes, it also has the potential to mediate adaptive immunity. However, in individuals with impairment of the adaptive arm of the immune system, such as AIDS patients, organ transplant recipients, the young, and the very old, the same recruited monocytes could amplify the infection. Understanding how macrophages function in propagating disease and how the life cycle of microsporidians can influence their responses is critical in developing antimicrosporidial compounds or anti-inflammatory intervention strategies. Studies to define host recognition of the parasite, signaling pathways, and subsequent cytokine profiles are under way.
Acknowledgments
This study was supported by NIH grant RR020159-01, by Louisiana Board of Regents grant LEQSF (2004-7)-RD-A-10, and by LSU FRG.
We thank Elizabeth Didier and Lisa Bowers for donating the E. cuniculi III strain and for their assistance, Andrew Whitehead and Jen Roach for their technical expertise, Earl Weidner for his knowledge and continuous support, and the staff at Our Lady of the Lake Blood Center for providing buffy coats. We also thank Dominique Homberger, James Moroney, and Karin Peterson for their critical review of the manuscript. We acknowledge the contributions of Nicole Hazard and our undergraduate researchers.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 18 December 2006.
REFERENCES
- 1.Boldorini, R., G. Monga, A. Tosoni, E. S. Didier, M. Nebuloni, G. Costanzi, G. Mazzucco, and J. M. Orenstein. 1998. Renal Encephalitozoon (Septata) intestinalis infection in a patient with AIDS. Post-mortem identification by means of transmission electron microscopy and PCR. Virchows Arch. 432:535-539. [DOI] [PubMed] [Google Scholar]
- 2.Carlson, J. R., L. Li, C. L. Helton, R. J. Munn, K. Wasson, R. V. Perez, B. J. Gallay, and W. E. Finkbeiner. 2004. Disseminated microsporidiosis in a pancreas/kidney transplant recipient. Arch. Pathol. Lab. Med. 128:e41-e43. [DOI] [PubMed] [Google Scholar]
- 3.Didier, E. S. 1998. Microsporidiosis. Clin. Infect. Dis. 27:1-7. [DOI] [PubMed] [Google Scholar]
- 4.Didier, E. S. 2005. Microsporidiosis: an emerging and opportunistic infection in humans and animals. Acta Trop. 94:61-76. [DOI] [PubMed] [Google Scholar]
- 5.Didier, E. S., P. J. Didier, D. N. Friedberg, S. M. Stenson, J. M. Orenstein, R. W. Yee, F. O. Tio, R. M. Davis, C. Vossbrinck, N. Millichamp, et al. 1991. Isolation and characterization of a new human microsporidian, Encephalitozoon hellem (n. sp.), from three AIDS patients with keratoconjunctivitis. J. Infect. Dis. 163:617-621. [DOI] [PubMed] [Google Scholar]
- 6.Didier, E. S., P. J. Didier, K. F. Snowden, and J. A. Shadduck. 2000. Microsporidiosis in mammals. Microbes Infect. 2:709-720. [DOI] [PubMed] [Google Scholar]
- 7.Didier, E. S., M. E. Stovall, L. C. Green, P. J. Brindley, K. Sestak, and P. J. Didier. 2004. Epidemiology of microsporidiosis: sources and modes of transmission. Vet. Parasitol. 126:145-166. [DOI] [PubMed] [Google Scholar]
- 8.Franzen, C. 2004. Microsporidia: how can they invade other cells? Trends Parasitol. 20:275-279. [DOI] [PubMed] [Google Scholar]
- 9.Franzen, C., A. Muller, P. Hartmann, and B. Salzberger. 2005. Cell invasion and intracellular fate of Encephalitozoon cuniculi (Microsporidia). Parasitology 130:285-292. [DOI] [PubMed] [Google Scholar]
- 10.Gazzard, B. G. 1988. HIV disease and the gastroenterologist. Gut 29:1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill, E. E., and N. M. Fast. 2006. Assessing the microsporidia-fungi relationship: combined phylogenetic analysis of eight genes. Gene 375:103-109. [DOI] [PubMed] [Google Scholar]
- 12.Goetz, M., S. Eichenlaub, G. R. Pape, and R. M. Hoffmann. 2001. Chronic diarrhea as a result of intestinal microsposidiosis in a liver transplant recipient. Transplantation 71:334-337. [DOI] [PubMed] [Google Scholar]
- 13.Grant, A. D., and K. M. De Cock. 2001. ABC of AIDS. HIV infection and AIDS in the developing world. BMJ 322:1475-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerard, A., M. Rabodonirina, L. Cotte, O. Liguory, M. A. Piens, S. Daoud, S. Picot, and J. L. Touraine. 1999. Intestinal microsporidiosis occurring in two renal transplant recipients treated with mycophenolate mofetil. Transplantation 68:699-707. [DOI] [PubMed] [Google Scholar]
- 15.Hale-Donze, H., T. Greenwell-Wild, D. Mizel, T. M. Doherty, D. Chatterjee, J. M. Orenstein, and S. M. Wahl. 2002. Mycobacterium avium complex promotes recruitment of monocyte hosts for HIV-1 and bacteria. J. Immunol. 169:3854-3862. [DOI] [PubMed] [Google Scholar]
- 16.Huffnagle, G. B., R. M. Strieter, L. K. McNeil, R. A. McDonald, M. D. Burdick, S. L. Kunkel, and G. B. Toews. 1997. Macrophage inflammatory protein-1alpha (MIP-1alpha) is required for the efferent phase of pulmonary cell-mediated immunity to a Cryptococcus neoformans infection. J. Immunol. 159:318-327. [PubMed] [Google Scholar]
- 17.Huffnagle, G. B., R. M. Strieter, T. J. Standiford, R. A. McDonald, M. D. Burdick, S. L. Kunkel, and G. B. Toews. 1995. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J. Immunol. 155:4790-4797. [PubMed] [Google Scholar]
- 18.Khan, I. A., and M. Moretto. 1999. Role of gamma interferon in cellular immune response against murine Encephalitozoon cuniculi infection. Infect. Immun. 67:1887-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan, I. A., M. Moretto, and L. M. Weiss. 2001. Immune response to Encephalitozoon cuniculi infection. Microbes Infect. 3:401-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan, I. A., J. D. Schwartzman, L. H. Kasper, and M. Moretto. 1999. CD8+ CTLs are essential for protective immunity against Encephalitozoon cuniculi infection. J. Immunol. 162:6086-6091. [PubMed] [Google Scholar]
- 21.Kim, H. S., E. H. Choi, J. Khan, E. Roilides, A. Francesconi, M. Kasai, T. Sein, R. L. Schaufele, K. Sakurai, C. G. Son, B. T. Greer, S. Chanock, C. A. Lyman, and T. J. Walsh. 2005. Expression of genes encoding innate host defense molecules in normal human monocytes in response to Candida albicans. Infect. Immun. 73:3714-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotler, D., and S. B. Heymsfield. 1998. HIV infection: a model chronic illness for studying wasting diseases. Am. J. Clin. Nutr. 68:519-520. [DOI] [PubMed] [Google Scholar]
- 23.Kotler, D. P., and J. M. Orenstein. 1994. Prevalence of intestinal microsporidiosis in HIV-infected individuals referred for gastroenterological evaluation. Am. J. Gastroenterol. 89:1998-2002. [PubMed] [Google Scholar]
- 24.Laing, K. J., and C. J. Secombes. 2004. Chemokines. Dev. Comp. Immunol. 28:443-460. [DOI] [PubMed] [Google Scholar]
- 25.Moretto, M., B. Durell, J. D. Schwartzman, and I. A. Khan. 2001. Gamma delta T cell-deficient mice have a down-regulated CD8+ T cell immune response against Encephalitozoon cuniculi infection. J. Immunol. 166:7389-7397. [DOI] [PubMed] [Google Scholar]
- 26.Muller, A., R. Bialek, A. Kamper, G. Fatkenheuer, B. Salzberger, and C. Franzen. 2001. Detection of microsporidia in travelers with diarrhea. J. Clin. Microbiol. 39:1630-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murdoch, C., and A. Finn. 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood 95:3032-3043. [PubMed] [Google Scholar]
- 28.Niederkorn, J. Y., J. A. Shadduck, and E. C. Schmidt. 1981. Susceptibility of selected inbred strains of mice to Encephalitozoon cuniculi. J. Infect. Dis. 144:249-253. [DOI] [PubMed] [Google Scholar]
- 29.Orenstein, J. M., and D. P. Kolter. 1999. Clinical syndromes associated with microsporidiosis, p. 258-292. In M. Wittner and L. M. Weiss (ed.), The microsporidia and microsporidiosis. ASM Press, Washington, DC.
- 30.Orenstein, J. M. 2003. Diagnostic pathology of microsporidiosis. Ultrastruct. Pathol. 27:141-149. [DOI] [PubMed] [Google Scholar]
- 31.Orenstein, J. M. 1991. Microsporidiosis in the acquired immunodeficiency syndrome. J. Parasitol. 77:843-864. [PubMed] [Google Scholar]
- 32.Orenstein, J. M., J. Chiang, W. Steinberg, P. D. Smith, H. Rotterdam, and D. P. Kotler. 1990. Intestinal microsporidiosis as a cause of diarrhea in human immunodeficiency virus-infected patients: a report of 20 cases. Hum. Pathol. 21:475-481. [DOI] [PubMed] [Google Scholar]
- 33.Orenstein, J. M., H. P. Gaetz, A. T. Yachnis, S. S. Frankel, R. B. Mertens, and E. S. Didier. 1997. Disseminated microsporidiosis in AIDS: are any organs spared? AIDS (London). 11:385-386. [PubMed] [Google Scholar]
- 34.Pease, J. E., and T. J. Williams. 2006. The attraction of chemokines as a target for specific anti-inflammatory therapy. Br. J. Pharmacol. 147(Suppl. 1):S212-S221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roeder, A., C. J. Kirschning, R. A. Rupec, M. Schaller, and H. C. Korting. 2004. Toll-like receptors and innate antifungal responses. Trends Microbiol. 12:44-49. [DOI] [PubMed] [Google Scholar]
- 36.Salat, J., P. Braunfuchsova, and J. Kopecky. 2001. Experimental infection of immunocompetent and immunodeficient mice with Encephalitozoon cuniculi. Folia Parasitol. (Prague) 48:249-254. [PubMed] [Google Scholar]
- 37.Shadduck, J. A., and J. M. Orenstein. 1993. Comparative pathology of microsporidiosis. Arch. Pathol. Lab. Med. 117:1215-1219. [PubMed] [Google Scholar]
- 38.Snowden, K., K. Logan, and E. S. Didier. 1999. Encephalitozoon cuniculi strain III is a cause of encephalitozoonosis in both humans and dogs. J. Infect. Dis. 180:2086-2088. [DOI] [PubMed] [Google Scholar]
- 39.Thomarat, F., C. P. Vivares, and M. Gouy. 2004. Phylogenetic analysis of the complete genome sequence of Encephalitozoon cuniculi supports the fungal origin of microsporidia and reveals a high frequency of fast-evolving genes. J. Mol. Evol. 59:780-791. [DOI] [PubMed] [Google Scholar]
- 40.Visvesvara, G. S. 2002. In vitro cultivation of microsporidia of clinical importance. Clin. Microbiol. Rev. 15:401-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber, R., R. T. Bryan, D. A. Schwartz, and R. L. Owen. 1994. Human microsporidial infections. Clin. Microbiol. Rev. 7:426-461. [DOI] [PMC free article] [PubMed] [Google Scholar]