

Abstract
Mucosal hyperplasia is a characteristic component of otitis media. The present study investigated the participation of signaling via the Jun N-terminal protein kinase (JNK) mitogen-activated protein kinase in middle ear mucosal hyperplasia in animal models of bacterial otitis media. Otitis media was induced by the inoculation of nontypeable Haemophilus influenzae into the middle ear cavity. Western blotting revealed that phosphorylation of JNK isoforms in the middle ear mucosa preceded but paralleled mucosal hyperplasia in this in vivo rat model. Nuclear JNK phosphorylation was observed in many cells of both the mucosal epithelium and stroma by immunohistochemistry. In an in vitro model of primary rat middle ear mucosal explants, bacterially induced mucosal growth was blocked by the Rac/Cdc42 inhibitor Clostridium difficile toxin B, the mixed-lineage kinase inhibitor CEP11004, and the JNK inhibitor SP600125. Finally, the JNK inhibitor SP600125 significantly inhibited mucosal hyperplasia during in vivo bacterial otitis media in guinea pigs. Inhibition of JNK in vivo resulted in a diminished proliferative response, as shown by a local decrease in proliferating cell nuclear antigen protein expression by immunohistochemistry. We conclude that activation of JNK is a critical pathway for bacterially induced mucosal hyperplasia during otitis media, influencing tissue proliferation.
Otitis media (OM) is the most common medical problem for which children see a physician. Despite otalgia and temporary hearing loss, a single episode of acute OM is not usually a serious concern. On the other hand, recurrent acute OM and chronic OM have been associated with numerous adverse long-term sequelae, including conductive and sensorineural hearing loss (6), impaired speech and language development (21), impaired academic achievement (3), and irreversible middle ear (ME) disease (39).
Hyperplasia of the ME mucosa is an important component of OM, involving substantial cell proliferation and differentiation (17). Hyperplasia contributes to the deleterious sequelae of OM, including the production of mucous secretions of ME effusions (36). Hyperplasia is also involved in fibrosis and other permanent damage that can occur in repeated and/or chronic OM (32). The regulation of mucosal hyperplasia in the ME is therefore of clinical significance.
Three major groups of distinctly regulated mitogen-activated protein kinase (MAPK) cascades are known to lead to altered gene expression and to tissue proliferation in mammals, including extracellular signal-regulated kinase 1/2 (ERK1/ERK2), Jun N-terminal protein kinase (JNK), and p38 MAPK. Recent studies in our laboratory investigated the roles of ERK and p38 in OM. It was found that both ERK and p38 can be activated in OM and that inhibition of either the ERK or the p38 pathway can reduce ME mucosal hyperplasia in vitro (24, 25). Others have found that p38 inhibition reduces bacterially induced mucin gene expression in human ME epithelial cell lines (14). However, in our earlier studies, ERK activation was not well correlated temporally with ME mucosal hyperplasia in an animal model of OM since it occurred very early and very late in OM (24). p38 activation also occurred primarily very early in OM, with peak activation 1 to 6 h after bacterial inoculation (25). Moreover, even at saturating levels of the MAPK kinase (MKK)/ERK inhibitor U0126 or the p38 inhibitor SB203580, mucosal growth was still observed in vitro (24, 25). This suggests that other pathways are involved in regulating hyperplasia.
JNK was originally identified as two protein kinases, p46 and p54, which specifically phosphorylate the transcription factor c-Jun on its N-terminal transactivation domain at serines 63 and 73 (11). Molecular cloning of JNK demonstrated that it is a member of the MAPK group of signaling proteins (5). Ten JNK isoforms are created by alternative splicing of mRNA transcripts derived from three genes (JNK1, JNK2, and JNK3) (9). JNK1 and JNK2 are expressed in many cell types, while JNK3 has a more limited pattern of expression and is largely restricted to neuronal cells, the heart, and the testes (22). In parallel with ERK1/ERK2, JNK appears to be essential for activator protein 1 gene activation induced by stress and exposure to various cytokines (13). The JNK signaling pathway has been implicated in a large variety of pathological conditions, including inflammatory disorders, neurodegenerative diseases, and metabolic disease. Studies of various cancer cell lines have revealed high levels of JNK activity, suggesting that JNK can mediate tissue proliferation (27). In addition, JNK has also been shown to influence apoptosis (18). The complexity of the JNK pathway provides multiple opportunities for the design of small-molecule inhibitors that might modulate signaling. JNK inhibitors have shown promise in animal models for the treatment of rheumatoid arthritis (10). The pharmaceutical industry is bringing JNK inhibitors into clinical trials for autoimmune, anti-inflammatory, and neurodegenerative diseases (2).
The family of Rho GTPases consists of at least 21 members in mammalian cells (29). Rac and Cdc42, two well-characterized members of the family, are known upstream activators of JNK in various cells (7, 8, 30, 35). The mixed-lineage kinases (MLKs) are also often found upstream of JNK (38). Specific inhibitors of Rac, Cdc42, and MLKs have also been developed (4, 23).
For this report, we investigated the role of the JNK signaling pathway in rat MEs infected with nontypeable Haemophilus influenzae (NTHI). In order to determine whether JNK signaling is involved in ME mucosal hypertrophy, activation of JNK was evaluated in a rat in vivo model of bacterial OM. In addition, specific inhibitors of the JNK pathway were used in an in vitro rat model of bacterially induced mucosal proliferation. Finally, persistent administration of a JNK inhibitor into the ME mucosa with a mini-osmotic pump was evaluated in vivo during bacterial OM in guinea pigs.
MATERIALS AND METHODS
In vivo assessment of JNK phosphorylation.
All experiments were performed according to National Institutes of Health guidelines on the care and use of laboratory animals and were approved by the institutional committee for animal experimentation. Rats weighing 250 to 300 g were anesthetized with a mixture of ketamine and xylazine at 100 mg/ml and acepromazine at 10 mg/ml, injected intramuscularly at a dose of 0.4 ml/100 g of body weight. Upon sedation, the animals were placed in a supine position, and a 3-cm vertical midline incision was made between each rat's mandible and clavicles. Middle-ear bulla exposure was obtained bilaterally with the aide of a dissecting microscope, and a 25-gauge syringe needle was used to fenestrate the center of the bulla bilaterally. The bullae of 24 male Sprague-Dawley rats were injected with 105 CFU/ml NTHI (strain 3655, biotype II) until the solution overflowed the fenestrations, a volume of about 50 μl. The original fascia covering the bulla was then used to recover the hole in the bone, and the incisions were stapled closed. Each animal was examined to guarantee that the tympanic membranes had not been ruptured during the injections. The animals were anesthetized and sacrificed, and the ME mucosae were dissected bilaterally from two to five rats at one of the following seven time points: 1, 6, 24, 48, and 72 h as well as 5 and 7 days after infection. The ME mucosae from three untreated control rats were also dissected bilaterally. Before Western blotting, the extracted ME mucosae were placed in radioimmunoprecipitation buffer and examined under a dissecting microscope. At 1 h, the ME mucosae were equivalent in thickness to the control specimens. At 6 h, the ME mucosae were somewhat thicker than control specimens, primarily due to edema. At 24 h and 48 h, the ME mucosae were hypertrophied and tolerated manipulation better than the control specimens. At 72 h, the ME mucosae were fragile, and it was more difficult to obtain intact specimens. By day 5, the thickness of the mucosae was reduced, and the fragility of the specimens decreased. On day 7, ME mucosal thickness was further recovered, and the tissue tolerated manipulation better than 5-day specimens did. The ME mucosae were immediately frozen at −70°C. The frozen tissues were homogenized in 50 μl of radioimmunoprecipitation buffer that contained 20 mM Tris (pH 7.4), 1 mM EDTA, 140 mM NaCl, 1% NP-40, 1 mM orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 50 mM sodium fluoride, and 10 μg/ml aprotinin, using a Potter-Elvhejlm homogenizer. The homogenates were centrifuged at 14,000 × g at 4°C, and the pellets were discarded. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). The samples were boiled for 5 min, and total protein extracts (10 μg per lane) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The separated proteins were transferred electrophoretically onto a polyvinylidene difluoride Western blotting membrane, and the membrane was then blocked with 5% dried nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature. The blot was incubated with a mouse monoclonal antibody (MAb) to phosphorylated JNK (pJNK; phosphorylated on Thr-183 and Tyr-185) (code G-7 [1:400]; Santa Cruz Biotechnology, Santa Cruz, CA) in TBS-T containing 5% dried nonfat milk. According to the manufacturer, the sensitivity to pJNK1 versus pJNK2 has not been quantified using this MAb. The membrane was washed three times with TBS-T and then incubated for 1 h with a goat anti-mouse horseradish peroxidase-conjugated secondary Ab (1:1,000) in TBS-T containing 5% nonfat dried milk. The membrane was then washed as before and visualized by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL). The blot was stripped and reprobed with a rabbit polyclonal Ab to total JNK (code FL; Santa Cruz Biotechnology, Santa Cruz, CA) (diluted 1:400). Densitometric analyses were performed using Scion imaging software.
Immunohistochemistry.
The localization of pJNK on the ME mucosa was detected immunohistochemically with a mouse MAb to pJNK (code G-7; Santa Cruz Biotechnology) (1:100). The ME mucosae were dissected bilaterally 48 h after the inoculation of NTHI, fixed with 4% paraformaldehyde in phosphate buffer for 30 min, embedded in an optimal cutting temperature compound, sectioned with a cryostat (7 μm), and stained with a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). All specimens were lightly counterstained with methyl green.
Tissue culture.
To evaluate ME mucosal signal transduction in vitro, we used our established model of bacterially induced mucosal proliferation (24, 25). In brief, the bullae of male Sprague-Dawley rats weighing 250 to 300 g were injected with NTHI in the manner described above. After 48 h, animals were decapitated, and the middle-ear bullae were rinsed to wash out effusion with warm phosphate-buffered saline (PBS). Each ME mucosa sample was immediately placed in a separate 60-mm Falcon petri dish coated with a thin layer of Sylgard 184 silicone elastomer. Culture medium, consisting of a mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium (3:1) supplemented with fetal calf serum (5%), hydrocortisone (0.4 μg/ml), isoproterenol (10−6 M), penicillin (100 U/ml), and streptomycin (100 μg/ml), was then added. The ME mucosae were divided into 1-mm2 tissue explants, using a Fine Science Tools diamond knife. The explants from each bullae were then individually transplanted, with the epithelium uppermost, into single wells of a 24-well Falcon cell culture plate containing 170 μl of culture medium. They were placed in an incubator at 37°C with 5% CO2 for 24 h and allowed to adhere to the culture plate surface. After 24 h, 300 μl of culture medium was added to each well, and the culture medium was changed in all wells with healthy, attached explants every day. Only explants that maintained a healthy appearance and remained firmly attached to the well surface throughout the entire duration of the study were used.
Inhibition of bacterially exposed mucosal explants with Clostridium difficile toxin B, CEP11004, and SP600125.
The bullae of six male Sprague-Dawley rats weighing 250 to 300 g and previously injected with NTHI were dissected, divided, and cultured in the manner described above. On day 1, all wells with healthy, attached explants were randomly divided into four groups. C. difficile toxin B (EMD Biosciences, La Jolla, CA) (50% inhibitory concentration [IC50], <1 nM) was added at 0 ng/ml, 0.1 ng/ml (0.37 pM), 1 ng/ml (3.7 pM), or 10 ng/ml (37 pM) in 300 μl of culture medium. Every day, all of the medium from each well was removed, and 300 μl of fresh culture medium was added with the appropriate concentration of C. difficile toxin B. All explants were maintained in culture for 10 days. Using the same procedures, explants were cultured with the JNK inhibitor CEP11004 (Cephalon, West Chester, PA) (IC50 = 150 to 300 nM) at 0, 10, 100, or 1,000 nM or the JNK inhibitor SP600125 (EMD Biosciences, La Jolla, CA) (IC50 = 0.1 to 10 μM, depending on cell type) at 0, 0.2, 2, or 20 μM. The first group served as a negative control, with the medium receiving a supplement of dimethyl sulfoxide (DMSO) alone at 1 μl/ml, the same concentration of DMSO used for all concentrations of CEP11004 and SP600125. Separate control groups were used for each inhibitor so that tissues from the same rats were included under all conditions to control for variation in responses to different inocula. For each individual inhibitor concentration, six to eight explants were photographed daily for 10 days with a room temperature (RT) digital color camera, and their surface areas were calculated using SPOT computer software calibrated to the appropriate magnification.
Cell viability was assessed in separate cultures by the trypan blue exclusion assay, with and without the highest dose of C. difficile toxin B, CEP11004, or SP600125 used. Briefly, after cells were collected by trypsinization (0.05% trypsin, 0.02% EDTA) from three cultures for each inhibitor, aliquots of the cell suspension (10 μl) were mixed with 0.4% trypan blue solution (10 μl) and placed on a hemocytometer. Data were compared using one-way analysis of variance, followed by Tukey-Kramer's t test post hoc to determine statistical differences. A P value of <0.05 was considered significant. Statview 5.0 was used for statistical analysis.
Inhibition of uninfected mucosal explants with C. difficile toxin B, CEP11004, and SP600125.
The bullae of 12 male Sprague-Dawley rats weighing 250 to 300 g and previously injected with PBS were dissected, divided, and cultured in the manner described above. The explants were divided into four groups. C. difficile toxin B was added at 0 ng/ml, 0.1 ng/ml (0.37 pM), 1 ng/ml (3.7 pM), or 10 ng/ml (37 pM) in 300 μl of culture medium. Using the same procedures, explants were cultured with the JNK inhibitor CEP11004 at 0, 10, 100, or 1,000 nM or the JNK inhibitor SP600125 (EMD Biosciences, La Jolla, CA) at 0, 0.2, 2, or 20 μM. The first group served as a negative control, with the medium receiving a supplement of DMSO alone at 1 μl/ml, the same concentration of DMSO used for all concentrations of CEP11004 and SP600125. For each individual inhibitor, 8 to 10 explants were evaluated. Cell viability was assessed in separate cultures by the trypan blue exclusion assay, with and without the highest dose of C. difficile toxin B, CEP11004, or SP600125 used, as described above.
Animal surgery.
Because of its surgical orientation, the ME of the rat is poorly suited to the application of biomaterials to the subepithelial compartment. For this reason, we employed the guinea pig, in which surgical access is feasible. Male albino Hartley guinea pigs weighing 300 to 350 g were anesthetized with a combination of ketamine (40 mg/kg of body weight), xylazine (10 mg/kg), and acepromazine (0.75 mg/kg) given intramuscularly. Upon sedation, the left bulla was exposed by retroauricular incision, and a small opening was drilled, with care taken not to perforate the ME mucosa. A fine-tipped microcatheter was made as previously described (28). The microcatheter consisted of three components, as follows. Ten millimeters of polymide tubing (inner diameter [i.d.] = 0.14 mm; outer diameter [o.d.] = 0.20 mm) (Microlumen, Tampa, FL) was inserted into the end of a 50-mm length of Silastic tubing (i.d. = 0.30 mm; o.d. = 0.64 mm) (Dow Corning, Midland, MI). The Silastic tube was connected to a 50-mm length of larger Silastic tubing (i.d. = 0.64 mm; o.d. = 1.19 mm) (Dow Corning, Midland, MI). The larger Silastic tube was connected to a mini-osmotic pump (200-μl capacity; 1.0 μl/h) (model 2001; DURECT Corporation, Cupertino, CA) during implantation. The microcatheter was filled with the same agent as the osmotic pump in all cases. The concentration of SP600125 was 1 mM. Control animals received only vehicle (PBS containing 5% DMSO). Prior to implantation, the osmotic pump was placed in sterile PBS at 37°C for 4 h, allowing it to be operable immediately upon implantation. The microcatheter was then inserted into the opening in the bulla under the ME mucosa and advanced 1 to 2 mm from the opening. Subepithelial delivery was chosen because we have found that penetration of reagents through the mucosal epithelial surface can be restricted. The microcatheter was secured to the surrounding bone of the bulla. The osmotic pump was placed in the subcutaneous pocket of the back. The incision was then closed. A 28 1/2-gauge syringe needle was then used to inject 105 CFU/ml NTHI (100 μl) through the left tympanic membrane. After the animals were sacrificed, the osmotic pumps were collected and the residual volume assessed to ensure that normal agent delivery had occurred.
Histology.
Animals were sacrificed 5 days after surgery. Following decapitation under deep anesthesia with a combination of ketamine (40 mg/kg of body weight), xylazine (10 mg/kg), and acepromazine (0.75 mg/kg) intramuscularly, the temporal bones were immediately removed and the bullae were perfused with 4% paraformaldehyde in phosphate buffer for 6 h. The bullae were decalcified in 8% EDTA for 2 weeks, dehydrated in graded ethanol, and embedded in paraffin. Sections (7 μm) were cut, stained with hematoxylin and eosin, and evaluated under a light microscope. The ME mucosa was examined, and any temporal bones in which the microcatheter was not in its proper placement in the subepithelial compartment were discarded. Each specimen was photographed with an RT digital color camera. For both the SP600125 and vehicle groups, seven ME mucosae were evaluated. Mucosal thickness was calculated at the end of the microcatheter and at a location approximately 500 μm from the catheter, using SPOT computer software calibrated to the appropriate magnification. The thickness data from the two locations were compared using the Wilcoxon signed rank test. A P value of <0.05 was considered significant. Statview 5.0 was used for statistical analysis.
Detection of apoptosis.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) was performed with a TACS TdT DAB kit (Trevigen, Gaithersburg, MD), following the manufacturer's instructions. Three guinea pigs were sacrificed 72 h after surgery and bacterial inoculation. Following decapitation under deep anesthesia with a combination of ketamine (40 mg/kg of body weight), xylazine (10 mg/kg), and acepromazine (0.75 mg/kg) intramuscularly, the temporal bones were immediately removed and the bullae were perfused with 4% paraformaldehyde in phosphate buffer for 1 h. The bullae were decalcified in 8% EDTA for 2 weeks, embedded in an optimal cutting temperature compound, and sectioned with a cryostat (7 μm). Sections were digested with proteinase K at a concentration of 20 μg/ml for 15 min. Endogenous peroxidase activity was quenched with 3% H2O2 for 5 min. The slides were immersed in terminal deoxynucleotidyltransferase (TdT) buffer. TdT, 1 mM Mn2+, and biotinylated deoxynucleoside triphosphates in TdT buffer were added to cover the sections and incubated in a humidity chamber at 37°C for 60 min. The slides were washed with PBS and incubated with streptavidin-horseradish peroxidase for 10 min. After being rinsed with PBS, the slides were immersed in DAB solution for 2 min. All specimens were lightly counterstained with methyl green. The ME mucosa was examined, and any temporal bones in which the microcatheter was not in its proper placement in the subepithelial compartment were discarded. Each specimen was photographed with an RT digital color camera. The total number of cells (TUNEL-positive cells plus unstained cells) was counted from a standard area (0.01 mm2) in the vicinity of the end of the microcatheter (catheter area), a standard area at a location approximately 100 μm from the catheter, a standard area at a location approximate 200 μm from the catheter, and a standard area at a location approximate 500 μm from the catheter, using SPOT computer software calibrated to the appropriate magnification. The ratio of TUNEL-positive cells to the total number of cells counted was determined, and this ratio was multiplied by 100. This was defined as the percentage of TUNEL-positive cells. The percentages of TUNEL-positive cells in the six locations were compared using one-way analysis of variance followed by Tukey-Kramer's t test post hoc to determine statistical differences. A P value of <0.05 was considered significant. Statview 5.0 was used for statistical analysis.
PCNA staining.
A Zymed PCNA staining kit (Zymed Laboratories, South San Francisco, CA) was used as a marker for cell proliferation, because proliferating cell nuclear antigen (PCNA) levels are elevated in actively proliferating cells during the S, G2, and M phases of cell mitosis. Tissue preparation was performed as outlined above for TUNEL, and staining and visualization were done according to the manufacturer's instructions. Briefly, the slides were processed through 3% H2O2 for 10 min to quench endogenous peroxidase activity. Sections were then blocked using the kit blocking solution and incubated with biotinylated mouse anti-PCNA Ab for 60 min at room temperature, followed by streptavidin-peroxidase incubation for 10 min and incubation for 2 min with the DAB chromogen solution. All specimens were lightly counterstained with methyl green. The percentage of PCNA-labeled cells was determined using the same methodology as that for the percentage of TUNEL-positive cells.
Cytotoxicity assay.
To assess the level of cytotoxicity of the dosage of SP600125 used in the mini-osmotic pump, the bullae of six male albino Hartley guinea pigs weighing 300 to 350 g were dissected, divided, and cultured in the manner described above. The explants were cultured with the JNK inhibitor SP600125 (1 mM) or with vehicle only (medium containing 5% DMSO). All explants were maintained in culture for 3 days. The explants were then exposed for 2 h to a 0.1% protease solution dissolved in PBS. Enzymatically dissociated cells were harvested, centrifuged at 1,000 × g for 5 min, and resuspended in Dulbecco's modified Eagle's medium. Aliquots of cell suspension (10 μl) were mixed with 0.4% trypan blue solution (10 μl) and placed on a hemocytometer. Data were compared using the Wilcoxon signed rank test. A P value of <0.05 was considered significant. Statview 5.0 was used for statistical analysis.
RESULTS
Levels of total JNK and pJNK.
Figure 1A illustrates the presence of total JNK and pJNK in the control ME mucosae and in the infected ME mucosae at various time points, as assessed by Western blotting. A pJNK signal was not detected in the control ME mucosae. pJNK1 (molecular mass, 46 kDa) was first observed 6 h after bacterial inoculation, was maximal between 24 h and 72 h, and returned to undetectable levels by 7 days. pJNK2 (molecular mass, 54 kDa) was first observed at 1 h and was also maximal from 24 h to 72 h after bacterial inoculation. pJNK2 decreased at 5 to 7 days but remained well above the control level. Figure 1B shows band density ratios of pJNK to total JNK. The ratio of pJNK1 to total JNK1 showed a high degree of phosphorylation at 6 h, a maximum between 24 h and 72 h, and a decline from 5 to 7 days. The ratio of pJNK2 to total JNK2 was maximal between 6 h and 48 h and moderate from 72 h to 7 days.
FIG. 1.

Bacterial OM activates the JNK signaling pathway in rat ME mucosa. Western blotting (A) shows the effects of bacterial inoculation on total JNK and pJNK. Pools of multiple samples were equalized for total protein at various postinfection times. pJNK2 was slightly induced at 1 h, whereas pJNK1 was induced 6 h after the bacterial inoculation. Both isoforms peaked at 24 to 72 h. Lane C, control. (B) Band density ratios of pJNK1 to total JNK1 and pJNK2 to total JNK2 for the blot shown in panel A. The ratio of pJNK1 to total JNK1 showed strong phosphorylation at 6 h, maximal phosphorylation between 24 h and 72 h, and gradually weaker phosphorylation from days 5 to 7. The ratio of pJNK2 to total JNK2 showed much higher levels of phosphorylation, with maximal activation between 6 h and 48 h and less activation between 72 h and 7 days. (C) Representative photograph of immunostaining for pJNK showing no immunolabeling in a control ME mucosa. (D) Immunolabeling of nuclei (inset) was seen in many mucosal epithelial and stromal cells 48 h after bacterial inoculation, at the peak of JNK activation. Bar = 10 μm.
Immunohistochemistry.
Immunohistochemical analysis at the peak of phosphorylation revealed the presence of extensive pJNK in rat ME mucosae 48 h after bacterial inoculation. Immunoreactivity to pJNK was observed in the nuclei of both stromal and epithelial cells (Fig. 1D). No immunostaining was observed when the primary Ab was omitted (data not shown).
Inhibition of bacterially induced ME mucosal hyperplasia by C. difficile toxin B, CEP11004, or SP600125.
Figure 2 shows the effects of C. difficile toxin B, CEP11004, or SP600125 on the surface area of cultured ME mucosal explants from bacterially inoculated rats. We have previously shown that ME mucosal explants from the MEs of rats infected 48 h before dissection with NTHI display permanently enhanced tissue proliferation (see Fig. 1 in reference 24), which can be used as a model for bacterial enhancement of ME mucosal growth (24, 25). All explants maintained a healthy appearance and remained firmly attached to the plate surface throughout the duration of the study. C. difficile toxin B (10 ng/ml [37 pM]) significantly inhibited the growth of mucosal epithelial cells (P < 0.05), decreasing the surface area by 53.8%. C. difficile toxin B treatment at 0.1 ng/ml (0.37 pM) or 1 ng/ml (3.7 pM) showed no effect on explant outgrowth. Similarly, 1,000 nM CEP11004 significantly inhibited explant outgrowth (P < 0.05), decreasing the surface area by 56.4%. No inhibition was observed for CEP11004 at 10 or 100 nM. Both 2 μM and 20 μM SP600125 significantly inhibited explant outgrowth (P < 0.05), decreasing the surface area by 34.6% and 92.2%, respectively. No inhibition was observed for SP600125 at 0.2 μM. Figure 3 shows the typical microscopic appearances of treated and untreated mucosal explants from infected rat ME epithelia on day 7 of culture. As determined with trypan blue daily between days 4 and 10, the viability of individual cells from trypsinized and dissociated explants, with and without the highest dose of C. difficile toxin B, CEP11004, or SP600125, was >95% for all cultures (n = 3/group). There were no statistical differences between control explants and those treated with any inhibitor.
FIG. 2.
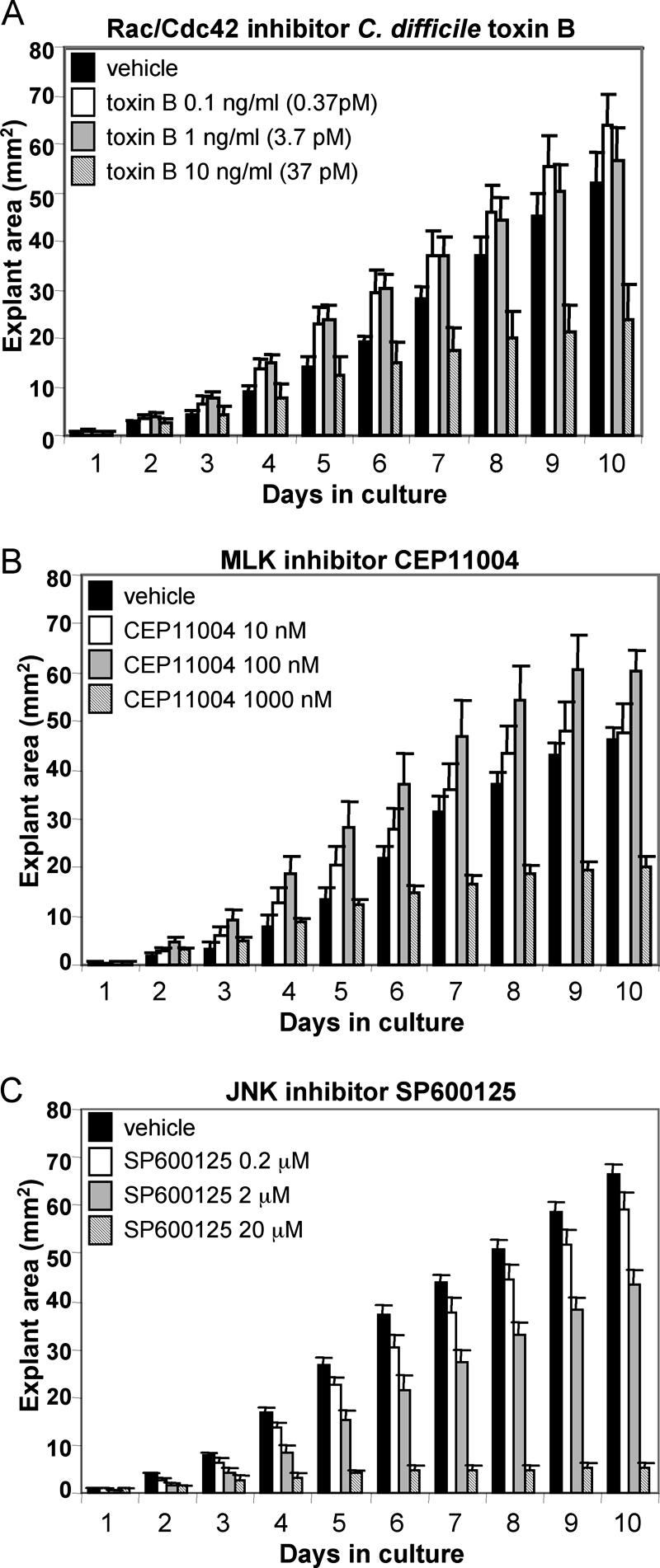
Surface area of mucosal outgrowth of hyperplastic explants from infected rat MEs. Mucosal explants (all harvested 48 h after bacterial inoculation) were cultured in the presence of various concentrations of C. difficile toxin B (0, 0.1, 1, or 10 ng/ml), CEP11004 (0, 10, 100, or 1,000 nM), or SP600125 (0, 0.2, 2, or 20 μM). The first group for CEP11004 or SP600125 served as a negative control, with the medium receiving a supplement of DMSO alone at 1 μl/ml, the same concentration of DMSO used for all concentrations of CEP11004 and SP600125. (A) With C. difficile toxin B, significant inhibition of growth was observed at 10 ng/ml (P < 0.05). (B) With CEP11004, significant inhibition of growth was observed at 1,000 nM (P < 0.05). (C) With SP600125, significant inhibition of growth was observed at 2 μM and 20 μM (P < 0.05). Values are means ± standard errors (SE) (n = 6 to 10 at each concentration).
FIG. 3.

Representative photographs of mucosal outgrowth of hyperplastic explants from infected rat MEs on day 7 of culture. Explants were maintained in normal culture medium alone (A) or with either 10 ng/ml C. difficile toxin B (B), 1,000 nM CEP11004 (C), or 2 μM SP600125 (D) for 7 days. An untreated mucosal explant from an uninfected ME is shown in panel E. Bar = 1 mm.
Inhibition of uninfected ME mucosae by C. difficile toxin B, CEP11004, or SP600125.
The growth of uninfected ME mucosae was also affected by JNK pathway inhibition, although to a lesser extent than that of infected mucosae. C. difficile toxin B at 10 ng/ml significantly inhibited the growth of explant cultures (P < 0.05), decreasing the surface area by 60.4%, compared with that of control specimens. As with infected mucosal explants, C. difficile toxin B treatment at 0.1 ng/ml (0.37 pM) or 1 ng/ml (3.7 pM) did not inhibit explant growth. CEP11004 at 1,000 nM significantly inhibited explant outgrowth (P < 0.05), decreasing the surface area by 73.0%. No inhibition was observed at 10 or 100 nM. In contrast to the case for infected mucosal explants, only 20 μM SP600125 significantly inhibited explant outgrowth (P < 0.05), decreasing the surface area by 56.4%. No inhibition was observed for SP600125 at 0.2 μM or 2 μM. As determined with trypan blue daily between days 4 and 10, the viability of cells from trypsinized explants, with and without the highest dose of C. difficile toxin B, CEP11004, or SP600125, was >95% (n = 3).
Inhibition of bacterially infected ME mucosae by SP600125 in vivo.
Figure 4 shows the typical microscopic appearances of the healthy guinea pig ME mucosa, bacterially inoculated ME mucosa, and bacterially inoculated ME mucosa treated with vehicle or SP600125 in vivo. Inhibition of mucosal hyperplasia by SP600125 was observed in a restricted region around the microcatheter end. Figure 5 shows a quantitative analysis of the effects of subepithelial delivery of vehicle or SP600125 on mucosal hyperplasia in bacterially inoculated guinea pig MEs in vivo. Continuous delivery of SP600125 (1 mM) to the subepithelial compartment locally inhibited epithelial (P < 0.017) and stromal (P < 0.018) hyperplasia, decreasing the thickness by 19.4% and 18.0%, respectively. In contrast, delivery of vehicle had no effect on mucosal hyperplasia.
FIG. 4.

Representative photographs of healthy mucosa of a guinea pig ME (A), mucosal hyperplasia in a bacterially inoculated ME (B), and mucosal hyperplasia influenced by local application of vehicle (PBS containing 5% DMSO) (C) or the JNK inhibitor SP600125 (D). For panel D, a mini-osmotic pump delivered 1 mM SP600125 through a microcatheter inserted into the ME mucosa at 1 μl/h for 5 days. Reduced hyperplasia of the ME mucosa is observed in the vicinity of the microcatheter end. On the other hand, vehicle alone showed no inhibitory effect on mucosal hyperplasia in the vicinity of the microcatheter end (C). The slides were stained with hematoxylin and eosin. Bar = 100 μm.
FIG. 5.
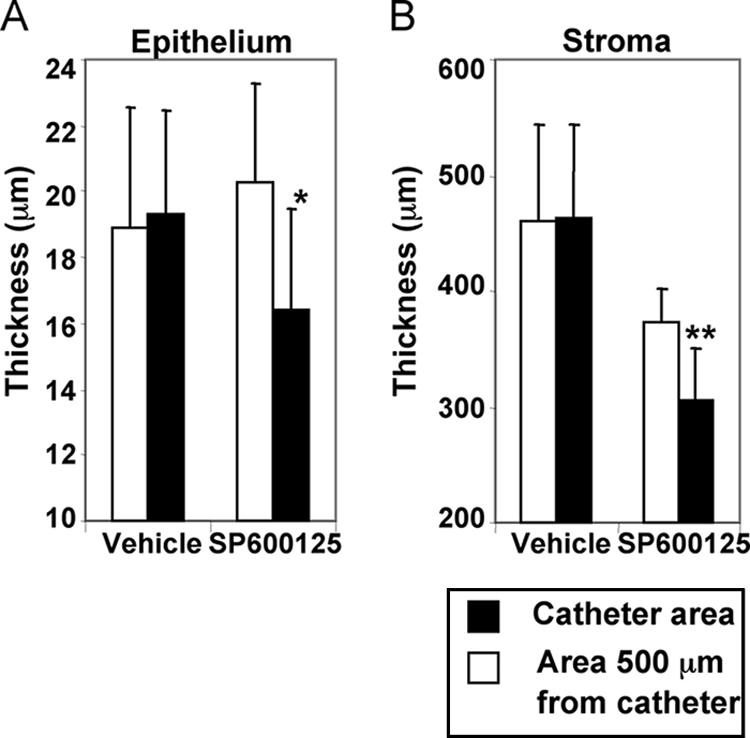
Average thickness of mucosae in bacterially inoculated guinea pig MEs in vivo. With focal delivery of SP600125, significant local inhibition of both epithelial (A) (*, P < 0.017) and stromal (B) (**, P < 0.018) thickness was observed at 1 mM compared with that in an area 500 μm from the catheter. On the other hand, vehicle (PBS containing 5% DMSO) showed no inhibitory effect on mucosal hyperplasia in the bacterially inoculated guinea pig in vivo. The mean thickness of four healthy mucosal epithelia was 9.95 ± 0.42 μm, and the outer diameter of the microcatheter was 200 μm in our study. Therefore, the bars in the graphs start from 10 μm for epithelium and 200 μm for stroma. Values are means ± SE (n = 7).
Inhibition of cell proliferation and increase in apoptosis by SP600125 in vivo.
Figure 6 shows the typical microscopic appearance of TUNEL staining and PCNA immunohistochemistry in bacterially inoculated ME mucosae treated with SP600125 or vehicle in vivo. An increase in TUNEL-positive cells and a decrease in PCNA immunoreactivity by SP600125 were observed in a restricted region around the microcatheter end. Decreased cell proliferation was also observed in a region 0 to 300 μm adjacent to the catheter. Apoptosis was increased only very near the catheter and involved primarily polymorphonuclear cells. In contrast, delivery of vehicle had no effect on TUNEL staining or PCNA immunoreactivity. Figure 7 shows a quantitative analysis of the effects of subepithelial delivery of SP600125 on mucosal hyperplasia in bacterially inoculated guinea pig MEs in vivo. Continuous delivery of SP600125 (1 mM) to the subepithelial compartment locally increased the percentage of TUNEL-positive cells only in the immediate catheter area, not in areas 100 μm, 200 μm, and 500 μm from the catheter (P < 0.05). In contrast, continuous delivery of SP600125 (1 mM) to the subepithelial compartment locally decreased the percentage of PCNA-labeled cells in the catheter area, and also at 100 μm and 200 μm, compared with that 500 μm from the catheter (P < 0.05). There was no significant difference in either variable at any location when vehicle alone was delivered (data not shown).
FIG. 6.

Representative photomicrographs of mucosal hyperplasia of a guinea pig ME, as influenced by local application of SP600125. A mini-osmotic pump delivered 1 mM SP600125 or vehicle (PBS containing 5% DMSO) through a microcatheter inserted into the ME mucosa at 1 μl/h for 3 days. TUNEL-positive cells were increased only in the vicinity of the microcatheter end compared with the nearby ME epithelium (A). These cells were primarily polymorphonuclear in morphology. On the other hand, PCNA immunoreactivity was reduced in a wide area surrounding the catheter end (B). Vehicle alone showed no effect on either TUNEL staining (C) or PCNA immunoreactivity (D). The sections of the catheter were dislocated (B) or lost (C and D) during the process of TUNEL staining or immunohistochemistry. Bar = 100 μm.
FIG. 7.
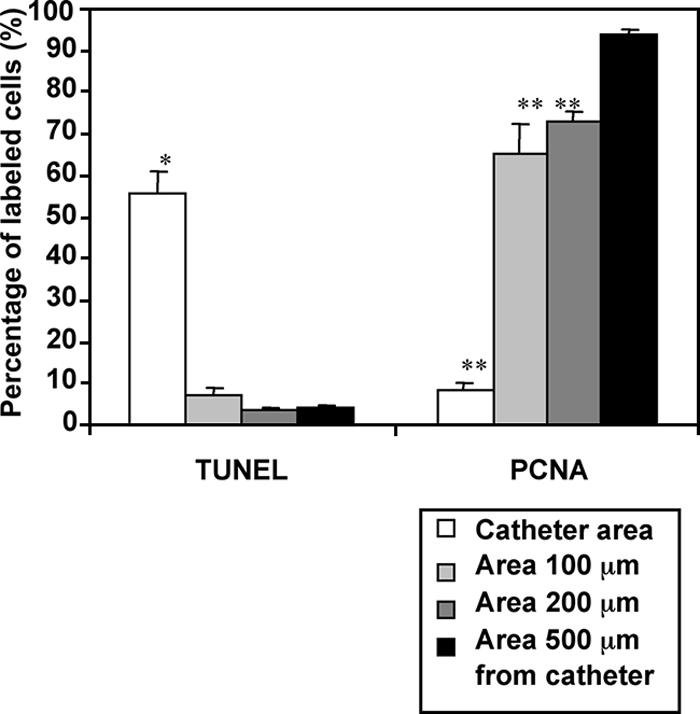
Percentages of TUNEL-positive and PCNA-labeled cells in bacterially inoculated guinea pig MEs in vivo. With focal delivery of SP600125, a significant local increase in the percentage of TUNEL-positive cells was observed compared with that in an area 500 μm from the catheter (*, P < 0.05). In contrast, there was no significant difference between the areas 100 μm, 200 μm, and 500 μm from the catheter. On the other hand, the percentages of PCNA-labeled cells were decreased locally in the catheter area and in areas 100 μm and 200 μm away compared with that in the area 500 μm from the catheter (**, P < 0.05). Values are means ± SE. Cells were counted from six locations in three different guinea pigs.
Cytotoxicity assay for 1 mM SP600125.
We observed no evidence of mucosal cell cytotoxicity by 1 mM SP600125 in the region at the end of the microcatheter supplied by the mini-osmotic pump. In mucosal explants, the percentage of viable cells detected by trypan blue exclusion 72 h after 1 mM SP600125 treatment was 89.6% ± 2.1% (n = 8). In the explants maintained in medium alone, the percentage of viable cells was 88.8% ± 1.8% (n = 8).
DISCUSSION
OM is a common disease and the leading reason for prescribing antibiotics during childhood (26). Mucosal hyperplasia is a major feature of OM (17) and can contribute to its symptoms and to irreversible ME disease (32, 36). It is therefore of interest to determine how mucosal hyperplasia is regulated in the ME. To our knowledge, this study is the first to provide evidence that ME mucosal hyperplasia involves signaling by the JNK MAPK cascade.
Our Western blotting and cytochemical data indicate that inactive JNK is present in the healthy ME mucosa and that JNK activation occurs during OM. Moreover, the time course of JNK activation is well related to the kinetics of mucosal hyperplasia. As previously reported, the rat ME mucosa is maximally thickened by 4 days after NTHI inoculation, and it recovers substantially by day 8 (19). In our data, pJNK was induced prior to ME mucosa growth, but with a roughly parallel time course. This suggests that JNK phosphorylation may promote mucosal hyperplasia induced by bacteria in the ME. We have previously reported that ME mucosae harvested 48 h after bacterial inoculation show a higher rate of growth in culture than explants harvested at earlier or later time points (24). It can be speculated that this may be related to the peak of JNK activation that occurs in vivo at 48 h.
The presence of active JNK, even when correlated with hyperplasia, is not sufficient to indicate a causative role. Altering hyperplasia by interfering with JNK regulation in vitro provides more compelling evidence. As noted above, the JNK pathway is complex, providing multiple opportunities for intervention. Upstream activation of JNK occurs via MKK4 and MKK7. JNK is phosphorylated preferentially on Tyr185 by MKK4 and on Thr183 by MKK7 (15). MKKs are activated in turn by MKK kinases, including MAP/ERK kinase kinases 1 to 4 (MEKK1-4), by MLKs, including MLK1-3, by leucine-zipper-bearing kinase, by apoptosis signal-regulated kinase 1, and by transforming growth factor beta-activated kinase 1 (7). Activated MLKs phosphorylate and activate MKK4 and MKK7 (40). Upstream of the MKK kinase enzymes is another layer of JNK pathway protein kinases, such as the family of Rho GTPases, in particular Rac and Cdc42 (7, 8, 30, 35). Even further upstream, lipopolysaccharide, a component of the outer membranes of gram-negative bacteria such as NTHI, can stimulate the activation of JNK via Toll-like receptor 4 (1, 20). Specific inhibitors allowed us to intervene in three locations in the JNK phosphorylation cascade in vitro. Inhibition of Rac/Cdc42, MLKs, and JNK itself all reduced in vitro ME mucosal growth, implicating the activation of JNK via Rac/Cdc42 and MLKs in ME mucosal hyperplasia during OM.
The use of the in vitro model of mucosal growth allowed dissection of the JNK pathway with multiple inhibitors and dosages that would not be practical in vivo. Of course, it can be argued, as with any culture system, that in vitro responses might not be comparable to those for in vivo OM. However, inhibition of JNK by SP600125 also reduced mucosal thickness associated with OM in vivo. The reduction of mucosal hyperplasia in vivo therefore validates and extends our in vitro observations. Future studies will be required to determine the bacterial factors involved, perhaps using heat-killed bacteria or strains lacking particular virulence factors.
There are a variety of mechanisms by which JNK activation might influence mucosal growth in OM. The most likely possibilities would appear to be increased cell proliferation and decreased apoptosis, since JNK has been shown to influence both processes in other systems (18, 33). Our TUNEL staining and PCNA immunohistochemistry data following in vivo JNK inhibition (Fig. 6 and 7) implicate proliferation as opposed to apoptosis. The morphology of the mucosal cells in the region of reduced mucosal thickness also supported an effect of JNK on proliferation rather than cell death, since morphological signs of necrosis or apoptosis were not observed. These findings suggest that JNK activation in the proliferating ME mucosa exerts an important role in cell cycle progression of ME cells during bacterial OM. The relationship between JNK and cell proliferation is complex (e.g., see reference 16). However, fibroblasts lacking JNK show upregulation of p53 and p21 and, consequently, impaired cyclin D1- and E-associated kinase activities (31). Additional studies would be required to determine if a similar mechanism is involved in JNK cascade inhibition in ME mucosal cells.
The response of cultured ME mucosal explants to JNK cascade inhibitors influenced the growth of mucosal explants from both healthy and infected MEs (Fig. 2). The sensitivity of bacterially exposed explants to the JNK inhibitor SP600125 was higher than that of uninfected mucosal explants, indicating a greater role for JNK in bacterially stimulated growth. However, JNK activation via Rac/Cdc42 appears to be part of a final common pathway for mucosal growth, occurring in both bacterially induced growth and in growth stimulated by tissue culture.
While JNK activation appears to play a critical role in mucosal hyperplasia, mucosal growth was not completely suppressed by JNK inhibition in vivo. Since in vitro growth was also not completely suppressed by inhibition of the JNK cascade, this suggests that JNK-independent mechanisms contribute to mucosal hyperplasia during OM. Our previous studies implicated the ERK and p38 MAPK pathways in both the initiation and the resolution of bacterially induced ME mucosal hyperplasia (24, 25). Activated ERK1 and ERK2 were significantly elevated 1 to 6 h after bacterial inoculation. ERK phosphorylation was not significantly elevated 24 to 72 h after inoculation. However, high levels of ERK phosphorylation reoccurred on days 6 and 7, when OM was resolving. Activated p38 was also elevated 1 to 6 h after bacterial inoculation (25). Much lower levels of activated p38 were observed at 24 to 48 h. Thus, while the present study supports a role for JNK in the control of bacterially induced mucosal hyperplasia, ERK phosphorylation may be an early initiating event as well as one of the regulatory pathways involved in returning the ME mucosa to a resting state after the resolution of infection. p38 phosphorylation may also preferentially influence the early stages of bacterially induced mucosal hyperplasia. Of course, MAPK-independent pathways may also be involved.
In conjunction with recent studies, our results therefore suggest that mucosal hyperplasia in the ME epithelium depends not upon the activation of any one intracellular signaling pathway but on the combined actions of several. This is consistent with the case for endothelial cells, for which it has been found that ERK and big MAPK-1 are both involved in growth and cytoprotective functions (37, 41). JNK and p38 proteins play an important role in inflammatory stress responses and can also participate together in tissue proliferation (12, 34). p38 is involved in mucus production in ME cell lines (14). Inhibition of JNK activity may therefore be a potential approach to reduce the numerous adverse long-term sequelae of OM, including irreversible ME disease. However, given the involvement of other signaling pathways in OM, it is likely that a combination of inhibitors would be required to comprehensively reduce mucosal hyperplasia.
Acknowledgments
We thank Sharon Okamoto for her technical assistance. CEP11004 was generously supplied by Cephalon.
This work was supported by National Institutes of Health grant DC00129 and by the Research Service of Veterans Affairs.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 26 February 2007.
REFERENCES
- 1.Arndt, P. G., N. Suzuki, N. J. Avdi, K. C. Malcolm, and G. S. Worthen. 2004. Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-kinase and Syk-mediated pathways. J. Biol. Chem. 279:10883-10891. [DOI] [PubMed] [Google Scholar]
- 2.Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning, and D. W. Anderson. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 98:13681-13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett, K. E., M. P. Haggard, P. A. Silva, and I. A. Stewart. 2001. Behaviour and developmental effects of otitis media with effusion into the teens. Arch. Dis. Child. 85:91-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busch, C., and K. Aktories. 2000. Microbial toxins and the glycosylation of rho family GTPases. Curr. Opin. Struct. Biol. 10:528-535. [DOI] [PubMed] [Google Scholar]
- 5.Davis, R. J. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239-252. [DOI] [PubMed] [Google Scholar]
- 6.El-Sayed, Y. 1998. Bone conduction impairment in uncomplicated chronic suppurative otitis media. Am. J. Otolaryngol. 19:149-153. [DOI] [PubMed] [Google Scholar]
- 7.Fanger, G. R., N. L. Johnson, and G. L. Johnson. 1997. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 16:4961-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerwins, P., J. L. Blank, and G. L. Johnson. 1997. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J. Biol. Chem. 272:8288-8295. [DOI] [PubMed] [Google Scholar]
- 9.Gupta, S., T. Barrett, A. J. Whitmarsh, J. Cavanagh, H. K. Sluss, B. Derijard, and R. J. Davis. 1996. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15:2760-2770. [PMC free article] [PubMed] [Google Scholar]
- 10.Han, Z., D. L. Boyle, L. Chang, B. Bennet, M. Karin, L. Yang, A. M. Manning, and G. S. Firestein. 2001. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Investig. 108:73-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibi, M., A. Lin, T. Smeal, A. Minden, and M. Karin. 1993. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 7:2135-2148. [DOI] [PubMed] [Google Scholar]
- 12.Hoefen, R. J., and B. C. Berk. 2002. The role of MAP kinases in endothelial activation. Vasc. Pharmacol. 38:271-273. [DOI] [PubMed] [Google Scholar]
- 13.Ip, Y. T., and R. J. Davis. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 10:205-219. [DOI] [PubMed] [Google Scholar]
- 14.Jono, H., H. Xu, H. Kai, D. J. Lim, Y. S. Kim, X. H. Feng, and J. D. Li. 2003. Transforming growth factor-beta-Smad signaling pathway negatively regulates nontypeable Haemophilus influenzae-induced MUC5AC mucin transcription via mitogen-activated protein kinase (MAPK) phosphatase-1-dependent inhibition of p38 MAPK. J. Biol. Chem. 278:27811-27819. [DOI] [PubMed] [Google Scholar]
- 15.Lawler, S., Y. Fleming, M. Goedert, and P. Cohen. 1998. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8:1387-1390. [DOI] [PubMed] [Google Scholar]
- 16.Leppa, S., and D. Bohmann. 1999. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 18:6158-6162. [DOI] [PubMed] [Google Scholar]
- 17.Lim, D. J., and H. Birck. 1971. Ultrastructural pathology of the middle ear mucosa in serous otitis media. Ann. Otol. Rhinol. Laryngol. 80:838-853. [DOI] [PubMed] [Google Scholar]
- 18.Lin, A. 2003. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25:17-24. [DOI] [PubMed] [Google Scholar]
- 19.Magnuson, K., A. Hermansson, A. Melhus, and S. Hellstrom. 1997. The tympanic membrane and middle ear mucosa during non-typeable Haemophilus influenzae and Haemophilus influenzae type b acute otitis media: a study in the rat. Acta Otolaryngol. 117:396-405. [DOI] [PubMed] [Google Scholar]
- 20.Matsuguchi, T., A. Masuda, K. Sugimoto, Y. Nagai, and Y. Yoshikai. 2003. JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. 22:4455-4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maw, R., J. Wilks, I. Harvey, T. J. Peters, and J. Golding. 1999. Early surgery compared with watchful waiting for glue ear and effect on language development in preschool children: a randomised trial. Lancet 353:960-963. [DOI] [PubMed] [Google Scholar]
- 22.Mohit, A. A., J. H. Martin, and C. A. Miller. 1995. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14:67-78. [DOI] [PubMed] [Google Scholar]
- 23.Murakata, C., M. Kaneko, G. Gessner, T. S. Angeles, M. A. Ator, T. M. O'Kane, B. A. McKenna, B. A. Thomas, J. R. Mathiasen, M. S. Saporito, D. Bozyczko-Coyne, and R. L. Hudkins. 2002. Mixed lineage kinase activity of indolocarbazole analogues. Bioorg. Med. Chem. Lett. 12:147-150. [DOI] [PubMed] [Google Scholar]
- 24.Palacios, S. D., K. Pak, A. G. Kayali, A. Z. Rivkin, C. Aletsee, Å. Melhus, N. J. Webster, and A. F. Ryan. 2002. Participation of Ras and extracellular regulated kinase in the hyperplastic response of middle-ear mucosa during bacterial otitis media. J. Infect. Dis. 186:1761-1769. [DOI] [PubMed] [Google Scholar]
- 25.Palacios, S. D., K. Pak, A. Z. Rivkin, A. G. Kayali, D. A. Austin, C. Aletsee, Å. Melhus, N. J. Webster, and A. F. Ryan. 2004. The role of p38 MAPK in middle ear mucosa hyperplasia during bacterial otitis media. Infect. Immun. 72:4662-4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piglansky, L., E. Leibovitz, S. Raiz, D. Greenberg, J. Press, A. Leiberman, and R. Dagan. 2003. Bacteriologic and clinical efficacy of high dose amoxicillin for therapy of acute otitis media in children. Pediatr. Infect. Dis. J. 22:405-413. [DOI] [PubMed] [Google Scholar]
- 27.Potapova, O., M. Gorospe, F. Bost, N. M. Dean, W. A. Gaarde, D. Mercola, and N. J. Holbrook. 2000. c-Jun N-terminal kinase is essential for growth of human T98G glioblastoma cells. J. Biol. Chem. 275:24767-24775. [DOI] [PubMed] [Google Scholar]
- 28.Prieskorn, D. M., and J. M. Miller. 2000. Technical report: chronic and acute intracochlear infusion in rodents. Hear. Res. 140:212-215. [DOI] [PubMed] [Google Scholar]
- 29.Ridley, A. J. 2001. Rho family proteins: coordinating cell responses. Trends Cell Biol. 11:471-477. [DOI] [PubMed] [Google Scholar]
- 30.Russell, M., C. A. Lange-Carter, and G. L. Johnson. 1995. Direct interaction between Ras and the kinase domain of mitogen-activated protein kinase kinase kinase (MEKK1). J. Biol. Chem. 270:11757-11760. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber, M., A. Kolbus, F. Piu, A. Szabowski, U. Mohle-Steinlein, J. Tian, M. Karin, P. Angel, and E. F. Wagner. 1999. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 13:607-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuknecht, H. F. 1993. Pathology of the ear. Lea & Febiger, Philadelphia, PA.
- 33.Schwabe, R. F., C. A. Bradham, T. Uehara, E. Hatano, B. L. Bennett, R. Schoonhoven, and D. A. Brenner. 2003. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology 37:824-832. [DOI] [PubMed] [Google Scholar]
- 34.Surapisitchat, J., R. J. Hoefen, X. Pi, M. Yoshizumi, C. Ya, and B. C. Berk. 2001. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: inhibitory crosstalk among MAPK family members. Proc. Natl. Acad. Sci. USA 98:6476-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teramoto, H., O. A. Coso, H. Miyata, T. Igishi, T. Miki, and J. S. Gutkind. 1996. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 271:27225-27228. [DOI] [PubMed] [Google Scholar]
- 36.Tos, M., and K. Bak-Pedersen. 1997. Goblet cell population in the pathological middle ear and eustachian tube of children and adults. Ann. Otol. Rhinol. Laryngol. 86:209-218. [DOI] [PubMed] [Google Scholar]
- 37.Tseng, H., T. E. Peterson, and B. C. Berk. 1995. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ. Res. 77:869-878. [DOI] [PubMed] [Google Scholar]
- 38.Wang, L. H., C. G. Besirli, and E. M. Johnson, Jr. 2004. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu. Rev. Pharmacol. Toxicol. 44:451-474. [DOI] [PubMed] [Google Scholar]
- 39.Wright, C. G., and W. L. Meyerhoff. 1994. Pathology of otitis media. Ann. Otol. Rhinol. Laryngol. 163(Suppl.):24-26. [DOI] [PubMed] [Google Scholar]
- 40.Xu, Z., A. C. Maroney, P. Dobrzanski, N. V. Kukekov, and L. A. Greene. 2001. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol. 21:4713-4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan, C., M. Takahashi, M. Okuda, J. D. Lee, and B. C. Berk. 1999. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J. Biol. Chem. 274:143-150. [DOI] [PubMed] [Google Scholar]