

Abstract
A rapid two-step DNA extraction method and a multiplex PCR for the detection of dermatophytes in general and Trichophyton rubrum specifically were developed and evaluated with DNA extracted from pure cultures and from clinically diseased nails. DNA from the following dermatophytes was used: Epidermophyton floccosum, Microsporum audouinii, Microsporum canis, Microsporum gypseum, Microsporum nanum, Trichophyton mentagrophytes, Trichophyton rubrum, Trichophyton schoenleinii, Trichophyton soudanense, Trichophyton terrestre, Trichophyton tonsurans, Trichophyton verrucosum, and Trichophyton violaceum. Human DNA and DNA from the following nondermatophyte fungi were included as controls: Alternaria, Aspergillus niger, Candida albicans, Candida glabrata, Candida krusei, Malassezia furfur, Saccharomyces cerevisiae, and Scopulariopsis brevicaulis. A total of 118 nail samples received for routine microscopy and culture for dermatophytes were subsequently tested by the two PCRs separately and in a multiplex format. Using DNA extracted from pure cultures and the pan-dermatophyte PCR, the T. rubrum-specific PCR sequentially and in a multiplex format correctly detected all dermatophytes and additionally correctly identified T. rubrum. Comparison of the traditional diagnostic evaluation (microscopy and culture) of nail samples with PCR on DNA directly extracted from the nails showed excellent agreement between PCR and microscopy, but the number of samples with dermatophyte species identification was increased considerably from 22.9% to 41.5%, mainly due to the identification of T. rubrum by PCR in microscopy-positive but culture-negative samples. In conclusion, this 5-hour diagnostic test was shown to increase not only the speed but also the sensitivity of investigation for nail dermatophytosis.
Human-pathogenic dermatophytes are keratinophilic molds that infect human skin, nails, and hair. Three genera (Trichophyton, Microsporum, and Epidermophyton) of these organisms exist; however, their preferred sites for infections vary. Nail infections are mainly caused by T. rubrum, followed by T. mentagrophytes (1a, 19, 20, 25, 27, 29), in contrast to hair and skin infections, which may be caused by other dermatophyte species, including Microsporum spp. The prevalences of onychomycosis in European countries vary between 3 and 22% (8, 9, 27). In addition to dermatophytes, Candida and nondermatophyte molds may be recovered from clinically affected nails. In a study by Summerbell et al. in which 2,662 affected nails were examined, the following agents were isolated: T. rubrum (>70%), T. mentagrophytes (20%), Candida albicans (5.5%), and Scopulariopsis brevicaulis or nondermatophyte molds (1.6%) (26). While the recovery of an anthropophilic dermatophyte should always be regarded as representing a true pathogen, Candida and nondermatophyte molds may represent contaminants, colonizing agents, or a secondary infection due to local or systemic factors (2, 3, 20). As other conditions (for instance, psoriasis) may resemble onychomycosis and as onychomycosis requires long-term systemic antifungal treatment, the correct identification of causal fungi is mandatory (6, 28). The current diagnosis is based on detection of fungal elements by direct microscopy of the clinical specimens followed by in vitro culture and morphological identification of the fungus (2, 16, 18). Direct microscopic examination of skin and nail material is often sufficient for the diagnosis of a fungal infection but does not provide genus or species identification and hence does not differentiate unquestionably between dermatophytes and other molds. Furthermore, although rapid and economical, this technique gives false-negative results in 5 to 15% of the cases (5, 21, 22). The subsequent species identification is performed by culture and morphological examination of colonies. The culture is, however, negative in up to 40% of the microscopy-positive cases and is time-consuming due to the slow growth and sporulation and the need for additional physiological tests (21, 28). Therefore, the time required for species identification may vary from 10 to 15 days up to 3 to 4 weeks (16).
A simple, rapid, and specific method for the diagnosis of dermatophyte infections would obviously be a major improvement. Introduction of a PCR-based methodology would increase specificity, simplicity, and speed and potentially even reduce cost. For studies on species identification and typing, PCR (7, 11), PCR fingerprinting (4, 12), random amplification of polymorphic DNA (14, 17), PCR and restriction fragment length polymorphism analysis (12, 24), and arbitrarily primed PCR (16, 17) have all been applied. The main targets have been the following genes or DNA fragments: the ribosomal DNA region, DNA topoisomerase II genes, and the chitin synthase gene (11, 13). Recently, Kardjeva and colleagues presented a 48-h diagnostic method of onychomycosis involving a 14-step nail pretreatment and DNA extraction method and a subsequent T. rubrum-specific PCR combined with restriction fragment length polymorphism analysis and sequencing of the internal transcribed spacer region for the detection of other fungal agents (15). Such a methodology is, however, difficult to implement in a routine laboratory receiving large numbers of nail specimens.
In this paper we present an alternative multiplex PCR-based method especially developed for the detection of dermatophyte nail infections. By a two-step extraction procedure followed by a single multiplex PCR and electrophoresis, the method enables the diagnosis of infection caused by any one of the dermatophytes (pan-dermatophyte) and in the case of T. rubrum infection even a genus and species identification. A two-step, 15-minute method for extraction of DNA directly from patient samples allows application of this method in routine diagnostic laboratories.
MATERIALS AND METHODS
Strains and clinical isolates.
Twelve fungal strains were purchased from the National Collection of Pathogenic Fungi (United Kingdom). Clinical isolates were obtained from the Mycology Laboratory of the Statens Serum Institute (SSI) (Denmark) (Table 1). All clinical isolates were identified by observation of macro- and micromorphology.
TABLE 1.
Microorganisms used in the study
| Microorganism | No. of reference strains (NCPF no.) | No. of clinical isolates |
|---|---|---|
| Dermatophytes | ||
| Epidermophyton floccosum | 1 (777) | 14 |
| Microsporum audouinii | 1 (436) | 5 |
| Microsporum canis | 1 (177) | 10 |
| Microsporum gypseum | 1 (40) | 2 |
| Microsporum nanum | 1 | |
| Trichophyton mentagrophytes var. interdigitale | 1 (780) | 10 |
| Trichophyton mentagrophytes var. mentagrophytes | 1 (224) | |
| Trichophyton rubrum | 1 (113) | 12 |
| Trichophyton schoenleinii | 1 (124) | |
| Trichophyton soudanense | 1 (800) | 13 |
| Trichophyton terrestre | 1 (602) | 8 |
| Trichophyton tonsurans | 1 (690) | 8 |
| Trichophyton verrucosum | 6 | |
| Trichophyton violaceum | 1 (794) | |
| Other fungi | ||
| Alternaria sp. | 1 | |
| Aspergillus niger | 2 | |
| Candida albicans | 3 | |
| Candida glabrata | 4 | |
| Candida krusei | 2 | |
| Malassezia furfur | 5 | |
| Saccharomyces cerevisiae | 2 | |
| Scopulariopsis brevicaulis | 1 |
Clinical nail samples.
One hundred eighteen nail samples received for routine examination at the Laboratory of Mycology at SSI were prospectively included. The only inclusion criterion was the presence of a sufficient amount of material for investigation by direct microscopy and culture as well as PCR analysis.
DNA preparation from dermatophyte cultures.
The strains and clinical isolates were cultured in 2 ml of Sabouraud liquid medium with cycloheximide and chloramphenicol (SSI Diagnostika, Denmark) and incubated with shaking for up to 8 days at 27°C. After harvest, the pellet was resuspended in 500 μl of lysis buffer (400 mM Tris-HCl [pH 8.0], 60 mM EDTA [pH 8.0], 150 mM NaCl, 1% sodium dodecyl sulfate) and left at room temperature for 10 min. Next, 150 μl of potassium acetate (pH 4.8) was added and tubes were vortexed and centrifuged (1 min, 12,000 × g). The supernatant was transferred to a new tube, and an equal volume of isopropyl alcohol was added. The DNA pellet was washed in 70% ethanol. The dried DNA pellet was dissolved in 50 μl of TE (10 mM Tris, 1 mM EDTA) buffer. Two microliters of the DNA was used in 20 to 50 μl of the PCR mixture. Reagents were, unless otherwise stated, purchased from Sigma (Germany).
DNA preparation from nail samples.
For DNA preparation (1), DNA from nail samples was extracted by a 10-min incubation of the nail sample in 100 μl of extraction buffer (60 mM sodium bicarbonate [NaHCO3], 250 mM potassium chloride [KCl] and 50 mM Tris, pH 9.5) in 95°C and subsequent addition of 100 μl anti-inhibition buffer (2% bovine serum albumin). After vortex mixing, this DNA-containing solution was used for PCR.
Pan-dermatophyte PCR.
Pan-dermatophyte PCR (1) was as follows. Based on the comparison (VectorNTI; InforMax, Inc.) of nucleotide sequences of different dermatophytes in the NCBI nucleotide database, a set of primers detecting a DNA fragment encoding chitin synthase 1, panDerm1 (5′GAAGAAGATTGTCGTTTGCATCGTCTC3′) and panDerm2 (5′CTCGAGGTCAAAAGCACGCCAGAG3′), was designed. Twelve dermatophyte reference strains, 89 clinical dermatophyte isolates, 22 nondermatophyte fungal isolates, and purified human DNA (Table 1) were tested. PCR mixtures consisted of 10 μl of PCR Ready Mix (Sigma, Germany), 0.2 μl of each primer (panDerm1 and panDerm2) at 100 μM, and 4 μl of DNA in a volume of 20 μl. PCR was performed in a MWG-Biotech thermal cycler. The time-temperature profile for PCR was 45 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s at 72°C, preceded by initial denaturation for 10 min at 95°C. The presence of specific PCR products of approximately 366 bp was examined using electrophoresis on a 1% agarose gel and staining with ethidium bromide.
Trichophyton rubrum-specific PCR.
On the basis of alignment (VectorNTI; InforMax, Inc.) of sequences of internal transcribed spacer 2 in the NCBI nucleotide database, universal (uni, 5′TCTTTGAACGCACATTGCGCC3′) and Trichophyton rubrum-specific (Trubrum-rev, 5′CGGTCCTGAGGGCGCTGAA3′) primers were designed. Each reaction was performed in a volume of 20 μl by the addition of 4 μl of DNA from microorganisms listed above, 0.2 μl of each primer (at 100 μM), and 10 μl of PCR ReadyMix (Sigma, Germany). The amplification was performed in a thermal cycler (MWG-Biotech, Germany) and consisted of one initial cycle of denaturation for 5 min at 94°C and 45 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s of extension at 72°C. After the thermal cycles, the amplicons were electrophoresed in a 2% agarose gel and stained with ethidium bromide. To standardize the procedure, different DNA concentrations and thermal cycles were tested (data not shown).
Multiplex PCR.
The multiplex PCR was performed using the two specific sets of primers described above (panDerm1 and panDerm2 primers and uni and Trubrum-rev primers). The reaction was performed under different conditions; 0.2 mM of each primer was used. The following time-temperature profile was chosen: one initial cycle of denaturation for 5 min at 94°C and 45 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s of extension at 72°C. After the thermal cycles, the amplicons were electrophoresed in a 2% agarose gel and stained with ethidium bromide. Specificity of the multiplex PCR was tested with DNAs from all of the strains listed in Table 1 and with human DNA. To standardize the procedure, different DNA concentrations and thermal cycles were tested (data not shown). The multiplex PCR (and separately the pan-dermatophyte and the T. rubrum-specific PCRs) were subsequently evaluated using 97 nail specimens received for routine analysis.
RESULTS
Evaluation of pan-dermatophyte and T. rubrum PCRs using DNA extracted from fungal cultures.
Extracted DNA from cultures of 12 reference dermatophyte strains, 89 clinical dermatophyte isolates, and 21 other fungi (Table 1) was used for evaluation of the pan-dermatophyte primers and the T. rubrum-specific primers separately as well as in a multiplex PCR format. A 203-bp PCR product corresponding to T. rubrum was observed for 13/13 T. rubrum DNA samples with the T. rubrum-specific PCR separately and in multiplex format, and specific 366-bp PCR products were obtained for 101/101 dermatophyte DNA samples with the pan-dermatophyte PCR alone and in multiplex format (examples of results of the pan-dermatophyte and T. rubrum-specific PCRs are presented in Fig. 1). No PCR products were detected by the pan-dermatophyte PCR, the T. rubrum-specific PCR, or the multiplex PCR for the 21 nondermatophyte fungal isolates or for three samples of human DNA (100% sensitivity and 100% specificity for all three PCR systems).
FIG. 1.

Example of Trichophyton rubrum-specific and pan-dermatophyte PCR product analysis. Lanes: 1 and 12, molecular size marker (fragment sizes, 501, 489, 404, 331, 242, 190, 147, 111, and 110 bp); 2 and 3, results of T. rubrum-specific PCR performed for T. mentagrophytes DNA (lane 2) and T. rubrum DNA (lane 3); 4 to 11, results of pan-dermatophyte PCR performed for Microsporum audouinii (lane 4), T. mentagrophytes var. mentagrophytes (lane 5), Trichophyton schoenleninii (lane 6), Trichophyton terrestre (lane 7), T. rubrum (lane 8), T. tonsurans (lane 9), Trichophyton soudanense (lane 10), and Epidermophyton floccosum (lane 11).
Clinical evaluation of pan-dermatophyte and T. rubrum PCRs using DNA extracted directly from nail samples.
By conventional diagnostics of the 118 nail samples, 25 yielded growth of T. rubrum, 1 of T. rubrum and T. mentagrophytes, 1 of Trichophyton tonsurans, 3 of Alternaria sp., 1 of Acremonium sp., 1 of Aspergillus sp., 1 of Candida sp., 2 of S. brevicaulis, and 1 of a yeast species not further identified. Sixty-four samples were microscopy and culture negative and 18 were positive by direct microscopy for hyphae and conidia but culture negative; the latter samples were regarded as fungus positive (but no genus or species identification could be established). Samples which were culture positive for nondermatophyte fungi were regarded as dermatophyte negative by conventional methodology in the comparison with PCR results. DNA from 118 clinical samples was extracted using the rapid two-step protocol. For each of the samples, again three sets of PCRs were performed (T. rubrum-specific PCR, pan-dermatophyte PCR and multiplex pan-dermatophyte plus T. rubrum PCR). All multiplex PCR results were in agreement with the single-PCR results, indicating no loss of sensitivity in the multiplex PCR setup.
Overall, 50/118 (42.4%) of the samples were dermatophyte positive by PCR and 45/118 (38.1%) were positive by traditional diagnostics, including samples positive by microscopy but negative by culture. Among 24 specimens which were microscopy and culture positive (T. rubrum), 21 (87.5%) were confirmed by PCR as T. rubrum positive, 2 were PCR negative (8.3%), and 1 reported as T. mentagrophytes and T. rubrum positive by conventional examination was pan-dermatophyte positive but T. rubrum PCR negative. Of 64 specimens negative by conventional microscopy and culture, 49 (76.6%) were confirmed by PCR as negative but 15 (23.4%) were PCR positive (T. rubrum). Of 18 microscopy-positive but culture-negative specimens (the presence of hyphae was observed), 10 were T. rubrum PCR positive (55.6%), 7 were negative by PCR (38.9%), and the result for one sample was not possible to interpret (5.6%) (unspecific PCR products were synthesized). Two specimens which were negative by microscopic examination of the nail but T. rubrum positive in culture were negative by PCR (examples of results of the pan-dermatophyte and T. rubrum-specific PCRs are presented in Fig. 2). One specimen diagnosed by conventional examination as T. tonsurans was pan-dermatophyte and T. rubrum PCR positive. Finally, PCR results for nine specimens diagnosed by conventional examination as nondermatophyte species are presented in Table 2.
FIG. 2.
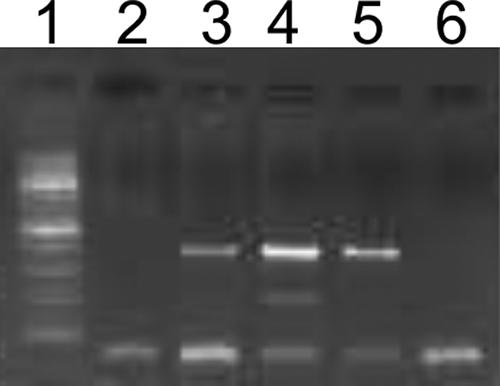
Example of Trichophyton rubrum-specific and/or pan-dermatophyte multiplex PCR product analysis. Lanes: 1, molecular size marker (100-bp DNA ladder); 2 to 6, results of multiplex PCR performed for DNA extracted directly from nail specimens diagnosed by conventional methods as negative (lane 2), M. audouinii (lane 3), T. rubrum (lane 4), T. mentagrophytes (lane 5), and Aspergillus sp. (lane 6).
TABLE 2.
PCR results for the nine clinical specimens with growth of a nondermatophyte fungus
| Routine microscopy result | Routine culture result | No. of isolates | PCR result |
|---|---|---|---|
| Negative | Acremonium | 1 | Negative |
| Positive | Alternaria | 1 | Negative |
| Negative | Alternaria | 1 | T. rubrum |
| Positive | Aspergillus | 2 | Negative |
| Positive | Candida sp. | 1 | Negative |
| Positive | S. brevicaulis | 1 | Negative |
| Positive | S. brevicaulis | 1 | T. rubrum |
| Positive | Yeasts | 1 | Negative |
To investigate whether the lack of PCR products in PCR-negative samples could be due to the presence of PCR-inhibitory substances in the samples, all PCR-negative specimens were spiked with T. rubrum DNA and subsequently retested in the multiplex pan-dermatophyte-T. rubrum PCR. A PCR product was produced in all cases (data not shown). All the T. rubrum-specific PCR products obtained from specimens not diagnosed as T. rubrum-positive samples by traditional methodology were sequenced (MWG Biotech, Germany), and the sequences of all of them matched that of T. rubrum reference strain NCPF 113. The pan-dermatophyte PCR product obtained from the DNA of the specimen diagnosed conventionally as T. rubrum and T. mentagrophytes was sequenced, and the sequence matched that of T. mentagrophytes reference strain NCPF 224.
A comparison of results obtained by conventional diagnostics and PCR is shown in Table 3. Overall, the number of positive samples was increased by 11% (45 [38.1%] versus 50 [42.2%] of 118 specimens were positive by the conventional and PCR methodologies, respectively). Furthermore, due to the presence of a considerable number of microscopy-positive but culture-negative samples, the percentage of samples with a species identification was almost doubled by use of the PCR (49 out of 118 specimens were found to be T. rubrum positive by the PCR-based method, while only 27 out of 118 specimens were dermatophyte positive by culture).
TABLE 3.
Comparison of the results of conventional and PCR-based examinations of nail specimens
| Method | % (no./total)
|
|
|---|---|---|
| Dermatophyte | Species-specific identification | |
| Conventional | 38.1 (45/118) | 22.9 (27/118) |
| PCR based | 42.4 (50/118) | 41.5 (49/118) |
| Difference in detection by PCR-based method | 4.3 | 18.6 |
DISCUSSION
Day-to-day detection of dermatophyte infection in nails is obviously a major improvement in the diagnosis of tinea unguium, allowing antifungal treatment to be instituted promptly upon correct diagnosis and at the same time restricted to those with dermatophyte infections. Although the species distributions vary between different parts of the world, T. rubrum is in most surveys reported to be the major pathogen in tinea unguium, accounting for 63 to 89% of the infections (1a, 9, 10, 19, 23, 27). At the same time, dermatophytes belonging to the less terbinafine-susceptible genus Microsporum are unanimously reported to be very rare agents of onychomycosis, and the detection of dermatophyte DNA in a nail specimen will therefore represent infection with a terbinafine-susceptible dermatophyte in the vast majority of cases and thus provide sufficient information to guide the clinician despite a lack of species identification.
The clinical evaluation of single and multiplex PCR detection of any dermatophyte and of T. rubrum specifically in nail specimens showed increased sensitivity compared to conventional diagnosis (Table 3). In this comparison, samples positive by microscopy but negative by culture were regarded as dermatophyte positive by traditional diagnostics, though we cannot rule out the possibility that some of these cases may represent nondermatophyte infections. It is not uncommon, however, to obtain negative culture results from patients with dermatophytosis, due to difficulties associated with sampling (insufficient material or use of nail clippings instead of subungual material) or to prior medical treatment, etc., and cases with positive microscopy but negative culture should therefore always be investigated further (5). On one occasion the species identifications obtained by conventional culture and PCR were conflicting (T. tonsurans by culture and T. rubrum by PCR). The fact that the T. rubrum PCR was negative when applied to the T. tonsurans reference strain as well as to all the clinical control T. tonsurans isolates tested initially raises the question of whether this was a case of misidentification by conventional identification or a double infection with T. tonsurans and T. rubrum. As nail infections in Denmark caused by T. tonsurans are extremely rare, especially among Danes (as in this case), the former explanation is the more likely in our opinion; however, the isolate was not stored and thus no further examinations were possible. One specimen was by conventional methodology diagnosed as a mixed infection with T. rubrum and T. mentagrophytes, but the PCR yielded solely a pan-dermatophyte PCR product. This was sequenced and the sequence matched that of T. mentagrophytes, in agreement with the culture result. Several explanations for this apparent lack of detection of the T. rubrum isolate in this case exist. (i) Although the specimens used for conventional and PCR testing derived from the same patient, they are not exactly the same material and the T. rubrum may not have been present in the specimen used for PCR. (ii) This may be a case of contamination of the culture plates by T. rubrum. (iii) The sensitivity of the T. rubrum PCR may be insufficient in cases of mixed infections. The facts, however, that the T. rubrum-specific primers target a multicopy gene, in contrast to the pan-dermatophyte primers, and that the T. rubrum PCR was also negative when the sample was run in a single-PCR setup suggest that the sensitivity of the T. rubrum PCR should not be inferior to that of the pan-dermatophyte PCR. However, examination of additional samples from cases of documented mixed infections is necessary to evaluate this further.
The interpretation of the detection of nondermatophyte molds in nail specimens is controversial. Such findings may reflect the presence of mold elements in the nail specimen due to contamination, transient colonization or infection of a traumatized or otherwise diseased nail, or contamination in the laboratory. Therefore, at least repeated recovery of identical mold species is typically required before a pathogenic role is considered, and even in these cases the recovery may represent an infection which is secondary to an underlying pathological nail condition. The finding in this study that two nails yielded molds by culture but T. rubrum by PCR may reflect overgrowth by the rapidly growing contaminating or colonizing mold or true double infection.
Although dermatophyte and/or T. rubrum identification in the nail specimens has been attempted using a range of molecular methods, only one recently published study involved DNA extraction directly from nail specimens without prior culture (15). The extraction method described, however, was a multistep procedure involving 14 steps and thus was labor-intensive and per se associated with an increased risk of contamination. The application of a two-step, 15-min procedure for extraction of DNA directly from nail specimens and a multiplex PCR-based diagnosis of any dermatophyte and/or T. rubrum with increased sensitivity compared to conventional diagnostic procedures allow for the first time integration of a molecular biology-based method into the routine examination of nail dermatophytosis also for diagnostic laboratories receiving specimens on a larger scale. This brings hope that rapid, specific, and low-cost diagnoses of onychomycosis may become broadly available in the near future.
Footnotes
Published ahead of print on 31 January 2007.
REFERENCES
- 1.Brillowska-Dąbrowska, A. December 2006. DNA preparation from nail samples. Denmark patent WO2006133701.
- 1a.Dolenc-Voljc, M. 2005. Dermatophyte infections in the Ljubljana region, Slovenia, 1995-2002. Mycoses 48:181-186. [DOI] [PubMed] [Google Scholar]
- 2.Elewski, B. E. 1998. Onychomycosis: pathogenesis, diagnosis, and management. Clin. Microbiol. Rev. 11:415-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis, D. H., A. B. Watson, J. E. Marley, and T. G. Williams. 1997. Non-dermatophytes in onychomycosis of the toenails. Br. J. Dermatol. 136:490-493. [PubMed] [Google Scholar]
- 4.Faggi, E., G. Pini, and E. Campisi. 2002. PCR fingerprinting for identification of common species of dermatophytes. J. Clin. Microbiol. 40:4804-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gentles, J. C. 1971. Laboratory investigations of dermatophyte infections of nails. Sabouraudia 9:149-152. [DOI] [PubMed] [Google Scholar]
- 6.Hainer, B. L. 2003. Dermatophyte infections. Am. Fam. Physician 67:101-108. [PubMed] [Google Scholar]
- 7.Harmsen, D., A. Schwinn, E. B. Brocker, and M. Frosch. 1999. Molecular differentiation of dermatophyte fungi. Mycoses 42:67-70. [DOI] [PubMed] [Google Scholar]
- 8.Hay, R. J. 2001. The future of onychomycosis therapy may involve a combination of approaches. Br. J. Dermatol. 145(Suppl. 60):3-8. [PubMed] [Google Scholar]
- 9.Heikkila, H., and S. Stubb. 1995. The prevalence of onychomycosis in Finland. Br. J. Dermatol. 133:699-703. [DOI] [PubMed] [Google Scholar]
- 10.Ilkit, M. 2005. Onychomycosis in Adana, Turkey: a 5-year study. Int. J. Dermatol. 44:851-854. [DOI] [PubMed] [Google Scholar]
- 11.Kanbe, T., Y. Suzuki, A. Kamiya, T. Mochizuki, M. Fujihiro, and A. Kikuchi. 2003. PCR-based identification of common dermatophyte species using primer sets specific for the DNA topoisomerase II genes. J. Dermatol. Sci. 32:151-161. [DOI] [PubMed] [Google Scholar]
- 12.Kanbe, T., Y. Suzuki, A. Kamiya, T. Mochizuki, M. Kawasaki, M. Fujihiro, and A. Kikuchi. 2003. Species-identification of dermatophytes Trichophyton, Microsporum and Epidermophyton by PCR and PCR-RFLP targeting of the DNA topoisomerase II genes. J. Dermatol. Sci. 33:41-54. [DOI] [PubMed] [Google Scholar]
- 13.Kano, R., A. Hirai, M. Muramatsu, T. Watari, and A. Hasegawa. 2003. Direct detection of dermatophytes in skin samples based on sequences of the chitin synthase 1 (CHS1) gene. J. Vet. Med. Sci. 65:267-270. [DOI] [PubMed] [Google Scholar]
- 14.Kano, R., Y. Nakamura, S. Watanabe, H. Takahashi, H. Tsujimoto, and A. Hasegawa. 1998. Differentiation of Microsporum species by random amplification of polymorphic DNA (RAPD) and Southern hybridization analyses. Mycoses 41:229-233. [DOI] [PubMed] [Google Scholar]
- 15.Kardjeva, V., R. Summerbell, T. Kantardjiev, D. Devliotou-Panagiotidou, E. Sotiriou, and Y. Graser. 2006. Forty-eight-hour diagnosis of onychomycosis with subtyping of Trichophyton rubrum strains. J. Clin. Microbiol. 44:1419-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu, D., S. Coloe, R. Baird, and J. Pedersen. 2000. Application of PCR to the identification of dermatophyte fungi. J. Med. Microbiol. 49:493-497. [DOI] [PubMed] [Google Scholar]
- 17.Liu, D., L. Pearce, G. Lilley, S. Coloe, R. Baird, and J. Pedersen. 2002. PCR identification of dermatophyte fungi Trichophyton rubrum, T. soudanense and T. gourvilii. J. Med. Microbiol. 51:117-122. [DOI] [PubMed] [Google Scholar]
- 18.Mahoney, J. M., J. Bennet, and B. Olsen. 2003. The diagnosis of onychomycosis. Dermatol. Clin. 21:463-467. [DOI] [PubMed] [Google Scholar]
- 19.Monod, M., S. Jaccoud, C. Zaugg, B. Lechenne, F. Baudraz, and R. Panizzon. 2002. Survey of dermatophyte infections in the Lausanne area, Switzerland. Dermatology 205:201-203. [DOI] [PubMed] [Google Scholar]
- 20.Mugge, C., U. F. Haustein, and P. Nenoff. 2006. Causative agents of onychomycosis—a retrospective study. J. Dtsch. Dermatol. Ges. 4:218-228. [DOI] [PubMed] [Google Scholar]
- 21.Petrini, B., and M. L. von Rosen. 2002. Optimal dermatophyte diagnosis requires both microscopy and culture. Lakartidningen 99:4084. [PubMed] [Google Scholar]
- 22.Rippon, J. W. 1988. Medical mycology: the pathogenic fungi and the pathogenic actinomycetes. W. B. Saunders Co., Philadelphia, PA.
- 23.Romano, C., C. Gianni, and E. M. Difonzo. 2005. Retrospective study of onychomycosis in Italy: 1985-2000. Mycoses 48:42-44. [DOI] [PubMed] [Google Scholar]
- 24.Shin, J. H., J. H. Sung, S. J. Park, J. A. Kim, J. H. Lee, D. Y. Lee, E. S. Lee, and J. M. Yang. 2003. Species identification and strain differentiation of dermatophyte fungi using polymerase chain reaction amplification and restriction enzyme analysis. J. Am. Acad. Dermatol. 48:857-865. [DOI] [PubMed] [Google Scholar]
- 25.Singh, D., D. C. Patel, K. Rogers, N. Wood, D. Riley, and A. J. Morris. 2003. Epidemiology of dermatophyte infection in Auckland, New Zealand. Australas. J. Dermatol. 44:263-266. [DOI] [PubMed] [Google Scholar]
- 26.Summerbell, R. C., J. Kane, and S. Krajden. 1989. Onychomycosis, tinea pedis and tinea manuum caused by non-dermatophytic filamentous fungi. Mycoses 32:609-619. [DOI] [PubMed] [Google Scholar]
- 27.Svejgaard, E. L., and J. Nilsson. 2004. Onychomycosis in Denmark: prevalence of fungal nail infection in general practice. Mycoses 47:131-135. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg, J. M., E. K. Koestenblatt, W. D. Tutrone, H. R. Tishler, and L. Najarian. 2003. Comparison of diagnostic methods in the evaluation of onychomycosis. J. Am. Acad. Dermatol. 49:193-197. [DOI] [PubMed] [Google Scholar]
- 29.Weitzman, I., and R. C. Summerbell. 1995. The dermatophytes. Clin. Microbiol. Rev. 8:240-259. [DOI] [PMC free article] [PubMed] [Google Scholar]