

Abstract
We have adapted our established Alu PCR assay for proviral DNA and PCR for total cellular DNA to a real-time PCR format and applied these to human immunodeficiency virus (HIV)-positive specimens collected for routine determination of the plasma viral load (pVL). In a cohort of five patients, measurements of integrated viral load (iVL) and cell-associated viral load (cVL) in CD4+ cells isolated by a single positive selection step were not indicative of HIV DNA levels in the circulation, and further analysis was performed on peripheral blood mononuclear cells (PBMC). In a cohort of 46 samples total cVL was quantitated in most samples, but iVL could be quantitated in only 47.8%, since in 26% iVL was undetectable and in 21.7% the results were invalid due to high levels of unintegrated HIV DNA. There was no correlation of cVL or iVL with pVL, CD4 count, or duration of successful antiretroviral treatment. Out of 26 patients with undetectable pVL, 4 patients failed therapy within the subsequent 12 months and had higher than average iVL, but this was not the case for cVL. Among nine patients with long-term undetectable pVL, no consistent decline in cVL or iVL was seen with time, and changes in cVL and iVL within a patient could be concordant or discordant. These results show that cVL and iVL can be coordinately measured in PBMC from clinical samples but do not correlate with pVL, CD4 counts, or length of suppressive antiretroviral therapy. Interestingly, a high iVL (but not a high cVL) in patients with undetectable pVL was associated with subsequent treatment failure.
Human immunodeficiency virus (HIV) infection in patients is largely managed by monitoring CD4 counts and plasma viral load (pVL). With successful antiretroviral therapy, pVL are often undetectable by either previously routine (sensitivity, <400 copies/ml) or more recent, more sensitive (<50 copies/ml) pVL assays. However, even patients who have had undetectable pVL for extended periods show a rapid rebound of the pVL upon withdrawal of therapy. Previous studies have provided evidence of continuing viral replication and presence of HIV DNA in tissue sites such as lymph nodes and circulating cells (e.g., resting or activated CD4+ T cells, CD4− T cells, and monocytes) in patients on successful therapy (6, 7, 9, 26). Calculation of the half-life of replication-competent virus that can be cultured from resting CD4+ T cells from patients on antiretroviral therapy also suggests a long-lived viral reservoir, lasting 6 months from initiation of treatment (22) or around 44 months with up to 7 years of suppressive therapy (12, 25). These studies demonstrate the presence of long-lived HIV reservoirs in patients on successful therapy, although other reservoirs in addition to circulating CD4+ T cells are important (8).
Quantitation of circulating reservoirs for HIV via analysis of HIV DNA levels has previously been suggested. Analysis of HIV DNA in peripheral blood mononuclear cells (PBMC) or CD4+ T cells from patients on antiretroviral therapy has shown that while HIV DNA levels correlate with pVL at the onset of therapy and also decline rapidly, HIV DNA cannot be eliminated and is detectable in most patients (15, 16). Cells harboring HIV DNA can still be detected following 9 years of highly active antiretroviral therapy (HAART) and continued suppression of pVL (10). Previous studies that have quantitated total cell-associated viral load (cVL) or total HIV DNA in HIV-positive samples have found no correlation of cVL with pVL or CD4 counts (14, 17, 19, 24, 29) except in specific situations such as structured treatment interruptions or at the onset of therapy (1, 15-17).
Total cell-associated HIV DNA is composed of unintegrated linear and unintegrated circular one-long terminal repeat (1-LTR) and 2-LTR forms and integrated proviral DNA. Unintegrated HIV DNA, particularly 2-LTR circles, has been suggested to be a marker of recent infection due to its labile nature (24), although stable unintegrated forms, including 2-LTR circles, have been shown to exist (4, 21), and its utility as a clinical marker of recent infection has been questioned (1). However, analysis and comparison of both (i) the total pool of cell-associated HIV DNA, where unintegrated DNA forms may reflect both recent and established infection events and integrated DNA forms more likely represent a stable archival reservoir, and (ii) specifically integrated proviral HIV DNA only may yield information on active infection in the circulating HIV reservoir and activity/infectivity of the unseen tissue reservoirs in transmitting virus to the circulating cell population. Fewer studies have directly compared both cVL and integrated viral load (iVL) from clinical samples (15).
We have adapted our previously established laboratory proviral DNA assay (18, 27, 28) to a real-time PCR format and developed it for analysis of both cVL and iVL in total PBMC from patients presenting for clinical pVL testing. We demonstrate that levels of cVL and iVL can be measured concurrently in clinical specimens and do not correlate with pVL, CD4 counts, or the duration of suppressive antiretroviral therapy. Among patients with long-term undetectable pVL we have observed concordant and discordant fluctuations in cVL and iVL over time and seen subsequent virological failure in four patients with higher than average iVL values.
MATERIALS AND METHODS
Isolation, extraction, and normalization of samples.
We obtained patient samples sent to the Serology Unit of Infectious Diseases Laboratories (IDL) at the Institute of Medical and Veterinary Science (IMVS) for routine clinical pVL monitoring. The plasma fraction was removed, and the pVL was quantitated by use of either the Roche Amplicor or Cobas Taqman HIV-1 test, with detection limits of 400 and 50 copies/ml, respectively. Total PBMC were extracted from the buffy coat layer from the same sample by Lymphoprep density gradient centrifugation (Nycomed). For initial comparison of PBMC and CD4+ cells, CD4+ cells were isolated from the total PBMC fraction by anti-CD4 magnetic bead isolation (Militenyi Biotec) as previously described (6).
Total cellular DNA was extracted from isolated cells using DNeasy extraction kits (QIAGEN). Samples were normalized by analysis of β-globin content using 10 pmol of primers β-glo 1 (CCACTTCATCCACGTTCACC) and β-glo 2 (GAAGAGCCAAGGACAGGTAC) and Quantitect SYBR green PCR mix in a reaction volume of 20 μl (QIAGEN). Reactions were carried out in a Rotorgene 3000 (Corbett Research) for 2 min at 50°C and 15 min at 95°C, followed by 35 cycles of 20 s at 95°C, 20 s at 58°C, and 20 s at 72°C (18). Reference samples with defined high and low copy numbers were used as controls in each assay to allow interassay comparison.
Analysis of total DNA (cVL).
Total cellular HIV DNA (cVL) was quantitated in samples corresponding to 10,000 cell equivalents. Standards were derived from total DNA extracted from a mix of chronically infected cell lines ACH-2, H3B, and 8E5 (18, 28) diluted in a background of 10,000 cell equivalents of total DNA extracted from uninfected Hut-78 cells. Reference sample pools with defined high and low copy numbers generated from a mix of chronically infected cell lines ACH-2, H3B, and 8E5, as standards, were used as controls in each assay to allow interassay comparison. Real-time PCRs were performed with 10 pmol of primers SS1 (CTAACTAGGGAACCCACTGC) and SS2a (GGGTCTGAGGGATCTCTAGT) directed against the HIV LTR using Quantitect SYBR green PCR mix in a reaction volume of 20 μl (QIAGEN). Reaction cycles were carried out as described above for β-globin PCR.
Analysis of proviral DNA (iVL).
Quantitation of proviral HIV DNA (iVL) was performed in samples corresponding to 50,000 or 100,000 cell equivalents using a first-round Alu PCR (18, 28) followed by a second-round nested PCR detecting the HIV LTR as described above for cVL. Standards were as described for cVL but diluted in a background of 50,000 or 100,000 cell equivalents of total DNA extracted from uninfected Hut-78 cells. Reference sample pools with defined high and low copy numbers generated from a mix of chronically infected cell lines ACH-2, H3B, and 8E5, as standards, were used as controls in each assay to allow interassay comparison. Alu PCR was performed with 25 pmol of primers Alu-164 (TCCCAGCTACTCGGGAGGCTGAGG) and HIV primer binding site 659 (PBS-659) (TTTCAGGTCCCTGTTCGGGCGCCAC) using 0.8 U of rTth DNA polymerase XL enzyme (Applied Biosystems) in a volume of 50 μl in the manufacturer's buffer supplemented with 1.2 mM Mg(OAc)2 and 200 uM deoxynucleoside triphosphates with a wax-bead-mediated hot start protocol. Reactions were carried out in a PE Applied Biosystems 2400 cycler as follows: 3 min at 94°C followed by 22 cycles of 30 s at 94°C, 30 s at 66°C, 5 min at 70°C, and a final 10-min extension at 72°C. PCR controls were performed without the Alu-164 PCR primer (Alu-minus PCR) to account for any second-round amplification of unintegrated HIV DNA. First-round Alu PCR mixtures were diluted 1/40 and subjected to a second-round real-time PCR for HIV DNA, as described above for cVL.
Statistical analysis.
Nonparametric statistics were employed in all analyses due to the nonnormal sample distribution. Correlations were assessed by Spearman's ρ test. Group comparisons were assessed by analysis of variance (ANOVA) with the Wilcoxon or Kruskal-Wallis test.
RESULTS
Establishment of cVL and iVL assays in a real-time PCR format.
We adapted our laboratory proviral HIV DNA assay (18, 27, 28) for measurements on patient samples in a real-time PCR format. The assay was established using total cellular DNA isolated from a mix of three chronically infected cell lines (H3B, ACH-2, and 8E5). Initial establishment of the assay also involved conversion of our previously used β-globin assay for normalization of DNA content (17) to a real-time PCR format. Amplification profiles, melt curves, and standard curves for β-globin PCR are shown in Fig. 1A. Total cell-associated HIV DNA (cVL) was quantitated by real-time PCR for early-stage reverse transcription products (strong-stop DNA). Amplification profiles for standard curves are shown in Fig. 1B. Proviral HIV DNA (iVL) was quantitated by first-round Alu PCR followed by second-round real-time PCR analysis of strong-stop DNA, as described for Fig. 1B, on a 1/40 dilution of the first-round product (Fig. 1C). Under these conditions both reactions yielded linear amplification curves and melt profiles characteristic of a single product. Agarose gel electrophoresis yielded a single product of the expected size (109 bp). The specificity of the product was confirmed by Southern analysis of the PCR product with an HIV LTR probe (Fig. 1D). Comparison of the PCR signal obtained from Alu and Alu-minus PCRs, where the first-round amplification omitted the Alu primer, showed an average difference of 10 threshold cycle (CT) values (Table 1). A quantifiable signal was seen only from Alu-minus PCR on the 2,000-copy-number standard, yielding Alu-minus copy number values of approximately 4. These results confirm the specificity of the assay and DNA standards for amplification of integrated HIV DNA and show approximately 500-fold discrimination between integrated (Alu PCR) and unintegrated (Alu-minus PCR) DNA.
FIG. 1.

cVL and iVL can be quantitated by real-time PCR. (A) β-Globin DNA was quantitated from total DNA extracted from a mix of three chronically infected cell lines, as used for HIV standards, and subjected to real-time PCR quantitation as described in Materials and Methods. Real-time amplification curves (cycle number versus normalized florescence), derived standard curves (concentration versus CT) and melt analysis (oC versus derivative of fluorescence/derivative of time) of PCR products are shown. (B) Cell-associated HIV DNA was quantitated from total DNA extracted from a mix of three chronically infected cell lines, diluted in a background of uninfected DNA from 10,000 cell equivalents, and subjected to real-time PCR quantitation of a region of the HIV LTR as described in Materials and Methods. Real-time amplification curves, derived standard curves and melt analysis of PCR products are shown. (C) Integrated HIV DNA was quantitated from total DNA as described for panel A and diluted in a background of uninfected DNA from 50,000 cell equivalents. DNA was amplified in a first-round PCR with Alu and PBS-659 primers followed by dilution and second-round PCR amplification of the HIV LTR, as described for panel A and in Materials and Methods. Real-time amplification curves, derived standard curves, and melt analysis of PCR products are shown. (D) Gel analysis of PCR products. Second-round PCR products were subjected to agarose gel electrophoresis, stained with ethidium bromide, and photographed. Lane 1, molecular weight markers; lane 2, PCR product. DNA was then subjected to Southern analysis using an HIV-specific LTR probe and analyzed by autoradiography (lane 3).
TABLE 1.
Validation of detection of integrationa
| Standard (no. of copies) |
CT value
|
||
|---|---|---|---|
| Alu PCR | Alu-minus PCR | ΔCT | |
| 2,000 | 16.89 ± 0.29 | 27.24 ± 0.83 | 10.35 |
| 500 | 18.53 ± 0.32 | 30.48 ± 0.63 | 11.95 |
| 100 | 19.77 ± 0.33 | 32.04 ± 0.59 | 12.27 |
| 10 | 24.01 ± 0.29 | 33.36 ± 2.05 | 9.35 |
| 4 | 27.58 ± 1.14 | 33.78 ± 1.51 | 6.2 |
| Avg ΔCT | 10.02 ± 1.09 | ||
Alu and Alu-minus PCRs from HIV DNA integration standards assayed in a background of 50,000 cells were performed as described in Materials and Methods. CT values (mean ± SEM) and difference in CT value (ΔCT) are indicated.
Measurements in CD4+ cells selected by a single positive bead isolation procedure are not reflective of the total circulating HIV DNA pool.
CD4+ T cells are reportedly a major circulating reservoir of HIV, but other circulating cell types can also harbor significant amounts of HIV DNA (6, 7, 11, 26). Analysis of HIV DNA in purified CD4+ cells would likely result in a cleaner assay by reducing the amount of background cellular DNA. However, the detection of HIV DNA in non-CD4+ cells, such as CD8+ T cells and CD4− CD8− T cells (6), suggests that measurements in CD4+ cells may not represent all the circulating cells that harbor HIV DNA. Thus, we investigated whether HIV DNA in the circulating CD4+ cell fraction was representative of the total circulating (PBMC) pool to see if we could incorporate a simple positive bead selection step into our procedure and improve the performance of the assay without loss of the breadth of information obtained. Plasma samples were collected from five patients, total PBMC were extracted, and CD4+ cells were isolated by positive anti-CD4 magnetic bead selection. CD4+ cells selected were 99% CD4+, 99% CD3+, and CD14− based on previous characterization (6). The unselected cell population also was CD4+ with variable levels (at least 11%) and CD14+ (approximately 2%) and contained HIV DNA (6; unpublished data). Additionally, previous studies in our laboratory that aimed at generating a highly purified CD4− fraction from the population left following CD4+ isolation necessitated both CD4 and CD14 bead depletion steps (6). Thus, the population selected by positive CD4 bead selection was a highly pure CD4+ population but was not completely representative of the CD4+ population present in starting PBMC.
Total DNA was extracted and normalized for cell number by PCR quantitation of β-globin DNA and circulating cVL and iVL quantitated using the assays described above. Results are presented as the copy number from 105 PBMC compared with the copy number from the number of CD4+ cells present in 105 PBMC (obtained by normalizing for the percentage of CD4+ cells, which was measured as part of clinical care) (Table 2). Although we did not expect to achieve concordance of the absolute values for measurements in CD4+ and PBMC, the results should give the same relative values for different patients if both measurements reflect the same HIV DNA pool. However, comparison of the relative values for iVL or cVL between the five patients assayed in either cellular pool did not yield the same results. Measurements of the iVL in total PBMC showed the relative levels 1 < 2 ≤ 4 < 5 < 3, but the iVL in CD4+ cells showed 2 < 1 ≤ 3 < 5; the iVL for patients 2 and 3 is underestimated in the CD4+ fraction relative to the iVL values for other patients and for the same patients' PBMC. Measurements of cVL in total PBMC showed 1 < 2 < 5 < 4 ≤ 3, but cVL in CD4+ cells indicated 2 < 5 < 1 < 3 < 4. The cVL in CD4+ cells from patients 2, 3, and 5 is underestimated compared to the cVL in the other patients and the cVL obtained from the same patients' total PBMC. Thus, analysis of circulating HIV DNA in CD4+ cells selected by a simple positive bead selection method may fail to detect HIV DNA, particularly cVL in some circulating cell reservoirs that are not selected by our anti-CD4 magnetic bead protocol, and we chose to perform all further analysis on total PBMC.
TABLE 2.
Quantitation of cVL and iVL in CD4+ cells does not reflect measurements in total PBMC isolated from the same patient samplesa
| Patient ID | cVL
|
iVL
|
||
|---|---|---|---|---|
| PBMC | CD4+ | PBMC | CD4+ | |
| 1 | 414 ± 32 | 204 ± 5 | ND | 36.1 ± 0.9 |
| 2 | 1,584 ± 262 | 137 ± 34 | 3.1 | 5.9 ± 2 |
| 3 | 3,540 ± 40 | 916 ± 17 | 18.1 ± 1 | 37.4 ± 4 |
| 4 | 3,485 ± 180 | 8,054 ± 1,755 | 4.1 ± 0.2 | INV |
| 5 | 2,011 ± 401 | 165 ± 32 | 12 ± 0.3 | 62.8 ± 3 |
Values represent HIV DNA copy numbers/105 PBMC or equivalent numbers of CD4+ cells calculated to be in 105 patient PBMC, based on CD4 counts. ND, not detected or below the limit of the assay; INV, invalid (result from Alu-minus control PCR greater or equal to Alu PCR).
iVL and cVL can be quantitated in total PBMC from specimens collected for clinical pVL analysis.
We aimed to assess whether measurements of both cVL and iVL could be performed on the same sample collected for routine clinical pVL measurement. Specimens from 46 patients presenting to the IMVS Serology Unit for routine pVL determination were obtained with informed consent. Patient details are shown in Table 3. After removal of the plasma for the pVL assay, the buffy coat layer was obtained and total PBMC were isolated. Total DNA was extracted, the cell number was normalized by analysis of β-globin, and cVL and iVL were quantitated. Samples of defined high and low HIV DNA concentrations were assayed in each run, and coefficients of variation (CV) were determined to be 30.4% ± 8.0% and 6.8% ± 1.7%, respectively, for inter- and intra-assay variation in measurement of iVL and 21% ± 5.8% and 19% ± 3.9%, respectively, for inter- and intra-assay variation in measurement of cVL. Additionally, a single patient sample assayed in three different Alu PCRs yielded an interassay CV of 37%. Multiple patient samples (n = 19) assayed in duplicate cVL assays yielded an interassay CV of 23% ± 3%. Thus, variation in interassay measurement of patient samples was similar to that of the reference samples used.
TABLE 3.
Clinical details of patientsa
| Patient ID | pVL (copies/ml) | Time (mo) | % CD4+ CD3+ | CD4 count | cVL (copies/105 cells) | iVL (copies/105 cells) | Therapyb | Other drug therapyc | Therapy outcomed |
|---|---|---|---|---|---|---|---|---|---|
| 1 | <400 | 25 | 12 | 465 | 414 | ND | TRIZ, EFV | ||
| 2 | <50 | >81 | 33 | 971 | 1,584 | 3 | |||
| 3 | <50 | >16 | 26 | 327 | 3,540 | 18 | COM, K | Y | |
| 4 | <400 | 80 | 24 | 410 | 3,485 | 4 | |||
| 5 | <400 | 57 | 29 | 641 | 2,011 | 12 | COM, NVP | ||
| 6 | <400 | 29 | 8 | 196 | 32 | 14 | ABC, 3TC, NVP | ||
| <400 | 297 | ND | ND | ABC, 3TC, NVP | |||||
| <40 | 12 | 354 | 287 | 21 | ABC, 3TC, NVP | ||||
| 7 | <400 | >16 | 35 | 651 | 47 | 36 | 3TC, STAV, NVP | ||
| <40 | 767 | 87 | 66 | 3TC, EFV, ABC | |||||
| <40 | 36 | ND | 3TC, EFV, ABC | ||||||
| 8 | <400 | 53 | 26 | 532 | 5,110 | 10 | COM, NVP | Y | |
| <400 | 57 | 29 | 641 | 2,011 | 60 | COM, NVP | |||
| 9 | <400 | 77 | 27 | 327 | 11,608 | 3 | |||
| <400 | 80 | 24 | 410 | 3,485 | 4 | ||||
| 10 | <50 | 18 | 16 | 325 | 2,535 | 38 | COM, K | F | |
| 11 | 35,500 | 14 | 473 | 18 | 8 | ||||
| 12 | <50 | >24 | 26 | 575 | 170 | INV | EFV, COM | ||
| <40 | >30 | 29 | 612 | 99 | 16 | EFV, COM | |||
| 13 | <400 | 222 | INV | ||||||
| 14 | <50 | >69 | 18 | 173 | 164 | ND | |||
| 15 | 205,000 | 229 | INV | ||||||
| 16 | 800 | 30 | 457 | 382 | INV | COM, EFV | |||
| 17 | <400 | 68 | 10 | 243 | 9 | ND | 3TC, TDF, EFV | Y | |
| 18 | <50 | 21 | 527 | 47 | 76 | TRIZ, NVP | Y | ||
| 19 | <50 | >10 | 13 | 351 | 226 | 211 | TDF, 3TC, EFV | Y | F |
| 20 | <400 | 22 | 13 | 433 | 8,584 | INV | TRIZ, EFV | Y | |
| 21 | 87,000 | 13 | 103 | 129 | 36 | NVP, COM | Y | ||
| 22 | <400 | >24 | 38 | 26 | ND | COM, NVP | |||
| <40 | >18 | 36 | 1,270 | 73 | 6 | COM, NVP | |||
| 23 | <400 | 19 | 37 | 1,059 | 27 | 43 | AZT, 3TC, NVP | F | |
| 24 | 10,000 | 14 | 321 | 1,714 | INV | 3TC, STAV | Y | ||
| 25 | 1,700 | 22 | 403 | 6 | ND | TRIZ | |||
| 210 | 36 | 35 | TRIZ, ATAZ | F | |||||
| 26 | <50 | >78 | 22 | 437 | ND | ND | |||
| 27 | 89,000 | 14 | 112 | ND | 3TC, K, TDF | Y | |||
| 47,000 | 13 | 151 | 43 | ND | 3TC, K, TDF | Y | |||
| 28 | <50 | 22 | 31 | 357 | 152 | ND | COM, EFV | ||
| 29 | <400 | >78 | 8 | 278 | 156 | INV | |||
| 30 | 1,900 | 12 | 107 | 10 | INV | COM, EFV | Y | ||
| 110 | >11 | 14 | 287 | 9,458 | INV | EFV, TRIZ, RTV | |||
| >12 | 13 | 310 | ND | ND | COM, EFV | ||||
| 31 | 4,000 | 16 | 347 | 123 | INV | ||||
| 32 | <400 | 42 | 18 | 234 | 30,481 | 37 | |||
| <400 | 45 | 21 | 305 | 1,176 | 7 | ||||
| 33 | 800 | 26 | 531 | 247 | 102 | 3TC, K, TDF | |||
| 34 | <50 | >81 | 33 | 971 | 1,584 | 9 | |||
| 35 | <400 | 45 | 52 | 1,088 | 19 | ND | |||
| 36 | 2,300 | 30 | 540 | 10 | ND | TDF, 3TC, K | |||
| 37 | <400 | 35 | 19 | 307 | 10 | INV | AZT, 3TC, NVP | Y | |
| 38 | 27,000 | 41 | 628 | 88 | 84 |
ND, not detected or below the limit of the assay; INV, invalid (result from Alu-minus control PCR greater or equal to Alu PCR).
TRIZ, Trizivir; EFV, efavirenz; COM, Combivir; K, Kaletra; NVP, nevirapine; ABC, abacivir; 3TC, lamivudine; STAV, stavudine; TDF, tenofovir; RTV, ritonavir; ATZ, atazanavir.
A Y indicates that other drug therapy included antidepressant, antifungal, and antibacterial drugs.
An F indicates that at 12 months postmeasurement, treatment had failed.
cVL was detected in almost all samples regardless of pVL or CD4 count (Table 4), whereas iVL could only be reliably detected in 47.8% of samples. iVL detection was most efficient in samples with CD4 counts of >350 (55.6%) and was least efficient where the pVL was >400 copies/ml (30.8%). This was largely due to higher levels (38.5%) of invalid measurements caused by a high Alu-minus control, most likely representing the higher levels of unintegrated DNA and elevated levels of ongoing viral replication in patients with pVL of >400 copies/ml. However, the differences in iVL detection between different pVL or CD4 count groupings were small, and larger sample numbers, particularly for pVL of >400 copies/ml, need to be assayed to support any significant differences. More patients with pVL of <400 copies/ml had undetectable cVL than those with pVL of >400 copies/ml, although again differences and sample numbers were small. Patients with undetectable iVL were present in both pVL groupings (Table 4). Ranges and average values for cVL and iVL measurements are shown in Table 4.
TABLE 4.
Characteristics of cVL and iVL measurements in patient samples
| cVL or iVL measurement | % Detected | % not Detected | % Invalid | Avg viral load (copies/105 cells) ± SEM | Viral load range (copies/105 cells) |
|---|---|---|---|---|---|
| cVL | |||||
| Total | 93.6 | 6.3 | 0 | 444.3 ± 41.3 | 6.3-11,608.4 |
| With pVL of <400 copies/ml | 91.2 | 8.8 | 0 | 2,131 ± 966.8 | 8.5-11,608.4 |
| With pVL of >400 copies/ml | 100 | 0 | 0 | 958.5 ± 719.5 | 9.5-9,458 |
| With CD4 count of <350 | 94.1 | 5.9 | 0 | 3,599.5 ± 1,875.2 | 8.5-11,608.4 |
| With CD4 count of >350 | 92.6 | 7.4 | 0 | 861.2 ± 375.7 | 6.3-8,584.6 |
| iVL | |||||
| Total | 47.8 | 26.1 | 21.7 | 42.4 ± 10.1 | 3.1-211.7 |
| With pVL of <400 copies/ml | 52.9 | 23.5 | 23.5 | 27 ± 7.6 | 3.1-211.7 |
| With pVL of >400 copies/ml | 30.8 | 30.8 | 38.5 | 28.8 ± 11.6 | 7.8-102.2 |
| With CD4 count of <350 | 47 | 29.4 | 23.5 | 15 ± 4.5 | 3.1-37.7 |
| With CD4 count of >350 | 55.6 | 22.2 | 22.2 | 35.8 ± 9.9 | 4.1-211.7 |
iVL and cVL do not correlate with pVL or CD4 count, but iVL may reflect clinical outcome.
We next assessed whether cVL or iVL correlated with each other or with clinical parameters such as pVL and CD4 count. Using nonparametric paired statistical analysis, there was a significant positive correlation between cVL and iVL (R = 0.52, P = 0.0015) (Fig. 2A). In contrast, no correlation between pVL and cVL or iVL was found, and no significant difference was observed in cVL or iVL between low-pVL (<400 copies/ml) or high-pVL (>400 copies/ml) groupings (Fig. 2B and C). Similarly, no correlation between CD4 counts and cVL or iVL was observed, and no significant difference was observed in cVL or iVL between low-CD4-count (<350) or high-CD4-count (>350 copies/ml) groupings (Fig. 2D and E).
FIG. 2.

cVL and iVL values are not associated with pVL or CD4 count. (A) Correlation of cVL and iVL. (B) Relationship between cVL and pVL. Patients with pVL of <400 copies/ml (c/ml) for whom therapy failed within the subsequent 12 months are indicated by open circles. (C) Relationship between iVL and pVL. Patients with pVL of <400 for whom therapy failed within the subsequent 12 months are indicated by black circles. (D) Relationship between cVL and CD4 count. (E) Relationship between iVL and CD4 count. No significant relationships were observed for panels B to E by Spearman's ρ test or ANOVA. (F) Relationship between the cVL-to-iVL ratio and pVL or CD4 count. ANOVA and Wilcoxon testing showed a significant difference between the cVL-to-iVL ratio and CD4 count (P = 0.04). Values represent averages ± standard errors of the means (SEM) (two to four measurements). INV, invalid.
The ratio of cVL to iVL tended to be higher in patients with low pVL (<400 copies/ml; P = 0.25), perhaps suggesting that in the presence of effective nucleoside reverse transcriptase inhibitor (NRTI) and non-nucleoside reverse transcriptase inhibitor (NNRTI) therapy there is ongoing but restricted reverse transcription, resulting in relatively less integrated virus. The ratio of cVL to iVL was significantly higher in patients with low CD4 counts (<350; P = 0.04) (Fig. 2F), perhaps reflecting that circulating CD4 reservoirs harbor higher ratios of unintegrated to integrated HIV DNA compared to CD4+ cells. Of the patients analyzed the majority were on combination NRTI/NNRTI therapy, with only seven receiving protease inhibitors. Although there was an unequal distribution in treatment groups, no significant difference in cVL or iVL was seen between patients on NRTI/NNRTI therapy and those on regimes containing protease inhibitors (data not shown).
Measurement of cVL and iVL has the potential to be of value for patients with undetectable pVL (<400 or <50 copies/ml), where additional data reflecting ongoing viral replication could assist our understanding of progression of infection. For patients with undetectable pVL and for whom cVL (n = 37) and iVL (n = 26) were quantitated, pVL and CD4 counts were monitored for the subsequent 12 months. During this time therapy for four patients failed as measured by increased pVL (Fig. 2B and C; Table 3). Elevated cVL (average value, >2,000 copies/105 cells [Table 4]) was not associated with therapy failure with broad cVL for patients failing therapy, and only one out of nine patients with cVL of >2,000 copies/105 cells failing therapy in the subsequent 12 months. All four patients for whom therapy failed had higher than average iVL values (>27 copies/105 cells [average values are shown in Table 4]), including the patient with the highest iVL observed. Seven out of eleven patients with iVL of >27 copies/105 cells and 15/15 patients with iVL of <20 copies/105 cells were maintained on suppressive therapy, suggesting that patients with iVL of <20 copies/105 cells were less likely to fail therapy within the next 12 months than patients with higher iVL values. However, more patient numbers are needed to substantiate any prognostic value of iVL measurements and determine critical values above which therapy failure becomes probable.
Extended duration of successful therapy is not associated with lower iVL and cVL.
For those samples with undetectable pVL, we grouped patients based on the length of time that pVL had remained consistently undetectable (<400 or <50 copies/ml for <30 months, 30 to 60 months, or >60 months [5 years]). No significant difference in cVL or iVL was associated with with extended duration of undetectable pVL (Fig. 3A and B). However, there was a trend towards a higher ratio of cVL to iVL with increased duration of undetectable pVL (Fig. 3C) (P = 0.32), consistent with results shown in Fig. 2F and potential restricted reverse transcription, resulting in relatively less integrated virus than incomplete unintegrated DNA forms with long-term successful antiretroviral therapy.
FIG. 3.
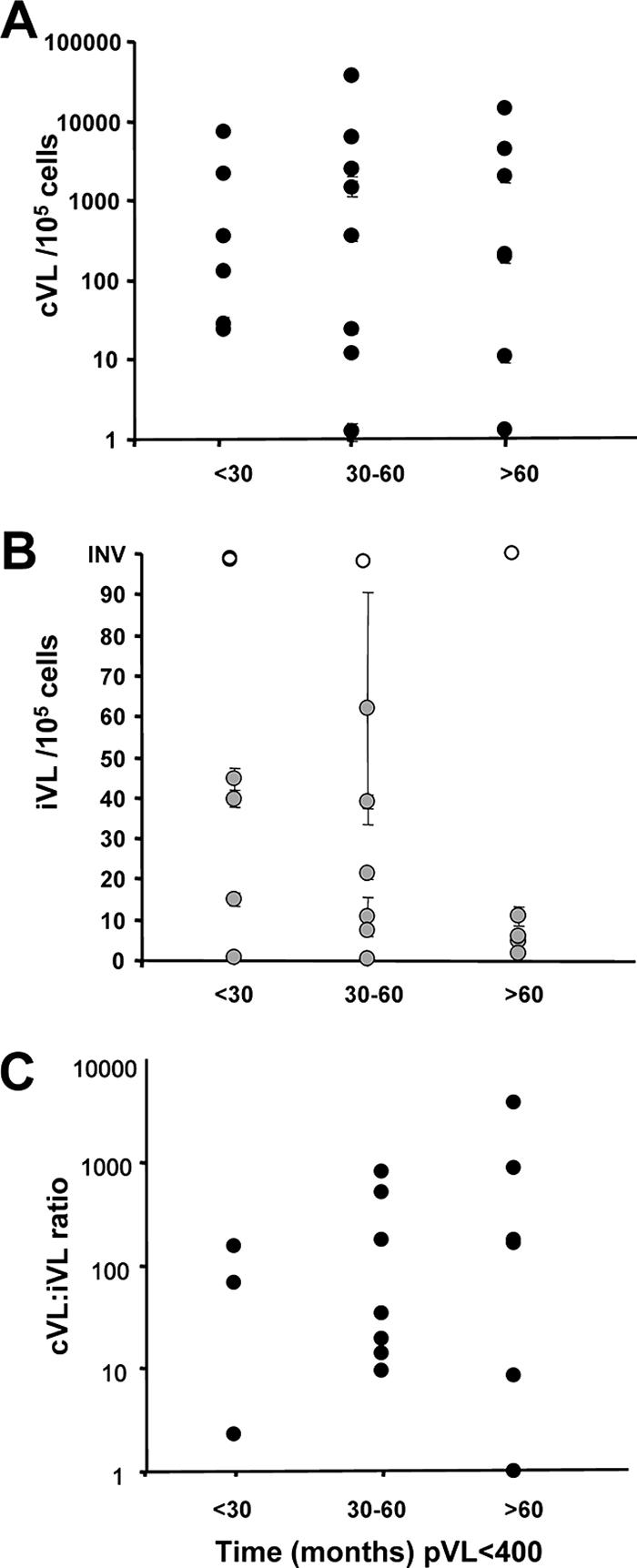
cVL and iVL levels do not differ with extended duration of undetectable pVL. Patients with pVL of <400 were grouped based on length of viral suppression prior to measurement of cVL or iVL. (A) cVL. (B) iVL. (C) cVL-to-iVL ratio. No significant difference between groups was observed by ANOVA and Kruskal-Wallis testing. Values represent averages ± SEM (two to four measurements). INV, invalid.
We also measured cVL and iVL in samples taken quarterly from nine patients with undetectable pVL. During the time when pVL was consistently suppressed, a consistent decline in cVL and iVL was not observed. Several patterns of cVL and iVL changes were observed: concordant cVL and iVL varying with time (Fig. 4A and B), discordant cVL and iVL declining and increasing with time, respectively (Fig. 4C), and discordant cVL and iVL declining or remaining constant with time (Fig. 4D). These results demonstrate variation in cVL and iVL without detectable changes in pVL.
FIG. 4.

cVL and iVL vary over time in samples taken serially from the same patient with undetectable pVL. Nine patients with pVL of <400 copies/ml were sampled at quarterly intervals at two or three time points, and cVL and iVL were quantitated. Results for four patients representing generalized concordant or discordant responses are presented. Values represent averages ± SEM (two to four measurements).
DISCUSSION
The persistence of HIV DNA in patients on HAART with long-term suppression of viremia has led to the proposal that monitoring of this reservoir may provide useful information for clinical management. We have previously reported the development and application of a laboratory assay to measure integrated proviral HIV DNA (18, 27, 28) and applied this to a limited number of cell-fractionated patient samples (6). In this study we have further adapted this assay to a real-time PCR format and to PBMC isolated from patient specimens that are collected at the clinic for routine pVL analysis. In our assay we utilize HIV DNA standards that are derived from a mix of three chronically infected cell lines. These cell lines harbor four defined integration sites that range in distance from 3.6 kb up to >20 kb from genomic Alu elements (18). At an average distribution of Alu elements of 5 to 8 kb and with HIV integration reported to occur close to Alu elements (23), our integration standards should encompass the maximum Alu integration distances and reflect the less efficient long-range PCR amplification likely to be encountered in an HIV patient sample. Under our assay conditions these integration standards perform well, giving good discrimination between integrated and unintegrated HIV DNA (Table 1). We prefer to use this standard over a random integrated DNA population (3, 20) with potentially more diverse integration sites because of the stability of the cell lines and the reliability of regenerating standard DNA preparations with defined integration sites for long-term future assays. The use of any integrated DNA standard would influence absolute iVL values, but the relative values between samples and the study conclusions would remain unchanged. The future adaptation of our assay to use an alternative random integrated DNA standard that would potentially yield a more precise iVL value needs to be considered and weighed against the technical ease and reliability of the use of stable chronically infected cell lines as used herein.
Using a single specimen per patient collected for pVL analysis, we measured both iVL and cVL in a patient cohort encompassing high, intermediate, and undetectable pVL. One limitation of using the 10-ml sample presented for routine pVL testing is that the majority of samples only yielded enough material for concurrent measurement of β-globin, cVL, and iVL if duplicate rather than multiple measurements were taken in each assay. iVL could not be reliably quantitated in all samples: 26% of samples were below the limit of detection of the assay, and 21.7% of samples yielded invalid measurements due to high levels of unintegrated DNA forms. Thus, the assay is also limited in its ability to detect integrated HIV DNA at low levels of cellular integration and high levels of unintegrated DNA. Similarly, in other studies iVL could not be detected in all samples and was undetectable in 3/11 (27%) of CD4+ lymphocyte samples and 10/11 (91%) of monocyte samples (5) and 5/23 (21%) of CD4+ lymphocyte samples (6). Undetectable iVL did not correlate with low pVL (<400 copies/ml). The use of the CD4+ cell fraction for analysis would potentially increase the percentage of cells containing HIV DNA and improve the iVL assay, but in our study measurements of HIV DNA in CD4+ cells selected by a simple positive bead isolation protocol did not reflect the total cell-associated HIV DNA content in PBMC (Table 2). Previous work in our laboratory suggested that a simple positive anti-CD4 magnetic bead selection protocol such as ours does not quantitatively isolate CD4+ cells, in particular low-level CD4+ cells such as CD14+ cells and potentially also HIV-infected cells with downregulated CD4 surface expression. The majority of the literature has suggested that CD4+ cells are an important circulating reservoir for HIV replication in patients on HAART (13), although work from our laboratory and others has detected significant amounts of HIV DNA in CD4− CD8− T cells (6, 7) or in monocytes/macrophages isolated from patients (11, 26), and other non-CD4 reservoirs that contribute to viral replication in the presence of HAART have been suggested (8). In combination these two points are likely reasons for the discrepancies between our measurements in the CD4+ and PBMC pools. Thus, our protocol measuring HIV DNA in total PBMC has favored using the simplest cell and DNA extraction procedure to obtain concurrent measurements of cVL and iVL in cells that are most representative of the circulating pool of HIV DNA.
In our study we have used real-time PCR measurement of total cell-associated DNA (cVL) and real-time Alu PCR for quantitation of integrated HIV DNA (iVL) in PBMC from 46 patient specimens. Other studies have developed real-time PCR-based assays for HIV DNA and applied these to patient samples, but several of these assays measure total cell-associated DNA consisting of integrated and unintegrated forms and are not specific for integrated or proviral DNA forms (10, 14, 17, 19). Analyzing total HIV DNA, previous studies have observed a correlation in the decline of cVL and pVL at the onset of HAART, but following this initial decline, during established HAART therapy, there is no association between cVL and pVL or CD4 count (1, 15, 16). Similarly, in our cross-sectional study we did not observe a correlation between cVL and pVL or CD4 counts.
Alu PCR methods with Southern detection of PCR products to specifically measure integrated HIV DNA have been described and applied to analyze circulating HIV cell reservoirs (9, 11) or in real-time PCR format for analysis of HIV replication in vitro (2, 20). Using an Alu PCR followed by quantitative gel analysis, iVL in PBMC taken from 10 patients after the introduction of HAART (15) and in CD4+ T-cell and monocyte fractions from 11 patients after 2 years of HAART treatment have been assessed (5). These reports, and the present study using a real-time Alu PCR quantitative format, found no correlation between iVL and either pVL or CD4 count.
In a cross-sectional study of patients with extended suppressed pVL we found no correlation between declining cVL or iVL and time, similar to the results found by Ibanez et al. (15) after a shorter period of HAART and Izopet et al. (16), who demonstrated an initial decline in iVL after 15 months of HAART but no further decline after 15 to 24 months. In our study, even patients with consistent clinical suppression of pVL lasting longer than 5 years still harbored measurable circulating cell-associated and integrated HIV DNA. Previous reports from patients maintained on successful HAART for up to 30 months (13) or 2 years (5) also found no decline in the frequency of infected resting CD4+ T cells or total HIV DNA in CD4+ cells with time, and Chun et al. (10) have recently reported detectable total cell-associated HIV DNA in CD4+ T cells with up to 9 years of suppressed viremia. In a longitudinal analysis of nine patients in the present study with long-term suppression of pVL (all lasting >12 months and up to 80 months), we found no consistent decline in cVL or iVL in PBMC over time. Some patients showed concordant changes, with iVL and cVL varying together with time and supporting the correlation between these two parameters (Fig. 2A). Other patients, however, showed discordant changes, with declining cVL associated with increasing or static iVL. Thus, although there is a population correlation between iVL and cVL (Fig. 2A), the discordant changes in cVL and iVL within the same patient in some instances suggest that distinct factors can influence iVL and cVL, and the ratio of cVL to iVL might prove to be an important parameter to consider. Thus, our results extend previous reports of the presence of long-term HIV DNA in CD4+ T cells and demonstrate the existence, maintenance, and variation in levels of the detectable circulating reservoir of HIV DNA (cVL and iVL) in PBMC from patients with long-term viral suppression.
Our assay is potentially of most value for monitoring changes in patients with undetectable pVL during the phase of clinical latency. cVL levels were not related to the onset of clinical failure as measured by viremia or declining CD4 counts within the subsequent 12 months, and thus there is little prognostic value in measurements of cVL. However, all four patients in our cohort for whom therapy failed had higher than average iVL values, and therapy remained successful for all patients with lower than average iVL values. Although more data are needed to make a convincing and significant association between iVL and therapy failure, the results suggest that iVL measurements in the total HIV DNA pool have a potential prognostic value for clinical outcomes.
In conclusion, we have developed an assay that can be applied to routinely collected plasma samples to coordinately analyze pVL, iVL, and cVL from the same clinical specimen. We suggest that it may be better to use total PBMC as a pool for analysis than CD4+ cells to avoid missing the contribution of circulating CD4− or CD4low populations that may contain significant HIV DNA in some situations. Measurement of circulating total cell-associated DNA or integrated HIV DNA did not correlate with CD4 count, pVL, or duration of effective therapy. While both cVL and iVL varied with duration of suppressive therapy, and the ratio of these two measurements may be informative, results suggest that measurement of only iVL may be predictive of subsequent clinical outcomes.
Acknowledgments
We thank the members of IMVS, IDL Serology, particularly Sue Weir for collection of patient samples, Lyndall Daly for providing clinical data, and the patient volunteers for willingly providing specimens. We also thank Adrian Purins for maintenance of cell stocks and Thomas Sullivan for statistical analysis.
This work was supported by the Australian Centre for HIV and Hepatitis Research (ACH2).
Footnotes
Published ahead of print on 21 February 2007.
REFERENCES
- 1.Brussel, A., D. Mathez, S. Broche-Pierre, R. Lancar, T. Calvez, P. Sonigo, and J. Leibowitch. 2003. Longitudinal monitoring of 2-long terminal repeat circles in peripheral blood mononuclear cells from patients with chronic HIV-1 infection. AIDS 17:645-652. [DOI] [PubMed] [Google Scholar]
- 2.Brussel, A., and P. Sonigo. 2003. Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J. Virol. 77:10119-10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler, S. L., M. S. Hansen, and F. D. Bushman. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631-634. [DOI] [PubMed] [Google Scholar]
- 4.Butler, S. L., E. P. Johnson, and F. D. Bushman. 2002. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J. Virol. 76:3739-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calcaterra, S., G. Cappiello, A. Di Caro, A. R. Garbuglia, and A. Benedetto. 2001. Comparative analysis of total and integrated HIV-1 DNA in peripheral CD4 lymphocytes and monocytes after long treatment with HAART. J. Infect. 43:239-245. [DOI] [PubMed] [Google Scholar]
- 6.Cheney, K. M., R. Kumar, A. Purins, L. Mundy, W. Ferguson, D. Shaw, C. J. Burrell, and P. Li. 2006. HIV type 1 persistence in CD4−/CD8− double negative T cells from patients on antiretroviral therapy. AIDS Res. Hum. Retrovir. 22:66-75. [DOI] [PubMed] [Google Scholar]
- 7.Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y. H. Kuo, R. Brookmeyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183-188. [DOI] [PubMed] [Google Scholar]
- 8.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. Shawn Justement, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. [DOI] [PubMed] [Google Scholar]
- 9.Chun, T. W., D. Finzi, J. Margolick, K. Chadwick, D. Schwartz, and R. F. Siliciano. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1:1284-1290. [DOI] [PubMed] [Google Scholar]
- 10.Chun, T. W., D. C. Nickle, J. S. Justement, D. Large, A. Semerjian, M. E. Curlin, M. A. O'Shea, C. W. Hallahan, M. Daucher, D. J. Ward, S. Moir, J. I. Mullins, C. Kovacs, and A. S. Fauci. 2005. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J. Clin. Investig. 115:3250-3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler, A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94:13193-13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T. Pierson, K. Smith, J. Lisziewicz, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5:512-517. [DOI] [PubMed] [Google Scholar]
- 13.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295-1300. [DOI] [PubMed] [Google Scholar]
- 14.Gibellini, D., F. Vitone, P. Schiavone, C. Ponti, M. La Placa, and M. C. Re. 2004. Quantitative detection of human immunodeficiency virus type 1 (HIV-1) proviral DNA in peripheral blood mononuclear cells by SYBR green real-time PCR technique. J. Clin. Virol. 29:282-289. [DOI] [PubMed] [Google Scholar]
- 15.Ibanez, A., T. Puig, J. Elias, B. Clotet, L. Ruiz, and M. A. Martinez. 1999. Quantification of integrated and total HIV-1 DNA after long-term highly active antiretroviral therapy in HIV-1-infected patients. AIDS 13:1045-1049. [DOI] [PubMed] [Google Scholar]
- 16.Izopet, J., M. Cazabat, C. Pasquier, K. Sandres-Saune, E. Bonnet, B. Marchou, P. Massip, and J. Puel. 2002. Evolution of total and integrated HIV-1 DNA and change in DNA sequences in patients with sustained plasma virus suppression. Virology 302:393-404. [DOI] [PubMed] [Google Scholar]
- 17.Kabamba-Mukadi, B., P. Henrivaux, J. Ruelle, N. Delferriere, M. Bodeus, and P. Goubau. 2005. Human immunodeficiency virus type 1 (HIV-1) proviral DNA load in purified CD4+ cells by LightCycler real-time PCR. BMC Infect. Dis. 5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar, R., N. Vandegraaff, L. Mundy, C. J. Burrell, and P. Li. 2002. Evaluation of PCR-based methods for the quantitation of integrated HIV-1 DNA. J. Virol. Methods 105:233-246. [DOI] [PubMed] [Google Scholar]
- 19.Lillo, F. B., M. A. Grasso, S. Lodini, M. G. Bellotti, and G. Colucci. 2004. Few modifications of the Cobas Amplicor HIV Monitor 1.5 test allow reliable quantitation of HIV-1 proviral load in peripheral blood mononuclear cells. J. Virol. Methods 120:201-205. [DOI] [PubMed] [Google Scholar]
- 20.O'Doherty, U., W. J. Swiggard, D. Jeyakumar, D. McGain, and M. H. Malim. 2002. A sensitive, quantitative assay for human immunodeficiency virus type 1 integration. J. Virol. 76:10942-10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierson, T. C., T. L. Kieffer, C. T. Ruff, C. Buck, S. J. Gange, and R. F. Siliciano. 2002. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J. Virol. 76:4138-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramratnam, B., J. E. Mittler, L. Zhang, D. Boden, A. Hurley, F. Fang, C. A. Macken, A. S. Perelson, M. Markowitz, and D. D. Ho. 2000. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6:82-85. [DOI] [PubMed] [Google Scholar]
- 23.Schroder, A. R., P. Shinn, H. Chen, C. Berry, J. R. Ecker, and F. Bushman. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521-529. [DOI] [PubMed] [Google Scholar]
- 24.Sharkey, M. E., I. Teo, T. Greenough, N. Sharova, K. Luzuriaga, J. L. Sullivan, R. P. Bucy, L. G. Kostrikis, A. Haase, C. Veryard, R. E. Davaro, S. H. Cheeseman, J. S. Daly, C. Bova, R. T. Ellison III, B. Mady, K. K. Lai, G. Moyle, M. Nelson, B. Gazzard, S. Shaunak, and M. Stevenson. 2000. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat. Med. 6:76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siliciano, J. D., J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange, and R. F. Siliciano. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9:727-728. [DOI] [PubMed] [Google Scholar]
- 26.Sonza, S., H. P. Mutimer, R. Oelrichs, D. Jardine, K. Harvey, A. Dunne, D. F. Purcell, C. Birch, and S. M. Crowe. 2001. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS 15:17-22. [DOI] [PubMed] [Google Scholar]
- 27.Vandegraaff, N., R. Kumar, C. J. Burrell, and P. Li. 2001. Kinetics of human immunodeficiency virus type 1 (HIV) DNA integration in acutely infected cells as determined using a novel assay for detection of integrated HIV DNA. J. Virol. 75:11253-11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandegraaff, N., R. Kumar, H. Hocking, T. R. Burke, Jr., J. Mills, D. Rhodes, C. J. Burrell, and P. Li. 2001. Specific inhibition of human immunodeficiency virus type 1 (HIV-1) integration in cell culture: putative inhibitors of HIV-1 integrase. Antimicrob. Agents Chemother. 45:2510-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitone, F., D. Gibellini, P. Schiavone, and M. C. Re. 2005. Quantitative DNA proviral detection in HIV-1 patients treated with antiretroviral therapy. J. Clin. Virol. 33:194-200. [DOI] [PubMed] [Google Scholar]