

Abstract
The capabilities of genome-scale metabolic networks can be described through the determination of a set of systemically independent and unique flux maps called extreme pathways. The first study of genome-scale extreme pathways for the simultaneous formation of all nonessential amino acids or ribonucleotides in Helicobacter pylori is presented. Three key results were obtained. First, the extreme pathways for the production of individual amino acids in H. pylori showed far fewer internal states per external state than previously found in Haemophilus influenzae, indicating a more rigid metabolic network. Second, the degree of pathway redundancy in H. pylori was essentially the same for the production of individual amino acids and linked amino acid sets, but was approximately twice that of the production of the ribonucleotides. Third, the metabolic network of H. pylori was unable to achieve extensive conversion of amino acids consumed to the set of either nonessential amino acids or ribonucleotides and thus diverted a large portion of its nitrogen to ammonia production, a potentially important result for pH regulation in its acidic habitat. Genome-scale extreme pathways elucidate emergent system-wide properties. Extreme pathway analysis is emerging as a potentially important method to analyze the link between the metabolic genotype and its phenotypes.
Access to the complete genome of an organism provides a basis for studying cellular processes as a whole. Organism-level metabolic modeling is a “genome-enabled” science; without a sequenced genome, organism-level modeling is essentially impossible. The sequencing of entire genomes has given us a “parts list” for a cell, and now the challenge is to integrate those parts to understand the mechanisms and organization by which cells use these parts to achieve their phenotypic expressions. Extensive cataloging of biological components has enabled the reconstruction of genome-scale models of cellular metabolism (Karp et al. 1996; Selkov et al. 1998; Overbeek et al. 2000; Covert et al. 2001). Metabolism involves the production of mass, energy, and redox requirements for all cellular functions, and thus provides the driving force for cellular activity. As one of the most thoroughly studied aspects of cellular function, it affords the best opportunity for the development of methodologies to characterize and analyze systems-level cellular properties of genome-scale models.
A metabolic network consists of the group of reactions and transport processes associated with the production and depletion of cellular metabolites. Using genomic, biochemical, and physiological data, the metabolic pathways and transporters known to exist in an organism can be modeled as a network with a specific environment (Fig. 1A). Exchange fluxes that cross system boundaries are defined as input and output fluxes. The stoichiometric matrix concisely represents this information in mathematical form, with the rows corresponding to all the metabolites in the system and the columns representing all of the known biochemical reactions and transporters in the system.
Figure 1.

A sample biochemical reaction network (A) and the convex representation of its metabolic possibilities (B). The extreme pathways (EPs) in B correlate with those in A, serving to represent the relationship between extreme pathways of a biochemical reaction network and the characterization of the possible phenotypes.
Metabolic pathways are commonly used as a means of simplifying the description and analysis of metabolic networks. Pathways have different definitions in the literature. Most commonly, however, metabolic pathways are defined as a linked set of biochemical reactions, in which the product of one reaction is the reactant of the subsequent reaction in the chain (Bono et al. 1998; Ouzounis and Karp 2000; Karp 2001; Tsoka and Ouzounis 2001). With this definition, glycolysis, the Krebs cycle, and the pentose phosphate pathway are referred to as metabolic pathways. Segmenting a network into pathways is generally performed based on historical discovery, conceptual understanding, or certain heuristics. Extreme pathways, studied herein, are mathematically defined metabolic pathways calculated from a reconstructed metabolic network.
Extreme pathways differ from other pathway definitions in that they not only are sets of connected reactions but also describe the functions of an entire metabolic network. Extreme pathways are to heuristically defined pathways what traffic patterns are to roads: Although knowledge of a road network can assist in evaluating how to get from one location to another, a knowledge of traffic patterns (dependent on weather, time, and road conditions) can more completely assess the capability of traveling (Covert et al. 2001). Heuristically defined pathways are linked reaction sets (roads), whereas extreme pathways describe the fluxes of molecules undergoing each reaction (flow of traffic along the roads).
Extreme pathways are flux maps through a biochemical network that characterize the functioning of the network (Fig. 1A); in other words, extreme pathways account for the relative magnitude of the number of molecules undergoing each reaction in the cell. Extreme pathways have the following important characteristics: (1) they are generated for steady-state condition with no metabolite build-up allowed within the system, enabling the analysis of time-invariant properties related to the structure of the metabolic network; (2) they are a unique and minimal set of flux maps that can describe the network and are determined directly from the stoichiometric matrix; (3) extreme pathways, although contiguous, can have multiple inputs and outputs; and (4) the extreme pathways circumscribe all potential steady-state flux maps through the network.
Extreme pathways can be represented graphically as the edges of a convex cone (Fig. 1B). Because all valid solutions are non-negative linear combinations of the extreme pathways (property 4), the convex cone circumscribes all potential metabolic phenotypes (i.e., all valid steady-state solutions of the network must lie within the cone). Thus, the extreme pathways characterize the extreme functions of the network; the network cannot attain any steady-state yields greater than that of the maximum-yield extreme pathways. Extreme pathway analysis of metabolic networks presents a mathematically rigorous approach to systems-level analyses. At a systems-level, emergent properties develop that can be determined by looking only at large-scale interactions rather than at individual components. Many other methods have been used to study cellular metabolism, including flux balance analysis (Varma and Palsson 1994; Bonarius et al. 1997; Edwards et al. 1999), metabolic control analysis (Kacser and Burns 1973; Fell 1996), and various types of pathway analysis (Mavrovouniotis and Stephanopoulos 1990; Liao et al. 1996; Karp et al. 1999; Schilling et al. 1999; Schuster et al. 1999; Küffner et al. 2000; Schilling et al. 2000). A detailed review of the development of extreme pathway analysis has been published elsewhere (Schilling et al. 2000).
In this study, we analyzed the extreme pathway structure of the H. pylori reconstructed metabolic network. This pathogen inhabits the gastric lining of nearly one half of the world's population (Cover and Blaser 1996), with a disproportionately high occurrence of infection in developing countries (Bardhan 1997). It has received increasing interest for its role in various gastric-associated diseases, such as gastritis, peptic ulcers, and gastric cancer (Cover and Blaser 1996; Kelly 1998). The genome sequence of H. pylori was recently published for strains 26695 (Tomb et al. 1997) and J99 (Alm et al. 1999), enabling the reconstruction of its metabolic network and subsequent analysis. The in silico model used in this study is based on the genome sequence of H. pylori strain 26695 (Schilling 2000; C.H. Schilling, M.W. Covert, I. Famili, G.M. Church, J.S. Edwards, and B.O. Palsson, in review.).
Genome-scale extreme pathways were calculated for H. pylori using a previously described algorithm (Schilling et al. 2000). Previous work has also been performed in the analysis of the extreme pathways of the metabolism of H. influenzae for the production of individual amino acids (Papin et al. 2002). Herein, we present a genome-scale analysis of the H. pylori metabolic network. With this organism, we were able to analyze the production not only of a single biomass compound (as was performed in the H. influenzae study) but also of significant subsets of biomass constituents, approaching a more complete model of how a cell produces all of its biomass constituents simultaneously. We studied the production of the set of non-essential amino acids, the set of ribonucleotides, the production of individual amino acids under various conditions, and the effect of urea in amino acid production. These studies resulted in large numerical data sets that have been analyzed to provide physiologically important characterizations.
Definitions
A succinct definition of important terms that are used throughout the text is provided for clarity.
Core allowable inputs included alanine, arginine, adenine, phosphate, sulfate, oxygen, histidine, isoleucine, leucine, methionine, phenylalanine, valine, and thiamin. Allowable outputs included ammonia, carbon dioxide, and the target product (outputted amino acids, ribonucleotides). Figure 2 illustrates those inputs and outputs that were used by the model.
Figure 2.

Diagrammatic representation of the in silico strain of Helicobacter pylori and its used exchange fluxes. Other inputs from minimal medium (data not shown) were allowed, but only the inputs taken up by the system in this study are indicated. Carbon sinks include acetate, succinate, formate, and lactate. Target outputs include asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, lysine, proline, serine, threonine, tryptophan, tyrosine, the set of nonessential amino acids, and the set of nucleotides.
Case 1 is the core inputs and outputs with succinate as an additional allowed output; case 2, core inputs and outputs with succinate and acetate as additional allowed outputs; case 3, core inputs and outputs with succinate, acetate, and formate as additional allowed outputs; and case 4, core inputs and outputs with succinate, acetate, formate, and lactate as additional allowed outputs.
Exchange flux is the flux of a metabolite that crosses system boundaries. An input represents a substrate taken up by the cell, whereas an output can be thought of either as a molecule that is secreted or as a metabolite that is produced. An external state refers to the set of values for the exchange fluxes of a given extreme pathway. A flux map refers to the magnitudes of the fluxes through every reaction in a specific metabolic network, and the internal state is the flux map corresponding to an extreme pathway without the exchange fluxes. Pathway redundancy is a network property characterized by the degree to which multiple extreme pathways correspond to an identical external state (i.e., the number of internal states per unique external state), and the spectrum of yields is a rank-ordered representation of the yields for all pathways associated with the synthesis of the target product in this study.
Nitrogen fate is the final destination of the input nitrogen of the system, that is, whether it ends up in the target product or as secreted ammonia. Nonessential amino acids are the amino acids that can be produced metabolically in H. pylori (essential amino acids are constituents of the minimal medium). Target product is the output of interest, constrained to be positive, and yield is the moles of the target amino acid produced per mole of carbon that enters the system. In this study, carbon enters in the form of alanine (three carbon atoms) and arginine (six carbon atoms).
RESULTS
The extreme pathway structure of the reconstructed metabolic network of H. pylori was analyzed. The extreme pathways were calculated for the production of (1) the individual nonessential amino acids and ribonucleotides, (2) the equimolar set of nonessential amino acids, (3) the set of nonessential amino acids in physiologic Escherichia coli ratios (caused by the lack of data for amino acid composition in H. pylori), and (4) the equimolar set of ribonucleotides. These analyses resulted in large data sets, necessitating the development of an analytical framework to study them. The developed approaches elucidated important emergent properties of the defined metabolic network of H. pylori.
Minimal Medium and Outputs Considered
The in silico strain of H. pylori was allowed inputs that constituted the minimal medium for growth (Nedenskov 1994; Reynolds and Penn 1994; Schilling 2000; C.H. Schilling, M.W. Covert, I. Famili, G.M. Church, J.S. Edwards, and B.O. Palsson, in review). Urea and adenine were also allowable inputs for some cases. The H. pylori model was allowed up to seven outputs: the target product (i.e., amino acid, ribonucleotide), carbon dioxide, acetate, succinate, formate, lactate, and ammonia. Acetate, succinate, formate, and lactate have been previously determined as the significant carbon byproducts of amino acid metabolism in H. pylori (Stark et al. 1997). This combination of outputs allowed the in silico model to produce all of the amino acids. Figure 2 diagrams the exchange flux constraints implemented in this analysis. Substrates used by the model included alanine, arginine, adenine, oxygen, phosphate, and sulfate. Input fluxes that were allowed as part of the minimal medium, but which were never used for nonessential amino acid or ribonucleotide production, were histidine, isoleucine, methionine, phenylalanine, valine, and thiamin. These compounds are necessary for the cell and are taken up for growth, but do not participate in the biosynthesis of nonessential amino acids or ribonucleotides under tested conditions. Various cases with different allowed inputs or outputs were used in generating the extreme pathways for H. pylori. Each case is described in Definitions.
It has been established that adenine is not a necessary component of the medium in which H. pylori grows (Mendz 2001). For this study, however, it was included in the minimal medium for the synthesis of the ribonucleotides. The model was not allowed sufficient byproduct secretion to synthesize adenine de novo. Adenine was subsequently accounted for in the redundancy, nitrogen fate, and yield calculations for ribonucleotide synthesis.
Carbon-Nitrogen Substrates
It has been shown experimentally that in the presence of glucose and all amino acids, H. pylori completely consumed alanine, arginine, asparagine, aspartate, glutamine, glutamate, proline, and serine (Stark et al. 1997). Of this set, only alanine and arginine are components of the previously determined minimal medium. Our genome-scale extreme pathway analysis is in agreement with these data because the extreme pathways for amino acid and ribonucleotide production only used alanine and arginine from the available carbon and nitrogen sources.
An in silico analysis was performed to ascertain the degree of dependence on alanine and arginine as substrates for production of amino acids. Table 1 lists the number of extreme pathways for amino acid production that do not use either alanine or arginine as inputs. Only a few of these extreme pathways do not require alanine from the minimal medium. A notable exception is the extreme pathways associated with the production of proline. Production of proline always required the presence of arginine, but 11 of these pathways did not require alanine. On average, 25.3% of all of the extreme pathways for the production of the individual amino acids did not use arginine as an input. However, only 0.1% of the extreme pathways for individual amino acid production did not use alanine as an input. Thus, the extreme pathways necessary to characterize the metabolic network have a far greater dependence on alanine than arginine.
Table 1.
The Number of Extreme Pathways Associated with the Production of the Indicated Amino Acid that Do Not Use the Indicated Carbon and Nitrogen Substrate
| Amino acid produced | EPs without alanine input | EPs without arginine input |
|---|---|---|
| Asparagine | 0 | 103 (30.3%) |
| Aspartate | 0 | 155 (31.6%) |
| Cysteine | 0 | 248 (24.3%) |
| Glutamine | 1 (0.3%) | 75 (23.8%) |
| Glutamate | 1 (0.2%) | 140 (28.4%) |
| Glycine | 0 | 130 (34.5%) |
| Lysine | 0 | 152 (24.9%) |
| Proline | 11 (1.3%) | 0 |
| Serine | 0 | 103 (29.0%) |
| Threonine | 0 | 115 (24.5%) |
| Tryptophan | 0 | 534 (27.3%) |
| Tyrosine | 0 | 248 (24.6%) |
These calculations are performed under case 4 conditions. The percentages are based on the total number of extreme pathways for the production of an individual amino acid. EPs indicates extreme pathways.
Biomass Subsets and Nitrogen Fates
The metabolic network of H. pylori allowed the calculation of the extreme pathways for the production of multiple subsets of various constituents of the biomass. We computed the extreme pathways for the production of the set of nonessential amino acids in equimolar ratios. This equimolar set was also analyzed with urea as an allowable input of the metabolic network. The extreme pathways were also calculated for a different set of fixed ratios. The amino acid composition of H. pylori protein was not available, so we used the amino acid composition of E. coli protein (Neidhardt and Umbarger 1996) as an approximation. The relative amount of amino acids in H. pylori protein was represented to one significant figure (asn-2, asp-2, cys-1, gln-2, glu-2, gly-6, lys-3, pro-2, ser-2, thr-2, trp-1, tyr-1); more precise ratios were computationally unfeasible. Although the ratios used were not ideal, they did help elucidate the extent to which the extreme pathways changed as the ratio of amino acid demand changed. We also analyzed extreme pathways involved in the production of the ribonucleotide set in equimolar ratios.
Equimolar Amino Acid Production by H. pylori
Because the metabolic network was unable to produce glycine under the conditions represented in cases 1 and 2 (Table 2), the equimolar nonessential amino acid set had a flux of zero for these cases. However, with the addition of formate and lactate as allowable outputs in cases 3 and 4, respectively, the network was capable of producing the complete equimolar set of nonessential amino acids (Table 3). For the production of the individual amino acids in case 3, there were 6916 extreme pathways required to characterize the solution space. For the production of the equimolar amino acid set under otherwise identical conditions, there were 4991 extreme pathways needed to characterize the solution space. Thus, 1925 fewer extreme pathways were needed to characterize the solution space for the production of the set of amino acids than were needed to characterize the production of all of the amino acids independently. Under the system constraints in case 4, 6032 extreme pathways characterized the network for the production of an equimolar amino acid set, which was 2274 fewer pathways than those for the independent production of the amino acids under similar conditions. Thus, setting the amino acid composition that must be produced can be thought of as a type of network requirement that affects the number of extreme pathways needed to characterize the solution space.
Table 2.
Extreme Pathway Characteristics Associated with the Production of the Indicated Amino Acids
| Amino Acid | Case 1 | Case 2 | Case 3 | Case 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| no. of EP | no. of ES | no. of EP/ES | no. of EP | no. of ES | no. of EP/ES | no. of EP | no. of ES | no. of EP/ES | no. of EP | no. of ES | no. of EP/ES | |
| Asparigine | 217 | 105 | 2.1 | 295 | 154 | 1.9 | 295 | 154 | 1.9 | 340 | 193 | 1.8 |
| Aspartic Acid | 360 | 102 | 3.5 | 466 | 142 | 3.3 | 466 | 142 | 3.3 | 491 | 163 | 3.0 |
| Cysteine | 473 | 232 | 2.0 | 822 | 420 | 2.0 | 822 | 420 | 2.0 | 1022 | 612 | 1.7 |
| Glutamine | 249 | 101 | 2.5 | 290 | 140 | 2.1 | 290 | 140 | 2.1 | 315 | 164 | 1.9 |
| Glutamic Acid | 441 | 148 | 3.0 | 473 | 178 | 2.7 | 473 | 178 | 2.7 | 493 | 198 | 2.5 |
| Glycine | 0 | ··· | ··· | 0 | ··· | ··· | 348 | 149 | 2.3 | 377 | 173 | 2.2 |
| Lysine | 474 | 167 | 2.8 | 587 | 213 | 2.8 | 587 | 213 | 2.8 | 611 | 237 | 2.6 |
| Proline | 479 | 149 | 3.2 | 621 | 209 | 3.0 | 621 | 209 | 3.0 | 867 | 326 | 2.7 |
| Serine | 212 | 101 | 2.1 | 326 | 158 | 2.1 | 326 | 158 | 2.1 | 355 | 186 | 1.9 |
| Threonine | 275 | 136 | 2.0 | 432 | 206 | 2.1 | 432 | 206 | 2.1 | 469 | 242 | 1.9 |
| Tryptophan | 936 | 615 | 1.5 | 1431 | 1034 | 1.4 | 1431 | 1034 | 1.4 | 1958 | 1486 | 1.3 |
| Tyrosine | 584 | 302 | 1.9 | 825 | 477 | 1.7 | 825 | 477 | 1.7 | 1008 | 649 | 1.6 |
| Average | 392 | 196 | 2.4 | 547 | 303 | 2.3 | 576 | 290 | 2.3 | 692 | 386 | 2.1 |
The various cases refer to changes in the carbon sinks of the network (see Fig. 2). Case 1: succinate; Case 2: succinate, acetate; Case 3: succinate, acetate, formate; Case 4: succinate, acetate, formate, lactate. EP indicates extreme pathway in above table; and ES, unique external state.
Table 3.
Extreme Pathway Characteristics for the Production of Subsets of Biomass and the Individual Nucleotides
| Product | Case | No. of EP | No. of ES | No. of EP/ES |
|---|---|---|---|---|
| Equimolar amino acids | 3 | 4991 | 1969 | 2.5 |
| Equimolar amino acids | 4 | 6032 | 2825 | 2.1 |
| E. coli ratio amino acids | 4 | 5553 | 2481 | 2.2 |
| Equimolar ribonucleotides | 4 | 1325 | 1164 | 1.1 |
| ATP | 4 | 1111 | 914 | 1.2 |
| CTP | 4 | 550 | 476 | 1.2 |
| GTP | 4 | 377 | 320 | 1.2 |
| UTP | 4 | 550 | 475 | 1.2 |
EP indicates extreme pathways; ES, unique external state.
The yield plots for the equimolar production of the amino acids are presented for cases 3 and 4 (Fig. 3). A large number of the extreme pathways were found within a narrow range of yield values. For case 3, ∼66% of the extreme pathways were found between the dashed lines, representing only 17% of the range of capable yield values. For case 4, ∼57% of the extreme pathways were found within the dashed lines, also representing only 17% of the possible yield values. Although the yield values within this range represent distinct extreme pathways, these plots indicate the relatively narrow range of yield values that may be experimentally indistinguishable.
Figure 3.

Plot of carbon yield values for the production of equimolar amino acid sets. Cases 3 and 4 are presented. The pathway values between the two cases do not correlate with each other; both data sets are independently rank ordered in terms of increasing yield. The pathways with yield values between the two dashed lines (spanning 17% of the range of yield values) correspond to 66% and 57% of the total number of pathways for cases 3 and 4, respectively.
In the production of the equimolar set of nonessential amino acids, nitrogen has two potential systemic outputs: the amino acids and ammonia. Figure 4 (without urea input) shows how nitrogen is directed in the extreme pathways that have been calculated for equimolar nonessential amino acid production in case 3. The percentage of nitrogen directed to these outputs is presented as percentages of total nitrogen input into the system. Here, 88% of the extreme pathways require at least 80% of the total input nitrogen for ammonia production. Urea was added as an allowable input to see what effect it would have on the distribution of nitrogen. There were 1477 more extreme pathways with this additional input (Fig. 4, with urea input). However, the number of pathways with >80% of the nitrogen diverted to ammonia increased only slightly, from 88% to 89%, with the addition of urea as an input. The minimum and maximum values for both conditions remained unchanged.
Figure 4.

Nitrogen fate plots for equimolar amino acid production. Case 3 exchange flux constraints, with and without urea, are shown.
H. pylori Amino Acid Production in Physiologic E. coli Ratios
The relative ratios of the individual amino acids in the produced set affects the extreme pathways that are generated. Because data is not available for the amino acid composition of H. pylori, the amino acid composition of E. coli was used as an estimate. We computed the extreme pathways for the set of nonessential amino acids in E. coli ratios (Fig. 5 illustrates yield values and nitrogen fates). As shown, the shape of these curves resembles that for the equimolar production of the amino acids, although the number of generated extreme pathways changed. The range of the yield values was different for the production of the set of amino acids in the two situations (equimolar and E. coli ratios) because there were many more amino acids produced in the set with E. coli–based ratios (i.e., six glycine molecules per set instead of one per set).
Figure 5.

Carbon yield (A) and nitrogen fate (B) plots for the production of the nonessential amino acid set in Escherichia coli ratios.
Nucleotide Production
The nitrogen fates of the extreme pathways for the production of an equimolar ratio of ribonucleotides are shown in Figure 6. The shape of this curve was similar to that for the production of the amino acid set, although with a less pronounced shallow slope region. The percentage of nitrogen that could be incorporated into ribonucleotides was 42%.
Figure 6.

Carbon yield (A) and nitrogen fate (B) plots for the production of an equimolar set of the ribonucleotides in case 4.
The extreme pathways for the equimolar set of deoxyribonucleotides were not determined because of computational unfeasibility. In attempting to calculate the extreme pathways for synthesis of the deoxyribonucleotides, the network was unable to produce deoxythymidine triphosphate (dTTP) under the conditions that have been outlined above. The stoichiometric matrix was investigated, and further in silico experiments were performed to ascertain the reason. It was subsequently determined that the model was unable to produce dTTP without simultaneously producing glycine.
Extreme Pathway Redundancy
The degree of pathway redundancy was evaluated for the individual production of the nonessential amino acids in H. pylori (Table 2). There was an average of two internal states per unique external state. Previous work has been performed on the pathway redundancy of H. influenzae (Papin et al. 2002). In contrast with H. influenzae, there was relatively little variation in the degree of pathway redundancy among the individual amino acids in H. pylori. In H. pylori, the maximum value for the extreme pathway redundancy was 3.5 for the production of aspartic acid in case 1; the minimum value was 1.3 for the production of tryptophan in case 4. This result is strikingly different from the range of values in H. influenzae; in one tested condition for H. influenzae, the minimum value was 10 and the maximum value was 236 for the production of glutamine and tryptophan, respectively.
The numbers of extreme pathways per unique external state for the nonessential amino acid sets were found to be of the same order of magnitude as that of the average values for the production of the individual amino acids (Table 3). The pathway redundancy was approximately two for all the evaluated cases. In our previous study with H. influenzae, the addition of succinate as another carbon output caused the average pathway redundancy to increase from 37 to 52 internal states per unique external state. However, similar increases were not seen in H. pylori with the addition of carbon outputs (acetate, case 2; formate, case 3; and lactate, case 4).
The redundancy calculations for the production of ribonucleotides in H. pylori indicated a reduced degree of redundancy (Table 3). For the production of ATP, CTP, GTP, and UTP, there was an average of 1.2 extreme pathways per unique external state. For the production of the set of ribonucleotides in equivalent molar ratios, the ratio of extreme pathways per unique external state was 1.1. This indicates less redundancy in the defined metabolic network of H. pylori for the production of these metabolites than for the amino acids.
The average degree of pathway redundancy for the production of the individual amino acids in H. pylori was an order of magnitude less than that in H. influenzae. The degree of redundancy for the two organisms was evaluated under similar conditions (Fig. 7). The outputs included in the comparison were acetate, succinate, and the individual nonessential amino acids common to both organisms. These amino acids included: asparagine, aspartic acid, glutamine, lysine, proline, serine, threonine, and tyrosine. Even with similarly sized organisms, there was a vastly different degree of redundancy (Table 4). There was an average of two extreme pathways with equivalent external states in H. pylori. The same outputs for H. influenzae resulted in an average of 46 extreme pathways per external state. Although the differences in the minimal medium requirements for each of the organisms make direct comparisons difficult, this difference in pathway redundancy does indicate a much more rigid metabolic network architecture for H. pylori than for H. influenzae.
Figure 7.

The exchange flux constraints used for the comparison of the two organisms H. influenzae and H. pylori. Note that more inputs were allowed into both systems, but the indicated inputs were the only ones used by these in silico strains. The organisms had identical output fluxes, with the exception of ammonia as a necessary output for H. pylori. The individual amino acids included in the comparison were asparagine, aspartic acid, glutamine, lysine, proline, serine, threonine, and tyrosine.
Table 4.
Network Redundancy Comparison Between the Two Indicated in silico Organisms
| In silico organism | Genome size | ORFs in genome | Reactions in model | No. of EP/ES |
|---|---|---|---|---|
| H. influenzae | 1.83 Mb | 1740 | 461 | 46 |
| H. pylori | 1.67 Mb | 1590 | 381 | 2 |
Isozymes in the input file were not included in this study. The amino acid set included in this comparison includes asparagine, aspartic acid, glutamine, lysine, proline, serine, threonine, and tyrosine. EP indicates extreme pathways; ES, unique external state; and ORF, open reading frame.
DISCUSSION
This study presents the first genome-scale extreme pathway analysis for a reconstructed genome-scale metabolic network of H. pylori, and it details physiological, emergent, systems-level properties of this organism. This in silico study resulted in the establishment of important systemic properties of the reconstructed network. We found that (1) the metabolic network associated with the production of individual amino acids in H. pylori is redundant, but to a much lesser degree than that of H. influenzae; (2) the pathway redundancy for the simultaneous production of multiple amino acids was on the same order of magnitude as the redundancy in the production of the individual amino acids, but the redundancy in ribonucleotide production was nearly half of that seen in the amino acids; and (3) the network was unable to achieve a high level of conversion of input nitrogen to the set of either amino acids or nucleotides, and the inclusion of urea as an input did not significantly affect the percentage of input nitrogen secreted as ammonia.
The degree of extreme pathway redundancy for the individual amino acid production was much lower in H. pylori than previously calculated in H. influenzae (Papin et al. 2002). There were approximately two internal states per unique external state for the production of the amino acids in the H. pylori network, whereas H. influenzae was found to have an average of 46 extreme pathways per external state under similar conditions (minimal medium with acetate and succinate as carbon outputs). This result indicates that the reconstructed H. pylori network is much less metabolically redundant and therefore less robust than the H. influenzae network. In its very specific environmental niche (the human stomach), H. pylori are surrounded by a wealth of amino acids and other precursors that it needs for growth. The ratio of internal to external states in the H. pylori network for amino acid production was found to be close to two despite the addition of more allowable outputs. This result was also somewhat surprising because the pathway redundancy changed dramatically with the inclusion of succinate as an additional carbon sink in our previous study on H. influenzae. In this previous study, the degree of redundancy also varied widely for the production of each different amino acid, in contrast to the data on H. pylori presented herein. This lack of variance again shows a much less flexible and versatile metabolism, an interesting fact in view of the similar size and gene number of H. pylori and H. influenzae. An implication of these in silico results is that the metabolic network of H. pylori may be very specifically tailored to its environment.
The extreme pathway analysis of the amino acid and ribonucleotide sets showed similar degrees of pathway redundancy in H. pylori (Table 3). For the production of the nonessential amino acid sets, there were approximately two internal states per external state, similar to that seen in the production of the individual amino acids. The yield curves for the production of the amino acid and nucleotide sets in H. pylori all showed a similar shape (an inverted S-curve). Thus, there was a region on each of the carbon yield and nitrogen fate plots that had a shallow slope, where many extreme pathways corresponded to a relatively small range of the respective yields. These regions could potentially hold significant physiological meaning. Specifically, the metabolic network has an extremely large number of different ways to produce a relatively small band of yield values. It has been hypothesized that these regions correspond to flux values relevant to primary objectives of the organism (Papin et al. 2002), such as reproduction and growth. Further investigation into the genome-scale functioning of metabolic networks is needed to determine if this in silico–generated hypothesis is valid. Redundancy in the metabolism of ribonucleotides was reduced in comparison to that of the amino acids. For the production of the individual ribonucleotides, there were 1.1 internal states per external state. For the set of ribonucleotides, there were 1.2 internal states per external state. This degree of redundancy indicates a much more rigid metabolic network than that for amino acid production.
Full integration of input nitrogen into either the amino acids or the ribonucleotides was not possible in the reconstructed H. pylori network. A maximum of 40% of input nitrogen was used in equimolar amino acid synthesis; alternatively, a maximum of 42% of the input nitrogen could be incorporated into ribonucleotide synthesis. In the in silico H. pylori model, the excess nitrogen was necessarily diverted to ammonia production. Because of computational intractability, we were unable to calculate the extreme pathways for the full complement of biomass constituents. Therefore, as has been previously indicated (McGee et al. 1999), it is possible that nitrogen taken up from the amino acids is primarily incorporated into biomass when all constituents are taken into account, and that ammonia production for pH control comes predominantly from the breakdown of urea. However, these initial extreme pathway calculations, in which uncoupled protein production can use a maximum of only 50% of the input nitrogen for all cases tested, indicate that nitrogen input from arginine and alanine is potentially used to produce ammonia to some extent in vivo. Also, although 40% utilization of nitrogen in equimolar amino acid production and 42% nitrogen utilization in ribonucleotide synthesis were maximum values, the bulk of the pathways located in the plateau regions achieved much smaller nitrogen utilization. Surprisingly, the addition of urea as an input did not alter the shape of the curve or the percentage of pathways that use small amounts of nitrogen. This lack of change in the nitrogen fate plots indicates that the network was incapable of directing a greater percentage of input nitrogen to ammonia production with the addition of urea. Because ribonucleotide synthesis and amino acid synthesis directed at least 50% of the inputted nitrogen to ammonia production, it is therefore possible that the production of nonessential amino acids with ribonucleotides will maintain a similar nitrogen fate. These two metabolite groups represent a substantial amount of the biomass, and consequently, this characteristic may hold true for the complete growth of H. pylori.
The results obtained were based on a reconstructed genome-scale network using all the currently available information for H. pylori. However, until we have 100% open reading frame assignments, no metabolic network reconstruction is complete. In silico models, such as the one used herein, can be valuable tools in helping to guide research. For example, it was found that dTTP could not be produced without simultaneous production of glycine in the defined metabolic network. If the production levels of these two metabolites were measured in vivo and found not to correlate directly, it would be strong evidence that there remains at least one unknown reaction in the network that involves either or both of these metabolites. Such critical comparison between data-driven model predictions and experimental results is thus a valuable tool for our quest to obtain comprehensive in silico representations of living cells.
In summary, this study presents the first calculation of genome-scale extreme pathways for the production of large subsets of biomass components (i.e., the full amino acid set, the nucleotide set). It should be emphasized that extreme pathways are completely and uniquely determined by the structure of the metabolic network. They differ in this description from many other pathway definitions that are often arbitrary or intuitive groupings of contiguous reactions in the network. Extreme pathway analysis is a form of genome-enabled science that can be used to determine the capabilities of a reconstructed metabolic network. Thus, it is useful to compare the in silico analysis of extreme pathways with experimental data to determine the extent to which the known metabolic map can be used to reconstruct observed phenotypic behaviors. Future studies will focus on enabling the calculation of the extreme pathways for the complete biomass of H. pylori and for larger genomes. As the complexity of life increases dramatically with the addition of a relatively small amount of genes (Claverie 2001), apparently so do the numbers of extreme pathways that characterize a metabolic system, allowing cells the ability to fine-tune its metabolic phenotype. Extreme pathways allow us to analyze the fundamental structure of metabolic networks to evaluate the metabolic functions of a cell from a pathway perspective.
METHODS
Formulation of the H. pylori Metabolic Genotype
The metabolic network for H. pylori was reconstructed from reviews of biochemical literature and genome databanks following previously used methods (Edwards and Palsson 1999; Schilling 2000; Schilling and Palsson 2000; Covert et al. 2001). The complete listing of the reactions included in this H. pylori model can be found at http://gcrg.ucsd.edu/organisms/hpylori.html. Isozymes were not included in the in silico model used for this study. The selection between isozymes had no effect on the extreme pathway calculations made in this analysis. For any given pathway, the respective flux would be equivalent independent of the catalyzing enzyme per details in the aforementioned extreme pathway analysis algorithm (Schilling et al. 2000).
Extreme Pathways and Convex Analysis
The m × n stoichiometric matrix, S, of this reconstructed network includes all network metabolites (m rows) and all corresponding transport processes and metabolic reactions (n columns). The flux represents the amount of mass moving through the associated reaction. An exchange flux corresponds to a flux across the system boundary. An internal flux corresponds to a reaction within the system.
A metabolic network can be constrained by implementing simple thermodynamic principles regarding the irreversibility of reactions. If a reaction is reversible, it is decomposed into two reactions (for the forward and backward directions). Thus defined, all internal fluxes are constrained to be nonnegative. By decoupling the reversible reactions, we have
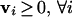 |
1 |
where v is a vector of all the fluxes in the metabolic network.
Another set of constraints on this system involves the conservation of mass, which in a steady state is described by the following flux balance equation:
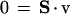 |
2 |
A linear basis for this system enables a characterization of the metabolic capabilities of an organism. Each solution to this equation would represent a steady-state set of flux values for the metabolic network. These correspond to a particular cell phenotype. Any solution to equation 2 can be reached with a combination of the linear basis vectors for the S matrix. However, this combination can be negative or positive, violating the thermodynamics of the chemical reactions. To overcome this difficulty, Schilling et al. (2000) developed an algorithm to evaluate a set of basis vectors, called extreme pathways, using convex analysis.
With a convex basis, every point in the space defined by equation 2 can be written as a nonnegative combination of the extreme pathways of the system (Equation 3).
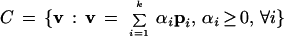 |
3 |
C represents the convex cone encompassing all possible solutions of the metabolic network; v represents the vector of fluxes for each reaction in the network; and αi and pi are the weights and the extreme pathways of the network, respectively. Every steady-state solution of the metabolic stoichiometric network can then be represented as a nonnegative combination of the extreme pathways. These extreme pathways are a unique and mathematically rigorous characterization of the solution space for the metabolic network.
WEB SITE REFERENCE
http://gcrg.ucsd.edu/organisms/hpylori.html; Complete listing of the reactions included in this H. pylori model.
Acknowledgments
We thank Marc Abrams, Dr. Tom Fahland, Iman Famili, Markus Herrgard, Jennifer Reed, and Sharon Smith for their contributions to this study. Support for this work is provided by grants from the National Science Foundation (BES98-14092, MCB98-73384), the National Institutes of Health (GM57089), and the Whitaker Foundation (Graduate Student Research Fellowship to J.P.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL palsson@ucsd.edu; FAX (858) 822-3120.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.218002. Article published online before print in April 2002.
REFERENCES
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Bardhan PK. Epidemiological features of Helicobacter pylori infection in developing countries. Clin Infect Dis. 1997;25:973–978. doi: 10.1086/516067. [DOI] [PubMed] [Google Scholar]
- Bonarius HPJ, Schmid G, Tramper J. Flux analysis of underdetermined metabolic networks: The quest for the missing constraints. Trends Biotech. 1997;15:308–314. [Google Scholar]
- Bono H, Ogata H, Goto S, Kanehisa M. Reconstruction of amino acid biosynthesis pathways from the complete genome sequence. Genome Res. 1998;8:203–210. doi: 10.1101/gr.8.3.203. [DOI] [PubMed] [Google Scholar]
- Claverie JM. Gene number: What if there are only 30,000 human genes? Science. 2001;291:1255–1257. doi: 10.1126/science.1058969. [DOI] [PubMed] [Google Scholar]
- Cover TL, Blaser MJ. Helicobacter pylori infection, a paradigm for chronic mucosal inflammation: Pathogenesis and implications for eradication and prevention. Adv Intern Med. 1996;41:85–117. [PubMed] [Google Scholar]
- Covert MW, Schilling CH, Famili I, Edwards JS, Goryanin II, Selkov E, Palsson BO. Metabolic modeling of microbial strains in silico. Trends Biochem Sci. 2001;26:179–186. doi: 10.1016/s0968-0004(00)01754-0. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J Biol Chem. 1999;274:17410–17416. doi: 10.1074/jbc.274.25.17410. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Ramakrishna R, Schilling CH, Palsson BO. Metabolic flux balance analysis. In: Lee SY, Papoutsakis ET, editors. Metabolic engineering. New York, NY: Marcel Deker; 1999. pp. 13–58. [Google Scholar]
- Fell D. Understanding the control of metabolism. United Kingdom: Portland Press, London; 1996. [Google Scholar]
- Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- Karp PD. Pathway databases: A case study in computational symbolic theories. Science. 2001;293:2040–2044. doi: 10.1126/science.1064621. [DOI] [PubMed] [Google Scholar]
- Karp PD, Ouzounis C, Paley S. Proceedings of the ISMB-96 Conference. 1996. HinCyc: A knowledge base of the complete genome and metabolic pathways of H. influenzae; pp. 116–125. , St. Louis, MO. [PubMed] [Google Scholar]
- Karp PD, Krummenacker M, Paley S, Wagg J. Integrated pathway-genome databases and their role in drug discovery. Trends Biotech. 1999;17:275–281. doi: 10.1016/s0167-7799(99)01316-5. [DOI] [PubMed] [Google Scholar]
- Kelly DJ. The physiology and metabolism of the human gastric pathogen Helicobacter pylori. Adv Microb Physiol. 1998;40:137–189. doi: 10.1016/s0065-2911(08)60131-9. [DOI] [PubMed] [Google Scholar]
- Küffner R, Zimmer R, Lengauer T. Pathway analysis in metabolic databases via differential metabolic display (DMD) Bioinformatics. 2000;16:825–836. doi: 10.1093/bioinformatics/16.9.825. [DOI] [PubMed] [Google Scholar]
- Liao JC, Hou SY, Chao YP. Pathway analysis, engineering and physiological considerations for redirecting central metabolism. Biotech Bioeng. 1996;52:129–140. doi: 10.1002/(SICI)1097-0290(19961005)52:1<129::AID-BIT13>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Mavrovouniotis ML, Stephanopoulos G. Computer-sided synthesis of niochemical pathways. Biotech Bioeng. 1990;36:1119–1132. doi: 10.1002/bit.260361107. [DOI] [PubMed] [Google Scholar]
- Mendz GL. Nucleotide metabolism. In: Mobley HL, et al., editors. Helicobacter pylori: Physiology and genetics. Washington, DC.: ASM Press; 2001. pp. 147–158. [PubMed] [Google Scholar]
- McGee DJ, Radcliff FJ, Mendz GL, Ferrero RL, Mobley HL. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J Bacteriol. 1999;181:7314–7322. doi: 10.1128/jb.181.23.7314-7322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedenskov P. Nutritional requirements for growth of Helicobacter pylori. Appl Environ Microbiol. 1994;60:3450–3453. doi: 10.1128/aem.60.9.3450-3453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Umbarger HE. In: Chemical composition of Escherichia coli. Escherichia coli and Salmonella: Cellular and molecular biology. Neidhardt FC, editor. 1996. , 13–16. ASM Press, Washington, DC. [Google Scholar]
- Ouzounis CA, Karp PD. Global properties of the metabolic map of Escherichia coli. Genome Res. 2000;10:568–576. doi: 10.1101/gr.10.4.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Larsen N, Pusch GD, D'Souza M, Selkov ES, Jr, Kyrpides N, Fonstein M, Maltsev N, Selkov E. WIT: Integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000;28:123–125. doi: 10.1093/nar/28.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papin, J.A., Price, N.D., Edwards, J.S., and Palsson, B.O. 2002. The genome-scale metabolic extreme pathway structure in Haemophilus influenzae shows significant network redundancy. J. Theor. Biol. (in press). [DOI] [PubMed]
- Reynolds DJ, Penn CW. Characteristics of Helicobacter pylori growth in a defined medium and determination of its amino acid requirements. Microbiology. 1994;140:2649–2656. doi: 10.1099/00221287-140-10-2649. [DOI] [PubMed] [Google Scholar]
- Schilling CH. “On systems biology and the pathway analysis of metabolic networks,” Ph.D. thesis. La Jolla, CA: University of California at San Diego; 2000. [Google Scholar]
- Schilling CH, Palsson BO. Assessment of the metabolic capabilities of Haemophilus influenzae Rd through a genome-scale pathway analysis. J Theor Biol. 2000;203:249–283. doi: 10.1006/jtbi.2000.1088. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Schuster S, Palsson BO, Heinrich R. Metabolic pathway analysis: Basic concepts and scientific applications in the post-genomic era. Biotechnol Prog. 1999;15:296–303. doi: 10.1021/bp990048k. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Letscher D, Palsson BO. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J Theor Biol. 2000;203:229–248. doi: 10.1006/jtbi.2000.1073. [DOI] [PubMed] [Google Scholar]
- Schuster S, Dandekar T, Fell DA. Detection of elementary flux modes in biochemical networks: A promising tool for pathway analysis and metabolic engineering. Trends Biotechnol. 1999;17:53–60. doi: 10.1016/s0167-7799(98)01290-6. [DOI] [PubMed] [Google Scholar]
- Selkov E, Jr, Grechkin Y, Mikhailova N, Selkov E. MPW: The metabolic pathways database. Nucleic Acids Res. 1998;26:43–45. doi: 10.1093/nar/26.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark RM, Suleiman MS, Hassan IJ, Greenman J, Millar MR. Amino acid utilisation and deamination of glutamine and asparagine by Helicobacter pylori. J Med Microbiol. 1997;46:793–800. doi: 10.1099/00222615-46-9-793. [DOI] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Tsoka S, Ouzounis C. Functional versatility and molecular diversity of the metabolic map of Escherichia coli. Genome Res. 2001;11:1503–1511. doi: 10.1101/gr.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A, Palsson BO. Metabolic flux balancing: Basic concepts, scientific and practical use. Biotechnology. 1994;12:994–998. [Google Scholar]