

Abstract
We report here the identification of a previously unknown transcription regulatory element for heat shock (HS) genes in Caenorhabditis elegans. We monitored the expression pattern of 11,917 genes from C. elegans to determine the genes that were up-regulated on HS. Twenty eight genes were observed to be consistently up-regulated in several different repetitions of the experiments. We analyzed the upstream regions of these genes using computational DNA pattern recognition methods. Two potential cis-regulatory motifs were identified in this way. One of these motifs (TTCTAGAA) was the DNA binding motif for the heat shock factor (HSF), whereas the other (GGGTGTC) was previously unreported in the literature. We determined the significance of these motifs for the HS genes using different statistical tests and parameters. Comparative sequence analysis of orthologous HS genes from C. elegans and Caenorhabditis briggsae indicated that the identified DNA regulatory motifs are conserved across related species. The role of the identified DNA sites in regulation of HS genes was tested by in vitro mutagenesis of a green fluorescent protein (GFP) reporter transgene driven by the C. elegans hsp-16–2 promoter. DNA sites corresponding to both motifs are shown to play a significant role in up-regulation of the hsp-16–2 gene on HS. This is one of the rare instances in which a novel regulatory element, identified using computational methods, is shown to be biologically active. The contributions of individual sites toward induction of transcription on HS are nonadditive, which indicates interaction and cross-talk between the sites, possibly through the transcription factors (TFs) binding to these sites.
[The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: L. Hillier.]
All living cells display a rapid molecular response to adverse environmental conditions, a phenomenon broadly termed as the heat shock (HS) response (Lindquist 1986; Bienz and Pelham 1987; Morimoto 1993). The HS response is characterized by increased expression of a set of proteins, the heat shock proteins (Hsps), which have been conserved in evolution (Lindquist and Craig 1988). The Hsps function as molecular chaperones in regulating cellular homeostasis and promoting survival (Hartl 1996).
In eukaryotes, the enhanced HS gene expression has been shown to be regulated by the heat shock transcription factors (HSFs), which acquire DNA binding activity in response to various kinds of stress (Wu 1995; Morimoto 1998; Morano and Thiele 1999; Pirkkala et al. 2001). A single HSF gene has been isolated from yeast Saccharomyces cerevisiae (Wiederrecht et al. 1988) and Drosophila melanogaster (Clos et al. 1990). Several members of the HSF family have been shown to exist in vertebrates and plants (HSF1–4) (Wu 1995; Nover et al. 1996; Morimoto 1998; Morano and Thiele 1999, Nakai 1999; Pirkkala et al. 2001) in which different HSFs are indicated to respond to various forms of stress. HSF1 in vertebrates is orthologous to HSF in the yeast and Drosophila and has been indicated as the HSF that mediates heat stress-induced expression of Hsps. HSF, in response to HS, binds to the DNA sites, commonly referred to as the heat shock elements (HSEs) characterized as multiples of the motif 5′-nGAAn- 3′ (Fernandes et al. 1994).
A few general transcription factors (TFs) are implicated to interact with the HSF for regulation of Hsp expression on HS. The HSF has been observed to interact with other general DNA binding factors, such as the TBP (TATA-box binding protein) and GAGA-factor (Mason and Lis 1997), which aids the binding of the HSF to the Hsp promoters. Both the TBP and GAGA-factor occupy the promoter regions before induction by HS and are thus positioned to facilitate HSF recruitment. There has also been evidence that HSF1 can interact with the STAT-1 (Signal Transducer and Activator of Transcription-1) TF to induce the expression of HS genes in human peripheral blood cells treated with the cytokine interferon-γ (IFN-γ) (Stephanou et al. 1999). However, apart from the HSEs, no other cis-elements are known to be specifically responsible for induction of the HS genes on heat stress.
We have been interested in studying the transcription regulatory mechanism in HS response. Gene expression patterns in C. elegans were determined, before and after HS, using DNA microarrays containing probes for approximately 12,000 genes from the C. elegans genome. We followed a general scheme for identification and computational validation of potential transcription regulatory elements responsible for heat stress induction (Fig. 1). Upstream promoter regions of the genes that were consistently up-regulated 1 and 4 hr after HS were analyzed using DNA pattern recognition programs. Two DNA motifs were found, the HSE (consensus: TTCTAGAA), which appears to represent the binding sites for the HSF, and HSAS (for heat shock associated site) (consensus: GGGTGTC), a previously unknown candidate regulatory motif. Statistical analyses and cross-species sequence comparison indicated that these motifs are significantly overrepresented in the promoter regions of the Hsps and are conserved across closely related species.
Figure 1.

Schema describing the steps for identification and validation of transcription regulatory elements from coregulated genes.
We determined the biological significance of the DNA motifs in regulation of HS genes using green fluorescent protein (GFP) technology. Two HSE and one HSAS site were predicted in the promoter region of hsp-16–2. To monitor hsp-16–2 promoter-dependent gene expression, a reporter construct was used that contained the hsp-16–2 promoter fused to a GFP coding sequence. Transgenic C. elegans animals containing this construct showed strong GFP induction on HS. When DNA sites corresponding to the two motifs were mutated, the promoter was no longer inducible by heat stress. Therefore, in addition to the HSE we identified a novel DNA element that plays a significant role in the transcriptional regulation of HS genes. It was observed that mutation of multiple DNA sites was required to eliminate heat stress-induced expression from the hsp-16–2 promoter, and the extent of expression induced by individual sites was non-additive. This indicates that an interaction between sites (possibly mediated through the TFs that bind to these sites) may be important for efficient transcription regulation of the HS genes under the HS condition.
RESULTS
Identification of HS Up-Regulated Genes in C. elegans
Five microarray hybridization experiments were performed with independently prepared mRNA: In two experiments, mRNAs were taken from the animals that were harvested 1 hr after the HS treatment, and in three experiments, animals were heat shocked for 2 hr, allowed to recover for 2 hr, then harvested. Genes that were induced in at least four of the five experiments and were overexpressed by an average factor of two or more over the five experiments compared with the normal non-HS worms were identified (Table 1).
Table 1.
Genes Up-Regulated on Heat Stress
| Gene | Average fold increase (5 experiments ± SD) | Average fold increase (2 × 1 hr experiments) | Average fold increase (3 × 4 hr experiments) | HSE sites? | HSAS sites? | Function |
|---|---|---|---|---|---|---|
| F44E5.5 | 52.1 ± 52.8 | 78.5 | 32.9 | + | + | Member of Hsp70- protein family |
| T27E4.2 | 51.2 ± 31.9 | 92.2 | 25.2 | + | + | Member of Hsp-16- protein family |
| F44E5.4 | 49.4 ± 39.2 | 85.5 | 25.3 | + | + | Member of Hsp70- protein family |
| F08G2.5 | 9.8 ± 8.8 | 5.5 | 12.6 | + | + | Predicted coding sequence, unknown function |
| C50F7.5 | 8.8 ± 6.2 | 2.3 | 13.1 | + | + | Predicted coding sequence, unknown function |
| T27F2.4 | 6.5 ± 5.1 | 2.4 | 9.2 | + | + | Predicted coding sequence, unknown function |
| Y38H6C.7 | 5.6 ± 3.2 | 7.5 | 4.3 | − | + | Predicted coding sequence, unknown function |
| H14N18.1 | 5.1 ± 3.1 | 4.5 | 5.4 | + | − | unc-33 (bag-2). Muscle function, HSP70 regulator |
| F58E10.4 | 4.7 ± 2.5 | 2.5 | 6.1 | + | + | aip-1. arsenite resistance |
| M05D6.1 | 4.6 ± 3.5 | 8.4 | 2.1 | + | + | Ser/Thr Kinase |
| F33H12.6 | 4.5 ± 2.4 | 6.0 | 3.6 | + | + | Predicted coding sequence, unknown function |
| F09B9.1 | 4.4 ± 4.5 | 2.4 | 5.8 | + | − | Predicted G-protein linked receptor |
| R07B1.4 | 4.3 ± 2.9 | 1.8 | 5.9 | + | + | Glutathione S-transferase |
| M01B12.1 | 4.1 ± 4.1 | 3.0 | 4.8 | + | − | Predicted coding sequence, unknown function |
| F30F8.4 | 3.8 ± 2.1 | 6.0 | 2.4 | − | − | Member of transposase protein family |
| D2013.9 | 3.8 ± 1.3 | 3.5 | 4.0 | + | − | Predicted coding sequence, unknown function |
| C12C8.1 | 3.6 ± 2.5 | 6.1 | 2.0 | + | − | Member of Hsp70- protein family |
| C30C11.4 | 3.2 ± 1.3 | 4.4 | 2.4 | + | + | Member of Hsp70- protein family |
| F53A9.2 | 3.2 ± 1.5 | 3.8 | 2.8 | + | + | Predicted coding sequence, unknown function |
| C25F9.2 | 3 ± 1.5 | 2.9 | 3.0 | − | + | Predicted coding sequence, unknown function |
| T27A3.4 | 2.9 ± 2.3 | 3.4 | 2.5 | − | − | Predicted coding sequence, unknown function |
| F41C3.2 | 2.4 ± 0.9 | 2.3 | 2.4 | − | + | Identified as sodium/phosphate transporter protein |
| T28H11.7 | 2.3 ± 1.4 | 2.1 | 2.5 | − | + | Predicted coding sequence, unknown function |
| F55A12.9 | 2.3 ± 0.6 | 2.6 | 2.0 | − | − | Predicted coding sequence, unknown function |
| W02D9.10 | 2.2 ± 0.4 | 1.8 | 2.5 | − | + | Predicted coding sequence, unknown function |
| R03D7.2 | 2.1 ± 0.9 | 2.1 | 2.1 | + | + | Identified as putative helicase |
| ZK1290.5 | 2 ± 0.5 | 1.7 | 2.3 | + | − | Identified as aldo/keto reductase. |
| C25D7.1 | 2 ± 1.0 | 1.7 | 2.2 | − | − | Predicted coding sequence, unknown function |
DNA Motifs Identified from Promoters of HS Genes
Because some amount of noise is frequently observed in DNA microarray experiments, we considered only those genes up-regulated by an average factor of four or more (Table 1) for the purpose of transcription regulatory element identification. Genes F44E5.5 and F44E5.4 share a common upstream region of 450 nucleotides (nt), hence to avoid redundancy, only the upstream of F44E5.5 was considered. Two DNA motifs were identified by the application of DNA pattern recognition programs, Consensus (Hertz and Stormo 1999) and ANN-Spec (Workman and Stormo 2000), on regions upstream (−500 to −1) of the up-regulated genes (Fig. 2). Given the relatively closely spaced gene distribution in C. elegans, the selected upstream regions are likely to contain relevant promoter elements. However, it is possible that the selected regions may exclude relevant motifs in genes with large promoters, long 5′UTRs, or membership in operons. We have used the translation start site (the −1 position) to select the candidate promoter regions because it is unambiguous, and because transcriptional start sites have not been determined for the large majority of C. elegans genes. One of the motifs (with a consensus sequence of TTCTAGAA) is the HSE, a well-known DNA binding site for the HSF; the other (with a consensus sequence of GGGTGTC) is called HSAS (for heat shock associated site). A thorough search of the Transfac database (Wingender et al. 2001; http://www.gene-regulation.de) and published literature indicated that the HSAS motif is novel and does not correspond to any known TF binding sites.
Figure 2.

DNA motifs identified from upstream regions of heat shock (HS) genes. Motifs identified by pattern recognition programs from the upstream regions (−500–−1) of HS genes up-regulated genes. Information content (I.C.) in bits (log2) and sequence logos for the DNA motifs are given. DNA logos were generated according to Schneider and Stephens (1990).
Determination of DNA Binding Probability and Cutoff Scores
A “site” corresponding to a particular motif in a sequence is simply a high-scoring subsequence that is obtained by the Patser program using the appropriate motif weight matrix as an input (see Methods). Weight matrices for the motifs were determined using the Consensus and ANN-Spec programs or were obtained from the Transfac database. From a consideration of the thermodynamics of protein–DNA interactions and the statistics of the scores (Stormo 1998; Stormo and Fields 1998), we expect that the score should be proportional to the free energy of binding. Therefore, at equilibrium, the probability of the protein binding to a site with a score, s, is simply:
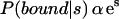 |
1 |
The exact proportionality factor depends on a number of things, including the availability of binding sites within the genome and the concentration of the TF in the nucleus, but because we only use it to rank different potential binding sites, we can ignore it. We also know that there are commonly multiple binding sites in the promoter region for a regulatory TF, so we calculate the probability that it will bind at any of those sites, referred to as the pp-value, as:
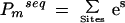 |
2 |
where m denotes the DNA binding motif for the TF. This treatment is likely oversimplified, given the known cooperative binding of TFs to promoter elements. Nevertheless, more complicated models have not proven more effective for the analysis presented here, and this simplified approach has produced meaningful results (see below).
For a given set of N sequences, the geometric mean of the pp-values is given by:
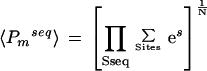 |
3 |
For a motif, m, the appropriate cutoff score for eliminating low scoring subsequences is calculated as follows. The candidate promoter regions (−500 to −1) of the 13 genes that were up-regulated by fourfold or more on heat stress and 3000 random genes were obtained from the C. elegans genome. Considering both strands of the DNA sequences, the sites scoring above a particular arbitrary threshold were determined. The geometric mean of the pp-values for the motifs were obtained for the two sequence sets. The arbitrary threshold value was gradually increased from zero to a certain high positive value, and the cutoff that maximized the difference of the log of geometric means of the pp-values from the two sets (DLGM = log <Pmseq>HS–log <Pmseq>Rand) was chosen to be the appropriate cutoff value for the motif.
For both HSE and HSAS, the cutoff values could be efficiently determined. For each of these motifs the DLGMs peaked at a certain threshold value before decreasing at higher thresholds. At low cutoffs, low scoring sites, which are present in substantial amounts in all sequences, are not eliminated. This results in a substantial number of sites being considered in the calculation of the pp-values, resulting in a small difference in the DLGM between the HS inducible and random promoters. As the threshold value is increased, the low scoring sites are eliminated leaving only the high scoring sites for calculation of the pp-values. For the HS regulatory motifs, the high scoring sites are expected to be more prevalent in the promoters of HS-inducible genes compared with random promoters, hence with increasing cutoffs, the DLGM value increases. As the cutoff is increased further, the high scoring sites are now ignored, eliminating those sites from pp-value calculation. Therefore for HSE and HSAS, the DLGM values decrease at high thresholds (at a very high cutoff value, where all sites are ignored, DLGM is zero). For several other motifs, the DLGMs remained low throughout the range tested and did not show a distinct maximum (Table 2).
Table 2.
Statistical Parameters for DNA Motifs
| DNA motif | More than fourfold expressed | More than twofold expressed | Random | |||
|---|---|---|---|---|---|---|
| 〈num. sites〉 | log(GM) | 〈num. sites〉 | log(GM) | 〈num. sites〉 | log(GM) | |
| HSE | 2.4 | 7.4 | 1.5 | 5.36 | 0.54 | 2.8 |
| HSAS | 1.3 | 5.88 | 1.11 | 5.03 | 0.52 | 2.7 |
| MSE | 0.07 | 0.81 | 0.18 | 1.13 | 0.13 | 0.76 |
| skn-1 | 0.46 | 2.5 | 0.41 | 2.62 | 0.17 | 1.2 |
| ces-2 | 0.0 | 0.0 | 0.07 | 0.32 | 0.04 | 0.23 |
| GATA | 0.3 | 1.85 | 0.29 | 1.71 | 0.24 | 1.56 |
| DNA motif | More than fourfold expressed | More than twofold expressed | ||
|---|---|---|---|---|
| ratio num. sites | diff. log (GM) | ratio num. sites | diff. log (GM) | |
| HSE | 4.44 | 4.6 | 2.78 | 2.56 |
| HSAS | 2.5 | 3.18 | 2.13 | 2.33 |
| MSE | 0.53 | 0.05 | 1.38 | 0.37 |
| skn-1 | 2.7 | 1.3 | 2.41 | 1.42 |
| ces-2 | 0.0 | −0.23 | 1.75 | 0.09 |
| GATA | 1.25 | 0.29 | 1.21 | 0.15 |
For both HSE and HSAS, we calculated the average number of sites per sequence scoring above the respective cutoff values, and the geometric mean of the pp-values for the −500 to −1 regions for genes that are up-regulated by fourfold or twofold, and a set of 3000 genes picked at random from the C. elegans genome (Table 2A). For the purpose of comparison, the same parameters for four other unrelated patterns are shown: MSE (consensus: CCCGCGGGAGCCCG), a muscle-specific transcription regulatory element (GuhaThakurta et al. 2002); GATA (consensus: ACTGATAA), a potential intestine specific regulatory motif (Egan et al. 1995; Fukushige et al. 1999); and two other DNA motifs, skn-1 and ces-2, taken from the Transfac database (http://www.gene-regulation.de). skn-1 represents the DNA binding site (consensus: TAATGTCATCCA) for the C. elegans skn-1 protein, which is a TF required for the correct specification of certain blastomere fates in early C. elegans embryos (Blackwell et al. 1994), and ces-2 represents the DNA binding site (consensus: ATTACGTAAT) for C. elegans protein ces-2, a TF that controls the cell death fate of individual cell types in programmed cell death (Metzstein et al. 1996). For C. elegans, skn-1 and ces-2 are the only two regulatory motifs for which weight matrices are available in the Transfac database. The ratios of the average number of sites per sequence for HS up-regulated genes to the random genes and the DLGMs are given in Table 2B. It can be seen that the DLGMs for HSE and HSAS are significantly higher compared with the other four unrelated motifs.
Nonparametric Mann-Whitney Analysis of Identified Motifs for HS Genes
We took the upstream regions (−500 to −1) of all 19,804 genes from the C. elegans genome, determined the DNA sites for a motif, m, above the cutoff using the Patser program, and calculated the pp-value for each of the sequences (equation 2). A combined pp-value for multiple motifs, M, can also be calculated for the upstream sequence of each gene in the C. elegans genome. For lack of more specific information regarding the mode of TF binding and interaction, we assumed that for up-regulation of genes on heat stress (1) relevant TFs (corresponding to the motifs being considered) need to bind to the upstream sequence, and (2) if there are multiple sites scoring above the cutoff for a particular motif, any one of those binding sites may be occupied by the corresponding TF. For a particular upstream sequence, the combined pp-value for multiple motifs is calculated by taking a product of individual pp-values (from equation 2) for the motifs:
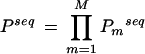 |
4 |
All (19,804) upstream sequences were sorted according to the decreasing log of the pp-value, ln (Pmseq) (equation 2), for individual motifs or combined pp-value, ln(Pseq) (equation 4), for multiple motifs.
Among the most commonly used biostatistical procedures is the comparison of two sample sets to infer whether differences exist between the two populations sampled. We have used the one-tailed Mann-Whitney nonparametric testing method (Zar 1974) to see whether the HS genes are placed significantly higher on the list of all genes sorted by the pp-values. We calculated the Mann-Whitney statistic (Mann and Whitney 1947; Zar 1974) for testing the null hypothesis, H0: Genes in a given set are placed no higher on the list of all genes sorted by the pp-value, compared with a random set of genes. The alternative hypothesis, HA was: Genes in a given set are placed higher on the list of all genes sorted by the pp-value, compared with random genes. For different lists generated by sorting the pp-values (as above), the Mann-Whitney statistic, U, was calculated for the HS genes up-regulated by a factor of four- or twofold. The U statistic can be used to determine z scores, which might be used to determine the probability for the null hypothesis. However, in our case the patterns were discovered using the same set of sequences that we are testing, and we do not have an independent test set. We use the z scores (Table 3) merely to measure the extent to which the identified motifs can help to distinguish the HS responsive genes from other genes and, in particular, to see if the HSAS motif increases the specificity of that observed for HSE alone. Not only are the z scores for the two identified motifs much larger than for other TF patterns, but only with the HSE–HSAS combination does the z score increase over the HSE z score alone.
Table 3.
Mann-Whitney Statistic for Nonparametric, One-Tailed Test of the Null Hypothesis, H0: Genes in a Given Set are Placed No Higher on the List of all Genes Sorted by the pp-value, Compared with a Random Set of Genes
| HSE | HSAS | MSE | skn-1 | ces-2 | GATA | |
|---|---|---|---|---|---|---|
| More than fourfold expressed | 4.8 | 3.5 | 0.1 | 1.2 | 0.6 | 0.3 |
| More than twofold expressed | 3.7 | 3.0 | 0.8 | 1.2 | 0.7 | 0.02 |
| HSE-HSAS | HSE-MSE | HSAS-skn-1 | MSE-ces-2 | |
|---|---|---|---|---|
| More than fourfold expressed | 5.0 | 4.1 | 2.9 | 0.4 |
| More than twofold expressed | 4.3 | 3.3 | 2.8 | 0.5 |
Conservation of Regulatory Sites across Related Species
Each of the HS up-regulated protein sequences was searched for potential orthologs in the C. briggsae sequences using the gapped-BLAST (Altschul et al. 1997) method. For a given C. elegans protein, the C. briggsae protein, with the lowest (<e−50) expectation value and a good alignment over the full length of the protein, was assumed to be the best candidate ortholog. C. briggsae genes CB024O08.9 and CB024O08.10 appeared to be orthologous to the strongly HS-inducible C. elegans genes F44E5.5 and F44E5.4, respectively, and were selected for comparisons of putative promoter regions.
The positions of both the HSE and HSAS sites in the upstream regions of these orthologous gene pairs were very similar. In C. elegans, the genes F44E5.5 and F44E5.4 are placed in opposite orientations on the genomic DNA. The gene structures and the distance between the genes are similar in both organisms (Fig. 3A). The two genes share the upstream intergenic DNA sequence of 450 nt. Comparisons of sequence similarities using the VISTA alignment tool (Mayor et al. 2000; http://www-gsd.lbl.gov/vista) show the expected strong conservation of exon sequences and reduced similarity in noncoding regions (Fig. 3B), as has been previously observed in comparisons of orthologous C. elegans and C. briggsae genes (Heschl and Baillie 1990; Maduro and Pilgrim 1996). The HSE and HSAS sites for these genes are all within the commonly shared upstream region and hence the sites for only one of these genes, F44E5.5, and the corresponding C. briggsae ortholog are shown (Fig. 3C). The pattern of the HSE and HSAS sites on the promoters of the two pairs of orthologous genes indicates that these elements are conserved across closely related species. This conservation can be more directly seen in a direct alignment of the C. elegans and C. briggsae upstream sequences (Fig. 3D) using the GLASS alignment algorithm originally developed for comparisons between human and rodent sequences (Batzoglou et al. 2000). We observe a similar situation with another orthologous gene pair, C. elegans gene T27E4.2 and C. briggsae gene G39L17.4. The pattern of HSE and HSAS sites are highly conserved in the upstream region of these genes, although in general the region has poor conservation, as observed from pair-wise sequence alignment performed with the GLASS method (data now shown).
Figure 3.

Comparison of the orthologous Caenorhabditis elegans and Caenorhabditis briggsae genes. (A) Structures of two Hsp70 protein family genes from C. elegans and corresponding orthologs from C. briggsae. The two top genes (lighter gray) are from C. elegans and the bottom two (dark gray) are from C. briggsae. (B) Sequence similarity comparison of C. elegans and C. briggsae genes shown in A using the VISTA alignment method. The percentage of sequence conservation over every 100 nt window is indicated. Note large sequence similarity decreases in noncoding regions. (C) Heat shock element (HSE) and heat shock associated site (HSAS) DNA site positions and strengths in the promoter regions of the divergently expressed Hsp70 genes shown in A. Vertical bars represent the DNA sites and horizontal lines the DNA sequences. Height of the vertical bars are proportional to the scores calculated by the Patser program. (D) Sequence alignment of promoter regions of Hsp70 genes shown in A, using the GLASS alignment algorithm. HSEs are indicated in red and HSAS elements in green. Also highlighted in blue is a well-conserved sequence also found in the hsp-16–2/−41 promoter region (see Fig. 4A).
GFP Expression Patterns of Mutated Promoter Constructs
A major advance in the attempts to localize gene expression and proteins has been the recent advent of GFP as a reporter molecule in living organisms (Chalfie et al. 1994). GFP is a protein from jellyfish that emits green fluorescence when excited by blue light, even when expressed in heterologous organisms.
The hsp-16–2 gene (gene id: Y46H3A.3) was one of approximately 7000 genes that was not represented in our DNA microarrays. The promoter region of hsp-16–2 was predicted to have two HSE sites and one HSAS site (Fig. 4A). The plasmid pPD122.18 has a GFP coding sequence (including four artificial nuclear localization signals) under the control of this promoter. DNA sites corresponding to the HSE and HSAS motifs were mutated in this plasmid and the GFP expressions driven by the wild-type and mutated promoters were observed before and after HS treatment (Fig. 4B). It was observed that mutation of individual sites still results in significant expression of GFP. However, mutagenesis of all three sites or two sites (either of the two HSEs, or one HSE and the HSAS) results in dramatically reduced expression of the GFP after HS. Transcription induction with one HSE and the HSAS is significantly higher compared with only one HSE (Fig. 5). Also, when the two HSEs are mutated, some amount of heat stress-induced GFP expression is still observed, which is eliminated when the HSAS site is mutated. These results show that both the HSE and the HSAS sites play a significant role in transcription regulation of HS genes in C. elegans. The amounts of transcription induced by the individual sites are nonadditive.
Figure 4.

Summary of mutated promoter construct experiments. (A) The hsp-16–2/−41 promoter sequence in pPD122.18. Candidate promoter elements identified in bold font; sequences above the promoter elements indicate sequences introduced in mutated derivatives. TATA boxes underlined, transcriptional starts (Candido et al. 1989) indicated with grey nucleotides. Nucleotides in outlined font represent conserved site also found in F44E5.4/5 promoter region (see Fig. 3D). (B) Schematic of promoter element mutants and corresponding expression in transgenic animals. Transgenic F1 roller animals were scored as “strong GFP” (green fluorescence protein) if >50 GFP+ nuclei were observed. The two non-GFP animals observed in the pPD122.18 and H1 mutated construct injections likely result from rare events when the reporter construct was not incorporated in the Rol marker-containing array. Transgenic animals were scored as “any GFP” if at least one GFP positive nucleus could be observed. Footnotes: a, GFP+ animals from these injections contained an average of 5.5 (predominantly neuronal) GFP positive nuclei; b, one GFP positive animal was observed with 10 GFP+ head neurons; c, one head neuron in one animal was GFP positive.
Figure 5.

Effect of HSAS element on reporter transgene expression. First generation (F1) transgenic animals containing a GFP reporter construct with either both HSE2 and HSAS mutated (HSE1 only) or HSE2 mutated (HSE1 + HSAS) were heat shocked at 35°C, returned to 20°C for 18 hr, then imaged for GFP expression. This digitally merged differential interference contrast/epifluorescence image shows the requirement for the HSAS element for strong transgene expression when the HSE is mutated. Size bar = 50 μm.
DISCUSSION
Identification of a New Transcription Regulatory Element
We have identified a set of genes reproducibly induced by HS in C. elegans using DNA microarray hybridization. These initial experiments involved a limited set of hybridizations and a single developmental stage and used microarrays containing probes for ∼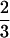 of the predicted C. elegans genes. Therefore, it is likely that we have only identified a subset of all the C. elegans genes induced by HS. Nevertheless, these studies have enabled us to identify a novel HS-responsive promoter element by computational DNA pattern recognition methods followed by statistical analysis. The role of this element (HSAS), along with the other well-known element (HSE), has been shown for one of the Hsps in vivo by mutational analysis using GFP reporter constructs introduced into transgenic animals.
of the predicted C. elegans genes. Therefore, it is likely that we have only identified a subset of all the C. elegans genes induced by HS. Nevertheless, these studies have enabled us to identify a novel HS-responsive promoter element by computational DNA pattern recognition methods followed by statistical analysis. The role of this element (HSAS), along with the other well-known element (HSE), has been shown for one of the Hsps in vivo by mutational analysis using GFP reporter constructs introduced into transgenic animals.
This is one of only a few instances in which a completely novel cis-regulatory site, identified solely by computational DNA pattern recognition methods, has been supported by experimental evidence (e.g., Chen et al. 1995; Hughes et al. 2000; and McCue et al. 2001). Traditionally, time-con-suming experimental methods such as systematic sequence deletions and mutations have been used to identify cis-regulatory regions and sites responsible for regulation of a particular gene. We note that the elements we have identified, being individually neither necessary nor sufficient for full reporter expression, would be difficult to identify using standard molecular techniques. With the advent of techniques like SAGE (Velculescu et al. 2000) or DNA microarrays (DeRisi et al. 1997; Kim et al. 2001) cohorts of coregulated genes can be easily identified. Because the genes that show similar expression profiles are assumed to have similar transcriptional mechanisms governing their expression, DNA pattern recognition methods should be a very useful way of identifying the cis-elements governing the expression of a set of coregulated genes (Tavazoie et al. 1999, GuhaThakurta and Stormo 2001).
Statistical Significance of the Identified Motifs
One important concern regarding DNA pattern recognition methods for regulatory element identification is the significance and specificity of individual motifs. Because the ANN-Spec program takes into consideration a background sequence set and discriminates against commonly occurring motifs in the background, it is designed to yield only those DNA patterns that are specific toward the training set (Workman and Stormo 2000). However, statistical validation of the motifs identified by the pattern recognition programs is useful for selecting the most promising candidate motifs for further experimental verification. We find that several parameters like the pp-values and the Mann-Whitney z scores (Tables 2 and 3) are useful measures of the specificity of an identified DNA motif. It is expected that if a set of genes are regulated by a common cis-regulatory motif, then the upstream promoter regions of those genes will either contain high scoring site for that motif or contain multiple sites (clusters of sites) (Wagner 1999) or both. In either case, the pp-values (equation 2) should be significantly higher for the coregulated genes compared with other genes. With HSE or HSAS, the mean pp-values or the Mann-Whitney z scores are substantially higher for the HS genes.
Cross-Species Conservation of Regulatory Elements
One method that is very useful for identifying conserved DNA motifs is cross-species comparative sequence analysis (Hardison et al. 1997; Wasserman et al. 2000; Cliften et al. 2001). We identified the C. briggsae orthologs of several C.elegans Hsp genes using BLAST searches. On the basis of BLAST alignments, we determined that genes CB024O8.9 and CB024O8.10 were orthologous to genes F44E5.5 and F44E5.4. The DNA sites corresponding to HSE and HSAS are remarkably similar in the upstream regions of these orthologous genes. Analysis of the upstream region of T27E4.2 and its likely C. briggsae ortholog G39L17.4 also revealed conservation of HSE and HSAS sites (data not shown). As more sequences become available from C. briggsae and other related organisms, comparative sequence analysis and phylogenetic footprinting (Wasserman and Fickett 1998; Wasserman et al. 2000) will become a powerful tool for identification of DNA regulatory motifs and adding confidence to the motifs identified by DNA pattern recognition methods. Interestingly, the human small HS genes HSPB2 and CRYAB, like the C. elegans HS gene pairs T27E4.2/T27E4.8 and F44E5.4/F44E5.5, are also closely linked and transcribed divergently, sharing a putative promoter region of <1 kb (Iwaki et al. 1997). This promoter region contains four possible HSAS sites, in addition to two classic HSE sites. It is therefore possible that the HSAS element also plays a role in HS-dependent gene expression in other non-nematode species.
Identification of HSE Motif
Our identification of a motif corresponding to the classic HSE was not unexpected, as C. elegans contains a single gene (Y53C10A.3) that encodes a likely ortholog of HSF. Double-stranded RNA inhibition (RNAi) of Y53C10A.3 expression reduces HS induction of an hsp-16–2/GFP reporter transgene (C.D. Link, unpubl.), supporting the view that the identified HSE motif does function in a typical HSF-dependent HS induction mechanism.
Mode of TF-DNA Interaction in the Hsp Promoters
A promoter element is organized in a hierarchical manner: Individual binding sites are organized in specific arrays to form ‘promoter modules’, which are substructures of the functional promoter, and the complete promoter element is composed of specifically organized promoter modules (Arnone and Davidson 1997; Yuh et al. 1998). Hence, individual binding site detection, although important, is not sufficient for understanding the regulatory mechanism and elucidation of complete promoter function (Werner 2000). The following discussions illustrate our efforts toward determining the relationship between the HSE and HSAS sites with the goal of understanding the regulatory mechanism of the Hsps.
In addition to the mutations of HSE and HSAS sites, we did three preliminary experiments in which we implanted either a single HSE, two closely paired HSEs, or a single HSAS in a “virgin” promoter, devoid of any known heat-inducible elements. These experiments did not attempt to replicate the spacing of these elements. In no case did we observe induced expression from the promoter on HS. This result indicates that (1) both HSE and HSAS may be required, (2) specific distances between the two or more HSEs or HSASs might be important for heat induced expression, or (3) additional sites remain to be identified. We note that the comparison of C. elegans and C. briggsae orthologous HS promoters identified a well-conserved stretch of nucleotides (AGAGACGCAGA) upstream of the HSAS that might represent such a site (Figs. 3D and 4A). However, this candidate site is not generally found among the HS-inducible genes identified in our gene expression analysis, perhaps because it has regulatory functions specifically in highly induced, divergently transcribed HS gene pairs such as F44E5.4/F44E5.5 and T27E4.2/T27E4.9. It is also possible that we may have missed additional sites by using rather stringent cutoffs that eliminated some low scoring sites that could be biologically functional. Weaker sites may also be placed at optimal distances from each other so that multiple TFs can bind the respective DNA sites and maintain intermolecular (TF–TF) interaction at the same time. This is observed in cooperative DNA binding by the TFs, in which the binding of a TF to its DNA site may be weak (which can happen when a DNA binding site is low scoring, i.e., does not conform well with the consensus), but cooperative binding with another TF, which binds a nearby DNA site, may be strong enough for stable TF–DNA complex formation.
It is interesting to note that single-site mutants do not affect the HS induction of hsp-16–2; however, double- and triple-site mutants have a dramatic effect on the expression of the gene (Fig. 4B). The extents of transcription induced by the individual sites are nonadditive (Fig. 4B), indicating an interaction between the sites, probably mediated through protein–protein interactions between TFs binding to those sites. If this is the case, it can be imagined that the TF binding sites would be located at certain distances where optimal TF–TF interactions can occur.
By use of DNA sites corresponding to HSE and HSAS motifs, we tried to build a consistent model for the organization of TF binding sites in the promoter regions of the Hsps, which would distinguish these genes from all other genes in the genome. We studied the strength, frequency of occurrence, and distances between the TF binding sites. However, we failed to obtain such a model. It is clear that many genes with high rankings for HSE, HSAS, or HSE + HSAS sites do not appear to be HS inducible in our gene expression analysis. Furthermore, our hsp-16–2 reporter mutagenesis studies indicate that individual HSE or HSAS elements are neither necessary nor sufficient for full HS inducibility. This indicates that, although we have identified a new HS regulatory element, our understanding of the regulatory mechanism governing the HS response is still incomplete. We note that the most strongly HS-inducible genes identified in our studies are arranged as divergently transcribed gene pairs (e.g., Fig. 3A); the contribution of this gene arrangement to HS regulation is unknown. The HSAS site identified in the hsp-16–2 promoter overlaps a 13-bp imperfect repeat previously indicated to be capable of forming a hairpin structure (Candido et al. 1989). It is also possible that other TFs are involved in the transcriptional pathway involving HSE and HSAS, which needs to be elucidated (some TF sites may be in further upstream regions that remain to be analyzed) or that the HSE and HSAS sites are organized in subtle patterns that remain difficult to identify computationally at this point. Involvement of an alternative transcription regulatory pathway, which uses a different set of TFs, is also a distinct possibility. We intend to address these complex issues of TF–DNA interaction further with computational and experimental means in the future.
METHODS
Sequences and Gene Annotations
All C. elegans sequences and their annotations were obtained from the WormBase web-site (http://www.wormbase.org). C. briggsae, a closely related nematode to C. elegans, is currently being sequenced at the Washington University Genome Sequencing Center, St. Louis, Missouri. We obtained the DNA and protein sequences for C. briggsae from the Washington University Genome Sequencing Center (http://genome.wustl.edu/gsc/Projects/C.briggsae) and L. Hillier (pers. comm.).
cDNA Microarray Experiments and Identification of C. elegans HS Genes
The microarray data were compiled from five independent HS experiments. These experiments were originally designed to investigate whether age-1 mutant animals, which have increased intrinsic thermotolerance (Lithgow et al. 1995), have an altered gene expression response after HS when compared with wild-type animals. Age-synchronous (4-d-old) populations of wild-type or age-1(hx546) animals were harvested as young adults, then split, and half of the populations were heat shocked at 35°C. and half of the animals were maintained at 20°C to generate a control population. Two HS regimes were used: ‘immediate response,‘ in which animals were harvested immediately after 1 hr of HS (two experiments: one wild-type and one age-1 population), and “recovery response,” in which animals were heat shocked for 2 hr, then allowed to recover for 2 h4 (20°C) before harvesting (three experiments: two wild-type and one age-1 population). Poly A+ RNA was prepared from these populations and reverse transcribed into Cy3- or Cy5-labeled cDNA; then HS and control cDNAs were cohybridized to glass-slide DNA microarrays containing probes for 11,917 known or predicted C. elegans genes, as previously described (Reinke et al. 2000). Relative HS-dependent expression changes for each gene was calculated from the ratios of Cy3 and Cy5 hybridization signals. No significant difference in HS-dependent gene expression was observed between age-1 and wild-type animals in this dataset. We therefore compiled the data from the five experiments to generate a list of genes that showed reproducible HS induction independent of genotype or HS regime (Table 1).
Identification of DNA Motifs
Two DNA pattern recognition programs, ANN-Spec (Workman and Stormo 2000) and Consensus (Hertz and Stormo 1999), were used to identify significant DNA patterns from the promoter regions (−500 to −1 relative to the translation start) of the HS genes up-regulated by an average factor of four or more over the five cDNA experiments (Table 1). Consensus and ANN-Spec are local multiple sequence alignment programs that run on a given set of sequences (training set) to identify conserved motifs commonly present in those sequences. Both the programs use weight matrix-based models (Stormo 2000) to represent ungapped DNA sequence motifs. Because the cis-regulatory sites in a set of similarly regulated sequences are expected to be conserved to a certain extent, the conserved motifs identified by these programs represent potential regulatory elements.
Consensus
The Consensus program (Hertz and Stormo 1999) uses a greedy algorithm and searches for a matrix with a low probability of occurring by chance or, equivalently, having a high information content. Version 6.c of Consensus was used and the top scoring result was reported. Different pattern lengths were tested, and both strands of the DNA were searched for motifs because TFs can bind to either strand. The patterns with high information content and the lowest expected frequency were considered.
ANN-Spec
ANN-Spec (Workman and Stormo 2000) uses a simple artificial neural network and Gibbs sampling (Lawrence et al. 1993) method to define DNA binding site patterns. The program searches for the parameters of a simple perception network (weight matrix) that maximize the specificity for protein (TF) binding to a positive sequence set (or training set) compared with a background sequence set. The use of background sequences allows the method to find patterns with greater discriminatory capability when compared with the original version of the Gibbs sampling method (Workman and Stormo 2000; GuhaThakurta and Stormo 2001). Binding sites in the positive data set are found with the resulting weight matrix and these sites are then used to define a local multiple sequence alignment. ANN-Spec Version 1.0 was used. A background sequence set of upstream regions from 3000 randomly picked genes was used. Different motif lengths were tried and both strands of the DNA were searched for motifs. Because of the nondeterministic nature of the algorithm, multiple training runs are performed (100), with each run iterating 2000 times. The results were sorted by their best attained objective function values. Weight matrices corresponding to the ten highest scoring runs were compared and if more than five of these top scoring ten runs give a motif with one consistent pattern consensus, that pattern is considered significant.
Calculation of “Site” Scores and Searching for “Sites” in Sequences
A position weight matrix (PWM) has previously been found to be a good model for describing protein binding sites in DNA (Stormo 2000). An l-long DNA binding site pattern may be described by a 4×l weight matrix, with four weights (for four DNA nucleotides) per pattern position. Let us assume each weight in the matrix is the binding energy contribution of each nucleotide at a particular pattern position. With the additional assumption that protein–DNA contacts at individual residue positions in the binding site are independent of each other (Berg and von Hippel 1987), the total binding energy (or score) for a TF molecule to a particular site is given by:
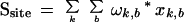 |
5 |
where, ω denotes the PWM weights, x denotes the inputs from the site (DNA bases at different positions), k ranges over the l positions of the site, and b ranges over all four DNA bases.
The Patser program (G.Z. Hertz and G.D. Stormo, unpubl.) allows one to score the words of a given sequence against a weight matrix. Once the weight matrices for regulatory motifs are obtained by Consensus, ANN-Spec, or from the Transfac database the matrices can be used as input for Patser to identify high scoring subsequences (or “sites”) in given sequences. Patser also calculates the p value (or probability) of observing a particular score or higher at a particular sequence position (Staden 1989).
Nonparametric Analysis with Mann-Whitney Statistics
Nonparametric or distribution-free tests may be applied in any situation in which actual measurements are not used, but instead the ranks of the measurements are used. The data may be ranked either from highest to lowest or from lowest to highest values. In our case, we have the pp-values for all C. elegans genes, based on the DNA binding site motifs, arranged in decreasing order. We use the nonparametric analog of the two sampled t test, commonly known as the Mann-Whitney test (Mann and Whitney 1947; Zar 1974).
We take the sorted list of pp-values calculated using either individual motifs or combinations of motifs. We then consider two sets of ranks of the HS up-regulated genes and that of the random genes. Because the labeling of the two samples as 1 and 2 is arbitrary, the Mann-Whitney statistic can be calculated as one of two ways:
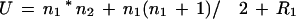 |
6a |
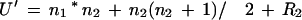 |
6b |
where, n1 and n2 are the two sample sizes and R1 and R2 are the summation of the ranks in the two samples. Usually, U and U‘ are different, and we take only the larger of the two values. It is known that the distribution of the Mann-Whitney statistic approaches normal distribution for large samples with a mean, μu, of n1n2/2. In our case the random sample size is large (3000), so we can use the above approximation. We observe that usually several ranks in the samples are tied; in such cases the standard error is given by:
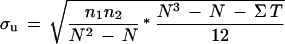 |
7 |
where, Σ T = ti3–ti, ti is the number of ties in a group of tied values, and N = n1 + n2. Therefore, if a U is calculated from data where either n1 or n2 is large, the significance of U can be determined by computing the test statistic:
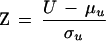 |
8 |
Recalling that the t distribution with infinite degrees of freedom is identical to the normal distribution, the critical value of Z is equal to the critical value of t∞.
Construction of Reporter Plasmids and DNA “Site” Mutation
The vector pPD122.18 (Fire lab 1999 expression vector kit, see http://ftp.ciwemb.edu/PNF:byName:/FireLabWeb/FireLabInfo/FireLabVectors/) was used for the construction of the mutated promoter constructs. This plasmid contains the entire promoter element for the hsp-16–2/16–41 gene pair from C. elegans oriented so that it drives the expression of a GFP coding sequence with four nuclear localization signals from the hsp-16–2 side. (Use of a nuclear-targeted GFP construct simplified quantitation of GFP induction, as it allowed simple counting of GFP+ nuclei.) The promoter of the hsp-16–2 gene contained two HSE and one HSAS sites (Fig. 4A). The two HSE sites had identical sequences corresponding to the HSE consensus, TTCTAGAA, whereas the HSAS site sequence was GGGTCTC. The DNA sites of interest were mutated in the promoter region using the Stratagene QuickChange protocol (Kunkel 1985; Nelson and McClelland 1992), which allows high efficiency mutagenesis. All these sites were altered by substituting noncomplementary bases at all positions (Fig. 4A), and the sequences of the altered sites were confirmed by DNA sequencing.
Microinjection of Vectors and Identification of HS Expression Pattern Using GFP
Injection mixtures were prepared with 100 ng/μL pPD122.18 (or promoter mutation derivative) and 100ng/μL pRF4 (dominant rol-6 marker). Approximately 20 N2 (wild-type) animals were injected for each construct; typically half of the injected animals segregated F1 roller animals. Pooled F1 progeny were propagated at 16°C for 5 d after the injection, heat shocked for 2 hr at 35°C, and then returned to 16°C. Rol F1 animals were recovered and mounted on slides for assaying GFP response 14–16 hr after the end of the HS. Animals were scored using a 40× objective on an Axioskop epifluorescence microscope. pPD122.18 F1 animals were observed carefully for GFP expression before and after the HS treatment; GFP expression was found to be completely HS dependent.
WEB SITE REFERENCES
http://ftp.ciwemb.edu; FireLabWeb.
http://genome.wustl.edu/gsc/Projects/C.briggsae; Washington University Genome Sequencing Center.
http://www.gene-regulation.de; Transfac database.
http://www-gsd.lbl.gov/vista; VISTA.
http://ural.wustl.edu; Stormo lab web site. Access to DNA motif finding program.
http://www.wormbase.org; WormBase.
Acknowledgments
We thank LaDeana Hillier for providing the updated protein and DNA sequences for C. briggsae and Alexander Poliakov for his help with generating the VISTA alignments. We thank Panayotis Benos, Ritesh Agrawal, and German Leparc for technical assistance, and Vadim Kapulkin and Gin Fonte for careful reading of this manuscript. Andrew Fire is thanked for kindly providing the plasmid vector pPD122.18. This work was supported by NIH grant HG00249 to GDS, NIH grant R25 GM62495–01 to LP, and NIH grant AG12423 to CDL.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL linkc@colorado.edu; FAX (303) 492-8063.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.228902.
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone MI, Davidson EH. The hardwiring of development: Organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins: Statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- Bienz M, Pelham HRB. Mechanisms of heat shock gene activation in higher eukaryotes. Adv Genet. 1987;24:31–72. doi: 10.1016/s0065-2660(08)60006-1. [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Bowerman B, Priess JR, Weintraub H. Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science. 1994;266:621–628. doi: 10.1126/science.7939715. [DOI] [PubMed] [Google Scholar]
- Candido EPM, Jones D, Dixon DK, Graham RW, Russnak RH, Kay RJ. Structure, organization, and expression of the 16-kDa heat shock gene family of Caenorhabditis elegans. Genome. 1989;31:690–697. doi: 10.1139/g89-126. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;11:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chen P, Ailion M, Stormo GD, Roth J. Five promoters integrate control of the cob/pdu regulon in Salmonella typhimurium. J Bacteriol. 1995;177:5401–5410. doi: 10.1128/jb.177.19.5401-5410.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, Gish WR, Waterston RH, Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. doi: 10.1101/gr.182901. [DOI] [PubMed] [Google Scholar]
- Clos J, Westwood JT, Becker PB, Wilson S, Lambert K, Wu C. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell. 1990;63:1085–1097. doi: 10.1016/0092-8674(90)90511-c. [DOI] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Egan CR, Chung MA, Allen FL, Heschl MF, Van Buskirk CL, McGhee JD. A gut-to-pharynx/tail switch in embryonic expression of the Caenorhabditis elegans ges-1 gene centers on two GATA sequences. Dev Biol. 1995;170:397–419. doi: 10.1006/dbio.1995.1225. [DOI] [PubMed] [Google Scholar]
- Fernandes M, O’Brian T, Lis JT. Structure and regulation of heat shock gene promoters. In: Morimoto RI, Tissieres A, Georgopolis C, editors. The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. pp. 375–393. [Google Scholar]
- Fukushige T, Hendzel MJ, Bazett-Jones DP, McGhee JD. Direct visualization of the elt-2 gut-specific GATA factor binding to a target promoter inside the living Caenorhabditis elegans embryo. Proc Natl Acad Sci. 1999;96:11883–11888. doi: 10.1073/pnas.96.21.11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta D, Stormo GD. Identifying target sites for cooperatively binding factors. Bioinformatics. 2001;17:608–621. doi: 10.1093/bioinformatics/17.7.608. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta D, Schriefer LA, Hresko MC, Waterston RH, Stormo GD. Proceedings of the 7th Pacific Symposium on Biocomputing. 2002;7:425–435. doi: 10.1142/9789812799623_0040. [DOI] [PubMed] [Google Scholar]
- Hardison R, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;7:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–578. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Heschl MF, Baillie DL. Functional elements and domains inferred from sequence comparisons of a heat shock gene in two nematodes. J Mol Evol. 1990;31:3–9. doi: 10.1007/BF02101786. [DOI] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Iwaki A, Nagano T, Nakagawa M, Iwaki T, Fukumaki Y. Identification and characterization of the gene encoding a new member of the alpha-crystallin/small hsp family, closely linked to the alphaB-crystallin gene in a head-to-head manner. Genomics. 1997;45:386–394. doi: 10.1006/geno.1997.4956. [DOI] [PubMed] [Google Scholar]
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenoharbditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Bogusky MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat shock response. Ann Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat shock response. Ann Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci. 1995;92:7540–7544. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M, Pilgrim D. Conservation of function and expression of unc-119 from two Caenorhabditis species despite divergence of non-coding DNA. Gene. 1996;183:77–85. doi: 10.1016/s0378-1119(96)00491-x. [DOI] [PubMed] [Google Scholar]
- Mann HB, Whitney DR. On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat. 1947;18:50–60. [Google Scholar]
- Mason PB, Lis JT. Cooperative and competitive protein interactions at the Hsp70 promoter. J Biol Chem. 1997;272:33227–33233. doi: 10.1074/jbc.272.52.33227. [DOI] [PubMed] [Google Scholar]
- Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, Derbyshire V, Lawrence CE. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein MM, Hengartner MO, Tsung N, Ellis RE, Horvitz HR. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans genes ces-2. Nature. 1996;382:545–547. doi: 10.1038/382545a0. [DOI] [PubMed] [Google Scholar]
- Morano KA, Thiele DJ. Heat shock factor function and regulation in response to cellular stress, growth, and different ion signals. Gene Expr. 1999;7:271–282. [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Cells in stress: Transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- ————— Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Nakai A. New aspects in the vertebrate heat shock factor system: Hsf3 and Hsf4. Cell Stress Chaperones. 1999;1:215–223. doi: 10.1379/1466-1268(1999)004<0086:naitvh>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M, McClelland M. Use of DNA methyltransferase/endonuclease enzyme combinations for megabase mapping of chromosomes. Methods Enzymol. 1992;216:279–303. doi: 10.1016/0076-6879(92)16027-h. [DOI] [PubMed] [Google Scholar]
- Nover L, Schart K-D, Gagliardi D, Vergne P, Czarnecka-Verner E, Gurley WB. The Hsf world: Classification and properties of plant heat stress transcription factors. Cell Stress Chaperones. 1996;4:86–93. doi: 10.1379/1466-1268(1996)001<0215:thwcap>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJ, Davis EB, Scherer S, Ward S, et al. A global profile of germline gene expression in C. elegans. Mol Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: A new way to display Consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. Methods for calculating the probabilities of finding patterns in sequences. Comput Appl Biosci. 1989;5:89–96. doi: 10.1093/bioinformatics/5.2.89. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Isenberg DA, Nakajima K, Latchman DS. STAT-1 interact and activate the transcription of the Hsp-70 and Hsp-90β gene promoters. J Biol Chem. 1999;274:1723–1728. doi: 10.1074/jbc.274.3.1723. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Information content and free energy in DNA-protein interactions. J Theor Biol. 1998;195:135–137. doi: 10.1006/jtbi.1998.0785. [DOI] [PubMed] [Google Scholar]
- ————— DNA binding sites: Representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Vogelstein B, Kinzler KW. Analyzing unchartered transcriptomes with SAGE. Trends Genet. 2000;16:423–425. doi: 10.1016/s0168-9525(00)02114-4. [DOI] [PubMed] [Google Scholar]
- Wagner A. Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics. 1999;15:776–784. doi: 10.1093/bioinformatics/15.10.776. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fickett JW. Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nat Genet. 2000;26:225–258. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- Werner T. Identification and functional modeling of DNA sequence elements of transcription. Brief Bioinform. 2000;1:372–380. doi: 10.1093/bib/1.4.372. [DOI] [PubMed] [Google Scholar]
- Wiederrecht G, Seto D, Parker CS. Isolation of the gene encoding the S. cervisiae heat shock transcription factor. Cell. 1988;54:841–853. doi: 10.1016/s0092-8674(88)91197-x. [DOI] [PubMed] [Google Scholar]
- Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, Schacherer F. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 2001;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman CT, Stormo GD. Ann-Spec: A method for discovering transcription factor binding sites with improved specificity. Pacific Symp Biocomput. 2000;5:464–475. doi: 10.1142/9789814447331_0044. [DOI] [PubMed] [Google Scholar]
- Wu C. Heat shock transcription factors, structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Yuh CH, Bolouri H, Davidson EH. Genomic cis-regulatory logic: Experimental and computational analysis of a sea urchin gene. Science. 1998;279:1896–902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical analysis. Englewood Cliffs, NJ: Prentice-Hall, Inc.; 1974. pp. 108–113. [Google Scholar]