

Abstract
Papillomavirus E2 is a sequence-specific DNA binding protein that regulates transcription and replication of the viral genome. The transcriptional activities of E2 are typically evaluated by transient transfection of nonreplicating E2-dependent reporters. We sought to address whether E2 activates transcription in an episomal context and its potential interaction with the chromatin remodeling proteins. Using an Epstein-Barr virus-based episomal reporter, we demonstrate that E2 stimulates transcription from an E2-dependent promoter in a chromatin context. This activation is enhanced by the presence of proteins associated with SWI/SNF complexes, which are ATP-dependent chromatin remodeling enzymes. We show that exogenous expression of the Brm ATPase enhances E2 activity in SWI/SNF-deficient cell lines and that the amino-terminal transactivation domain of E2 mediates association with the Brm complex in vivo. Using chromatin immunoprecipitation assays, we demonstrate that Brm enhances promoter occupancy by E2 in an episomal context. Our results demonstrate that E2 activates transcription from an episomal reporter system and reveal a novel property of E2 in collaborating with the Brm chromatin remodeling complex in enhancing transcriptional activation.
The double-stranded, closed circular DNA genomes of papillomaviruses naturally replicate as episomes that are associated with nucleosomal histone proteins (19). In eukaryotes, the packaging of DNA into nucleosomes and subsequent higher-order chromatin structures poses an obstacle to nuclear processes such as transcription, replication, and recombination during DNA repair. Transcription factors and other proteins gain access to nucleosome-bound DNA by using ATP-dependent chromatin modifying and remodeling enzymes. The first proteins implicated in chromatin remodeling were identified in yeast by characterization of defects in mating-type switching (SWI) and sucrose fermentation (SNF [for “sucrose nonfermenting”]) (5, 56, 75). The 2-MDa yeast SWI/SNF complex consists of 11 subunits, with functional homologues found in flies and mammals (59-61, 72, 82). Brahma (Brm), the regulator of Drosophila homeotic genes, was the first SWI/SNF relative to be discovered in higher eukaryotes (77). Humans have at least two genes that are closely related to Brm: hBrm, also known as hSNF2α, and Brm-related gene 1 (Brg1), also known as hSNF2β (9, 35, 54). These SWI/SNF ATPase subunits alone are sufficient for chromatin remodeling activity in vitro (62). Other subunits are thought to be required for stable complex assembly and/or targeting of transcription complexes to the promoter in vivo (55).
The E2 protein has a modular structure with a conserved N-terminal transactivation domain (TAD) of approximately 220 amino acids that serves as a platform for assembly of cellular transcription factors, including TBP, TFIIB, Gps2 (AMF1), and Brd4 (3, 7, 31, 58, 64, 69, 74, 89). The C-terminal 100 amino acids form the DNA binding and dimerization domain (DBD) (52, 63). The intervening nonconserved region is proposed to act as a “hinge” that separates the functional domains of the E2 protein.
Bovine papillomavirus type 1 (BPV1) E2 binds to the palindromic sequence ACCG(N4)CGGT (1), and human papillomavirus (HPV) E2 binds variations on this motif. The long control region (LCR) includes E2 binding motifs and cis elements for cellular factors that regulate the early viral promoters in a complex manner (24). These E2 binding sequences exhibit E2-dependent enhancer activity when present in two or more copies placed either upstream or downstream of a heterologous promoter (27, 73). E2 also activates transcription from E2-dependent promoters on replicating plasmids in Saccharomyces cerevisiae (39, 53). Because the E2 protein regulates viral transcription and replication, we hypothesized that it would interact with SWI/SNF complexes to overcome nucleosome repression.
BPV1 proteins are expressed and its genome is maintained as stable episomes in cultured murine C127 and NIH 3T3 cells. HPV genomes replicate in some epithelial cell-derived cell lines and in primary human keratinocytes but are less stable than the BPV genome in cultured mouse cell lines. HPVs are biologically and phylogenetically differentiated into two groups: “low-risk” types, which cause benign warts and rarely cancer, and “high-risk” types such as HPV-16 and HPV-18, which are associated with the development of cervical and other epithelial malignancies. BPV1 contains 17 E2 binding motifs present primarily within the LCR (44), while HPV-16 and HPV-18 contain four copies of the E2 binding sequences in their LCR (16, 51). During the early stages of infection, it is thought that the high-risk HPV E2 protein represses transcription of the viral E6 and E7 genes by binding to sites situated adjacent to the early promoter and TBP binding site. It was proposed that E2 represses this promoter by interfering with recruitment of the TFIID complex required for initiation of transcription (13-15, 28, 78). This model was supported by the report that introduction of E2 into HPV-induced cervical cancer cell lines led to activation of p53 and pRb (25), which are otherwise inactivated by E6 and E7, respectively (20). A truncated BPV E2 repressor (E2R) that initiates from a promoter embedded within the E2 open reading frame expresses the DBD but excludes a functional TAD and thus can act as a transcriptional repressor, although it does not suppress E6 and E7 expression in HeLa cells (26). Despite repressor functions, BPV and HPV E2 proteins, including those of HPV-16 and HPV-18, are potent transcriptional activators of artificial constructs with E2 binding sites inserted into a promoter. These activator and repressor functions may maintain viral homeostasis by regulation of viral gene expression (79). Nonetheless, the physiologic roles of BPV and HPV E2 transcriptional activation and repression in early infection are not well understood.
It was recently reported that HPV E2 represses transcription from integrated copies of the HPV-16 LCR but has no effect on transcription from episomal HPV-16 genomes in W12-derived cell lines (2). Therefore, to study the transcriptional activation function of E2 in an episomal model, we used a heterologous EBV-based episomal reporter system containing E2 binding sites fused to the simian virus 40 (SV40) minimal promoter. We show that E2 activates transcription in an episomal context and that this activation is enhanced by its interaction with the Brm-containing SWI/SNF chromatin remodeling complex.
MATERIALS AND METHODS
C33A, SW13, HeLa, and NIH 3T3 cells were cultured in Dulbecco modified eagle medium (Gibco) containing 10% bovine calf serum with penicillin and streptomycin. Flag-K804R Brm was cloned into pCG, transfected into SW13 cells, and selected by cotransfection with pBabe-puro, and a stable expression line was generated. FuGene 6 (Roche) was used for transfections at a ratio of 3 μl to 1 μg of DNA. Lipofectamine 2000 was used according to the manufacturer's instructions to transfect Brm and E2 plasmids. After 24 h, cells were washed in phosphate-buffered saline and lysed with 1× luciferase reporter lysis buffer (Promega). Lysates were assayed for luciferase activity with the Luminol substrate (Promega), and relative light units were normalized to 20 μg of protein.
The reporter plasmid containing four E2 binding sites fused to the SV40 minimal promoter was released from construct pGL2 and cloned into pREP4-Luc (K. Zhao, NIH). Flag-tagged human Brm with a Kozak sequence inserted upstream of the coding region was cloned into pCI-Neo (Promega). For coimmunoprecipitation experiments, cells were extracted with lysis buffer (50 mM Tris [pH 8.0], 100 mM NaCl, 20 mM NaF, 50 mM KH2PO4, 1% Triton X-100, 10% glycerol, and 0.1 mM dithiothreitol [DTT]) containing protease inhibitor cocktail. Lysates were sonicated, clarified, and resuspended in binding buffer (50 mM Tris-HCl [pH 8.0], 100 mM KCl, 0.1 mM EDTA, 0.2% NP-40, 2.5% glycerol, and 1 mM DTT). Brm (BD Biosciences), Flag M2 beads (Sigma), and E2 antibodies (58) were captured with protein A/G Sepharose. The beads were washed with wash buffer (100 mM Tris-HCl [pH 8.0], 100 mM NaCl, 0.5% NP-40), and bound proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (PALL), which were probed with the appropriate antibodies and developed with a Pierce chemiluminescence detection kit.
For chromatin immunoprecipitation (ChIP) assays, formaldehyde was added to the cell medium at a final concentration of 1% at 24 h posttransfection. Reactions were stopped with 125 mM glycine. After washes with phosphate-buffered saline, cells were sonicated. Chromatin extract containing 0.1% SDS, 1% Triton X-100, 0.1% sodium deoxycholate, 140 mM NaCl, and 10 mM Tris (pH 7.4) was incubated with the appropriate antibodies and protein G Sepharose. The beads were washed with buffer containing 0.1% SDS, 1% Triton X-100, 0.1% sodium deoxycholate, 140 mM NaCl, and 10 mM Tris (pH 7.4). Following a low-salt wash, beads were washed with high-salt buffer containing 0.1% SDS, 1% Triton X-100, 0.1% sodium deoxycholate, 300 mM NaCl, and 10 mM Tris (pH 7.4). The beads were rinsed with Tris (pH 8)-EDTA and suspended in elution/de-cross-linking buffer containing 100 mM NaHCO3,1% SDS, and 300 mM NaCl at 65°C for 4 h. Eluted DNA was extracted with phenol-chloroform isoamyl alcohol mix and was ethanol precipitated in the presence of 10 μg of glycogen. DNA samples were subjected to PCR amplification followed by agarose gel electrophoresis.
RESULTS
E2 activates transcription from an episomal reporter.
To address whether E2 activates transcription from a chromatin template in vivo, we constructed episomal and conventional nonreplicating (pGL2) reporters containing four high-affinity E2 binding sites upstream of a minimal SV40 promoter. The EBV-based episomal reporter system contains the EBV origin of replication and the EBNA1 coding region and has been used to address the chromatin remodeling activity of several transcription factors (17, 29, 46, 48, 84, 87). Synthetic E2 binding sites along with the SV40 minimal promoter were inserted upstream of the luciferase gene. The nonreplicating reporter does not contain a mammalian origin of replication, and regularly spaced nucleosomes do not assemble on this template. These reporter constructs were cotransfected into C33A, SW13, HeLa, and NIH 3T3 cells in the presence or absence of BPV1 E2 or HPV-16 E2 expression plasmids. C33A and SW13 cells are deficient for Brg1/Brm, while HeLa and NIH 3T3 cells express relatively high levels of Brg1 and Brm. In C33A cells, there was robust activation, of 135- and 100-fold, with BPV1 and HPV16 E2, respectively, from the nonreplicating reporter compared to basal reporter activity (Fig. 1). When an episomal reporter was used, moderate activations of 19- and 15-fold for BPV-1 and HPV-16 E2, respectively, were observed. In SW13 cells, although the activation by the E2 proteins was not as dramatic as in the C33A cell line, we observed a twofold increase in the ratio of activity for nonreplicating reporter compared to the episomal reporter (Fig. 1).
FIG. 1.

Papillomavirus E2 activates transcription in an episomal context. (Top) E2 binding site pREP4-based episomal and pGL2-based nonreplicating vectors with luciferase reporter gene. (Bottom) HPV-16 (HPV) or BPV1 (BPV) E2 and nonreplicating (NR) or episomal (Ep) reporters were transfected into C33A, SW13, HeLa, or NIH 3T3 cells. Luciferase activity was normalized to 20 μg of total protein. Each experiment was performed in duplicate and repeated three times.
In contrast, the inverse was found in HeLa cells. Transcriptional activation was more robust, at 57- and 88-fold for BPV1 and HPV-16, respectively, detected from the episomal reporter compared to 7- and 10-fold with a nonreplicating reporter, respectively (Fig. 1). In NIH 3T3 cells, E2 activation was also more pronounced with the episomal reporter than with nonreplicating reporter. Similarly, the SWI/SNF-positive, HPV-negative cell lines PC3 and H1299 also showed higher transcriptional activity with the episomal reporter than with the nonreplicating reporter (data not shown). In these experiments, equimolar amounts of episomal or nonreplicating plasmids were transfected into the cells, with the total amount of DNA held constant by carrier DNA. Cells were harvested at 24 h posttransfection to limit amplification of the replicating episomal reporter. Using PCR to detect the luciferase gene in Hirt extracts from the transfected cells, we found that comparable levels of both reporters were present (data not shown). We also assayed up to 72 h posttransfection, with results similar to the activities at 24 h, although there was slight decrease in induction in both episomal and nonreplicating reporters (data not shown), which may be due to progressive loss of the E2 expression plasmid. These results suggest that E2 activates transcription from a chromatin context in an episomal system and that this activation is enhanced by the presence of the endogenous SWI/SNF complexes.
Human Brm enhances the transcriptional activity of E2.
Robust transactivation of the episomal reporter compared to a nonreplicating reporter in HeLa cells and the opposite effect in C33A cells led us to predict that chromatin remodeling factors might act as cofactors in transcriptional activation by E2. If this were the case, E2 transcriptional activity might be enhanced in a Brm/Brg1-deficient cell line upon heterologous expression of a SWI/SNF factor such as Brm. To test this, SWI/SNF-deficient C33A cells were transiently transfected with the episomal reporter plasmid, BPV1 E2, and human Brm. In this cell line, Brm enhanced E2-mediated activation by 2.7- to 3.7-fold as a function of increasing amounts of transfected Brm (Fig. 2, top). The transactivation-defective mutant BPV1 E39G failed to show any activity itself or in the presence of Brm. A similar stimulatory response was observed in the Brm/Brg1-deficient cell line SW13; E2 transcriptional activity increased from 2- to 5.5-fold in SW13 cells with increasing amounts of transfected Brm (Fig. 2, bottom). To confirm that the transcriptional increase observed was not due to an increase in E2 transcript levels upon introduction of Brm, we assayed E2 RNA levels by reverse transcription-PCR. We did not observe any significant change in the levels of E2 transcripts (data not shown), which is in agreement with our previous results that the Brm protein does not significantly alter transcription from a nonreplicating plasmid (E2 is expressed from the CMV promoter). These data suggest that enhancement of E2 transactivation is facilitated by SWI/SNF factors.
FIG. 2.

Enhanced transactivation by E2 requires hBrm. Episomal E2 binding site reporter was cotransfected with either BPV1 E2 or Brm alone and E2 along with increasing amounts of hBrm (0.5, 1, and 1.5 μg) into the SWI/SNF-deficient cell lines C33A and SW13. Cells were harvested and assayed for luciferase activity and normalized to 20 μg of total protein.
E2 interacts with the Brm complex.
We reasoned that for enhanced transcriptional activation, E2 might physically interact with the Brm complex in vivo. Our attempts to create a stable cell line expressing wild-type Brm were unsuccessful, as overexpression of Brm proved to be lethal to SWI/SNF-deficient SW13 cells. This difficulty was overcome by creating a stable SW13 cell line expressing a Brm protein containing a C-terminal Flag epitope and a mutation (K804R) in the ATPase domain. The stable SW13 cells (Brm NTP-Flag) were transfected with BPV1 E2 and harvested after 24 h. The Brm complexes were immunoprecipitated with anti-Flag monoclonal antibody, and the bound proteins were analyzed for the presence of E2 by resolving the precipitates by SDS-PAGE and subsequent Western analysis using E2 antibodies. The immunoblotting results showed that E2 associated with the Brm complex (Fig. 3a). This interaction is specific, as control antibodies did not coprecipitate the E2 protein.
FIG. 3.
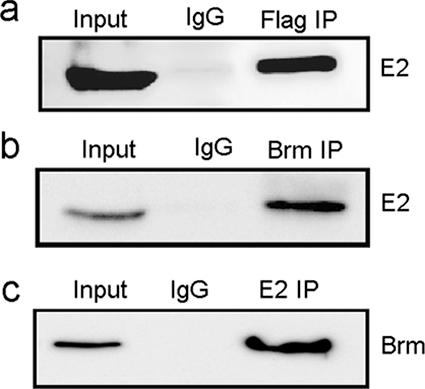
E2 associates with the Brm complex in vivo. (a) SW13 cells stably expressing Flag-tagged ATPase mutant Brm were transfected with BPV1 E2. The cell extract was immunoprecipitated with anti-Flag M2 beads or mouse immunoglobulin G (IgG), resolved by SDS-PAGE, and immunoblotted with E2 antibodies. (b) HeLa cells were transfected with E2 plasmid, the cell extract was immunoprecipitated with Brm or control mouse antibodies, and the immunoblot was reacted with E2 antibodies. (c) C33A cells were transfected with Flag-hBrm and E2, the cell extract was immunoprecipitated with rabbit E2 or control antibodies, complexes were resolved by SDS-PAGE, and the blot was probed with anti-Flag M2 antibodies.
Brm is a 180-kDa protein, and we had difficulties expressing it at detectable levels in transient transfection experiments. This was overcome by introducing a Kozak sequence at the translation initiation site of the Brm expression vector. This modified Flag-tagged Brm plasmid was introduced into C33A cells along with an E2 expression construct. E2 was immunoprecipitated with rabbit antiserum to E2, and the immunoprecipitate was resolved by SDS-PAGE and then immunoblotted with Flag antibody. The results show that E2 is in complex with Brm (Fig. 3c). Our unrelated control antibody did not coimmunoprecipitate the Brm complex, indicating that the E2-Brm immunoprecipitation is specific.
To test whether endogenous Brm complexes with E2, we used HeLa cells that constitutively express high levels of Brm in coimmunoprecipitation-Western blot experiments. HeLa cells were transfected with BPV-1 E2 and the Brm-E2 complexes were harvested 24 h posttransfection to avoid senescence due to the repression of E6/E7 transcription (25). Lysates were immunoprecipitated with a Brm monoclonal antibody and subjected to Western analysis for E2. Our results indicate that endogenous Brm associates with the BPV E2 protein and that this interaction is specific, as the control antibody did not immunoprecipitate E2 (Fig. 3b).
E2 associates with the Brm complex through its N-terminal domain.
Having demonstrated that E2 associates with the Brm complex, we proceeded to identify the domain of E2 required for this interaction. C33A cells were cotransfected with Flag-tagged Brm and either with full-length BPV E2 or a series of E2 deletion constructs (6, 89) (Fig. 4a). Cell lysates were subjected to immunoprecipitation with M2 beads to capture the Brm complexes and immunoblotted with E2 antibodies. Brm complexed with full-length E2, E2-TAD (1 to 216), and E2-Δ hinge constructs containing the transactivation domain of E2; however, it did not associate with E2R (160 to 410), E2 (55 to 410), or E2 (113 to 410) (Fig. 4a and b). These results imply that the E2-TAD is required for interaction with the Brm complex.
FIG. 4.
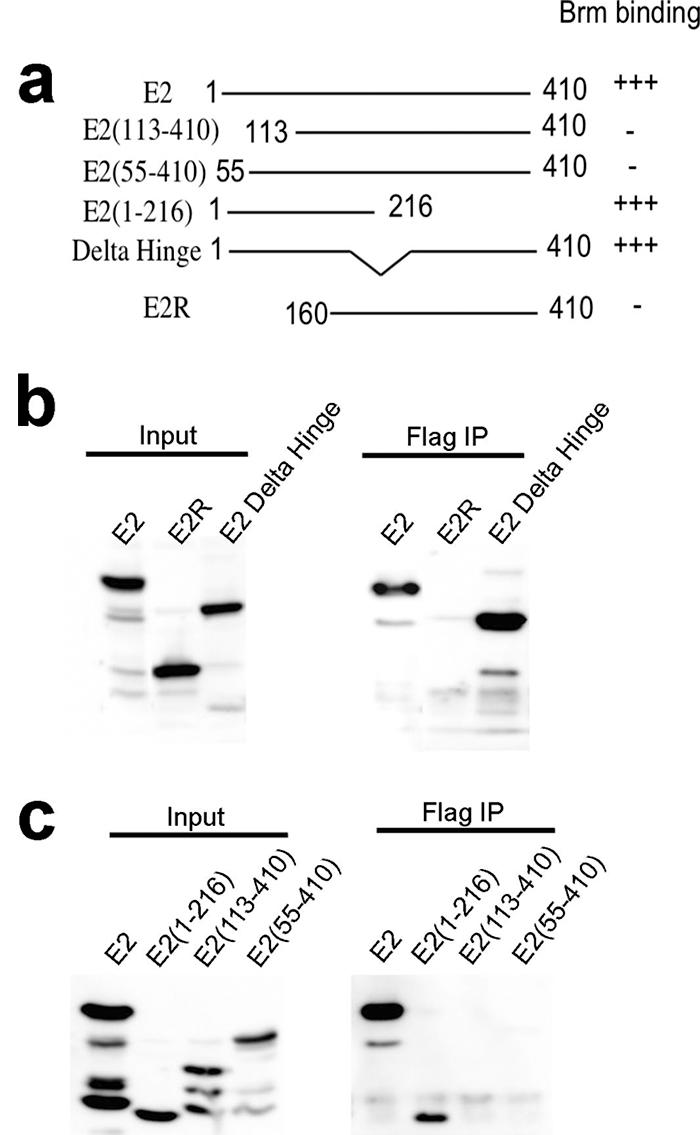
E2 association with the Brm complex requires the E2 N-terminal activation domain. (a) Diagram of BPV1 E2 deletion constructs. (b and c) Lysates from C33A cells cotransfected with hBrm and the series of E2 deletion expression vectors were subjected to immunoprecipitation (IP) with anti-Flag M2 beads, and the bound complexes were probed with E2 antiserum by immunoblotting.
Increased E2 recruitment to E2 binding sequence by Brm.
Our coimmunoprecipitation experiments demonstrate that E2 physically interacts with the Brm complex, and the reporter assays show that E2 enhanced transcription from episomal promoters. To address whether Brm increases the occupancy of E2 onto its cognate binding sequences on this episomal promoter, we performed ChIP assays with SW13 cells. Only trace levels of the 300-bp region of the episomal reporter encompassing the four E2 binding sites were amplified following ChIP assay with E2 antibodies (Fig. 5a, top). When both E2 and Brm were cotransfected into SW13 cells, there was increased E2-bound reporter DNA detected by PCR (Fig. 5a, bottom). To confirm the enhanced recruitment of E2 in the presence of Brm on E2 binding sites, we performed PCR with increasing volumes of input and immunoprecipitated DNA (Fig. 5b; compare the top and bottom panels). To test whether Brm-enhanced E2 recruitment to E2 binding sites requires the adjacent promoter and associated transcription factors, we performed ChIP assays using an episomal plasmid containing E2 binding sites but without the promoter region. Our results show that exogenous Brm increased the loading of E2 onto E2 binding sites (Fig. 5c and d). These results demonstrate that Brm facilitates E2 occupancy on its cognate binding sites.
FIG. 5.
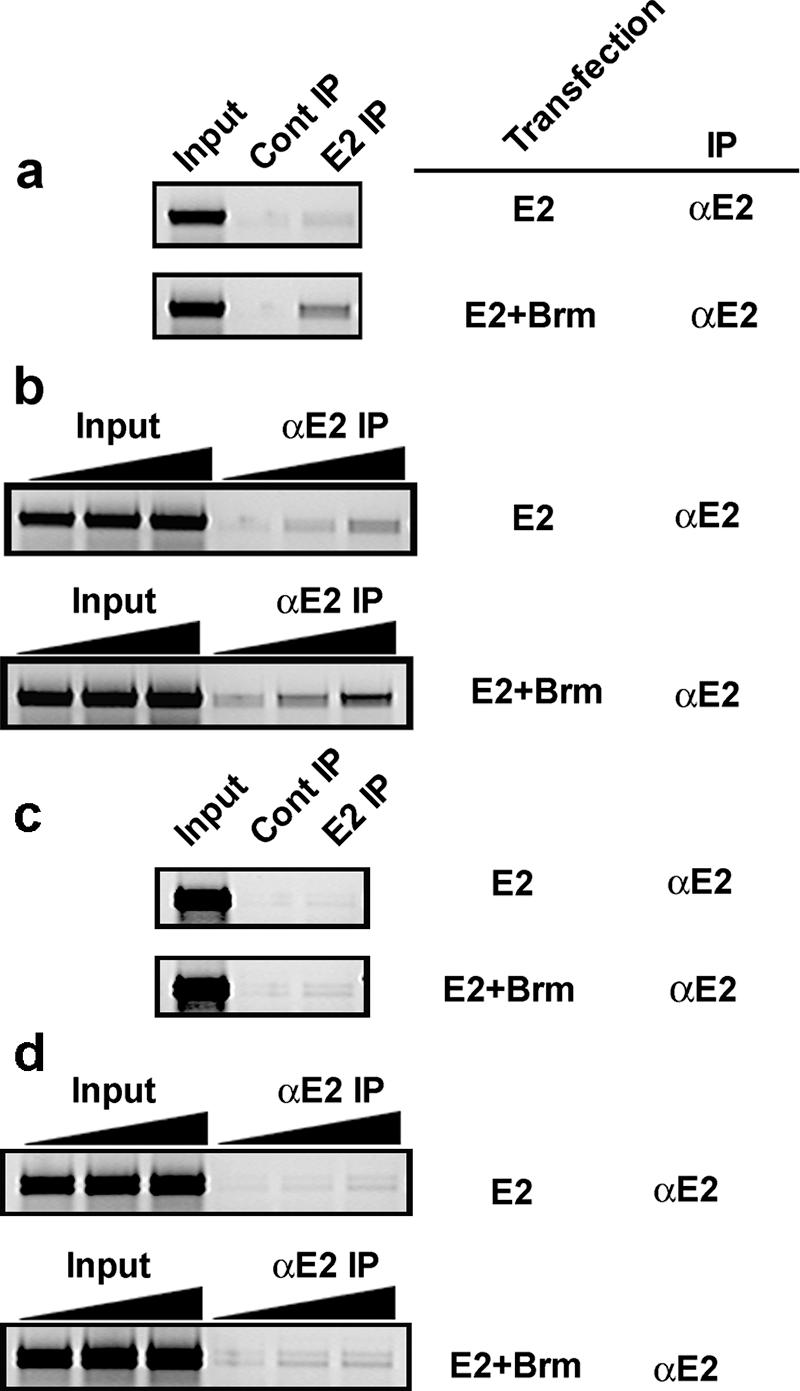
Brm increases the loading of E2 on E2 DNA binding sites. (a) The episomal reporter containing E2 binding sites was introduced into SW13 cells together with E2 alone or E2 plus hBrm. Formaldehyde was directly added to the medium at 24 h after transfection, and chromatin extract was subjected to E2 ChIP assay. (b) PCR was performed with increasing volumes (0.5 to 2 μl) of chromatin extracts for E2 ChIP assays, as described for panel a. (c) As in panel a, cotransfection of the episomal vector containing E2 binding sites but without the adjacent promoter sequences along with E2, in the presence or absence of exogenous hBrm, and subjected to E2 ChIP assay. (d) PCR was performed with increasing volumes (0.5 to 2 μl) of chromatin extracts and E2 ChIP assays, as described for panel c. Cont, control; IP, immunoprecipitation.
DISCUSSION
Viral transcription is programmed by E2 binding sequences and multiple regulatory elements present in the LCR. Transcription of the early LCR promoter is repressed by heterologous expression of E2, but the contribution of E2 transcriptional activation in the context of the genome has not been fully evaluated. While the transcriptional activation property of E2 is highly conserved (36, 80), this is typically studied by transient transfection of a nonreplicating E2-dependent reporter. One function of E2 proposed to be necessary for activation of transcription and replication is relief of nucleosome-mediated repression (42, 43, 88). Therefore, to address whether E2 can activate or repress transcription in the context of episomes, independent of secondary effects on PV protein expression and viral genome replication, we undertook a conventional approach in which E2 binding sites were positioned upstream of a minimal promoter and introduced into an EBV-based episomal luciferase reporter. EBV-based episomal luciferase reporter systems have been exploited to address the role of chromatin remodeling factors in transcription. These studies have shown that to assess the transcriptional activity of SWI/SNF remodeling factors, an episomal reporter system is more suitable than a nonreplicating reporter (17, 29, 46, 48, 84, 87). Using this assay system, we show that the BPV1 and HPV-16 E2 proteins activate transcription from an episomal promoter. This activation is enhanced in cells in which chromatin remodeling enzymes are abundant and is correspondingly reduced in those that are deficient.
Interestingly, with an episomal reporter system, E2 transcriptional activity increased relative to a nonreplicating reporter in the cell lines that express SWI/SNF factors. In HeLa cells, E2 showed robust activation with an episomal reporter compared to a nonreplicating reporter. HeLa cells have been extensively used to purify and characterize the components of SWI/SNF remodeling complexes (37, 38, 57, 70, 71, 83). In contrast, E2 showed weaker transcriptional activity on an episomal reporter than on a nonreplicating reporter in C33A cells. Correspondingly, C33A cells do not express detectable levels of Brm/Brg1 (54). We also observed a similar pattern of reduced activity with an episomal reporter compared to a nonreplicating reporter in SW13 cells. SW13 is commonly used as a SWI/SNF-deficient cell line (18, 46, 48). Importantly, heterologous expression of Brm enhanced the transcriptional activity of E2 from the episomal reporters in C33A and SW13 cells. We also observed relatively higher transcriptional activity of E2 on an episomal reporter than on a nonreplicating reporter in other SWI/SNF-positive cell lines, such as PC3 and H1299 (data not shown). A recent report indicated that the HPV-16 E2 protein is unable to activate transcription from an episomal HPV-16 genome present in the established cell line W12 (2). This outcome may also be complicated by the physiologic regulation of HPV E6 and E7, on which the cells depend for viability.
We show that E2 interacts with endogenous human Brm expressed in HeLa cells and Brm K804R expressed stably in SW13 cells. BPV and HPV E2 transcriptional activation correlates with levels of the Brm factor in transiently transfected Brg1/Brm-deficient cells. Our mapping results showed that the E2 amino-terminal TAD region is required for interaction with the Brm complex. It remains to be determined whether the association of E2 with Brm is direct or mediated by other factors. Brm interaction mapped to a large region of the E2 TAD, suggesting that multiple contacts and factors may be necessary for assembly of a stable Brm SWI/SNF complex. The E2 binding proteins Gps2 and hNAP-1 interact with p300 and probably connect E2 to this histone acetylase to modify chromatin access (7, 58, 65). TopBP1 is another candidate factor that may bridge E2 and Brm, as it interacts with the SWI/SNF remodeling complex (4, 47).
Viral proteins are known to interact with chromatin-modifying factors or remodeling complexes (45). HPV E7 interacts with the chromatin remodeling factor Brg1 and deregulates the transcriptional properties of Brg1 (40). Likewise, several viral proteins, such as the human immunodeficiency virus type 1 integrase, Tat, Tax, EBNA2, E1A, and SV40 large T protein, interact with chromatin-modifying factors in modulating their transcriptional properties (21-23, 50, 86, 90). Our coimmunoprecipitation results extend this list to papillomavirus E2. Interestingly, HPV E1 protein has been reported to directly interact with the nucleosomal protein H1 (76) and with the SWI/SNF chromatin remodeling component Ini1 (41). An E1 mutant defective for Ini1 binding failed to support replication of the viral genome. These observations suggest that E1 engages these enzymes during S phase to drive viral episome replication. E2 may also have a role in replication, as it was shown that E2 opposes the nucleosomal repression of the viral genome and facilitates its replication in vitro (42). Interestingly, E2 binding sites and additional elements that extend beyond the minimal replication origin that is otherwise sufficient for transient DNA replication are necessary for BPV genome maintenance (30). In an elegant study that investigated chromatin structure, nucleosome rearrangement was observed on the HPV-31 genome during epithelial cell differentiation and late gene expression was induced with histone deacetylase inhibitor (12).
SWI/SNF enzymes are proposed to disrupt histone-DNA contacts such that the histone octamer slides along the DNA, thereby mobilizing and repositioning the nucleosomes (33, 85). Although the precise mechanism by which SWI/SNF complexes disrupt nucleosome structure is still not fully resolved, recent data support the notion that these remodeling enzymes act as DNA translocating motors (67). Remodeling activity leads to enhanced accessibility of nucleosomal DNA to sequence-specific DNA binding proteins and facilitates numerous aspects of development, differentiation, cell cycle control, signaling, and transcriptional regulation (10, 11, 32, 38, 49, 66, 67, 81).
Numerous studies have shown that transcription factors recruit the SWI/SNF complex to promoter regions during transcriptional activation (8, 11, 34, 68). By ChIP assays, we show that Brm increases promoter occupancy by E2 on a chromatin template. These results suggest that E2 exploits the cellular Brm complex for its transcriptional activity. Moreover, our ChIP assays suggest that Brm enhances the occupancy of E2 on its binding sites in the absence of the minimal promoter. We do not know at this point whether the accessibility or stability of E2 protein is increased at the site of transcription by interaction with the SWI/SNF complex. Overall, our findings are in agreement with published reports and with the prediction that nucleosomes on the viral genome pose an obstacle to the transcription and replication machinery, an obstacle which is overcome by recruitment of the SWI/SNF complex by E2.
Acknowledgments
We thank K. Zhao (NIH) for pREP4-luciferase and Said Sif (Ohio State) for plasmids.
This work was supported by NIH grants R01 CA58376 to E.J.A. and R01 GM56244 to A.N.I.
Footnotes
Published ahead of print on 6 December 2006.
REFERENCES
- 1.Androphy, E. J., D. R. Lowy, and J. T. Schiller. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70-73. [DOI] [PubMed] [Google Scholar]
- 2.Bechtold, V., P. Beard, and K. Raj. 2003. Human papillomavirus type 16 E2 protein has no effect on transcription from episomal viral DNA. J. Virol. 77:2021-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson, J. D., R. Lawande, and P. M. Howley. 1997. Conserved interaction of the papillomavirus E2 transcriptional activator proteins with human and yeast TFIIB proteins. J. Virol. 71:8041-8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boner, W., E. R. Taylor, E. Tsirimonaki, K. Yamane, M. S. Campo, and I. M. Morgan. 2002. A functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 277:22297-22303. [DOI] [PubMed] [Google Scholar]
- 5.Breeden, L., and K. Nasmyth. 1987. Cell cycle control of the yeast HO gene: cis- and trans-acting regulators. Cell 48:389-397. [DOI] [PubMed] [Google Scholar]
- 6.Breiding, D. E., M. J. Grossel, and E. J. Androphy. 1996. Genetic analysis of the bovine papillomavirus E2 transcriptional activation domain. Virology 221:34-43. [DOI] [PubMed] [Google Scholar]
- 7.Breiding, D. E., F. Sverdrup, M. J. Grossel, N. Moscufo, W. Boonchai, and E. J. Androphy. 1997. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol. Cell. Biol. 17:7208-7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, R. C., S. Pattison, J. van Ree, E. Coghill, A. Perkins, S. M. Jane, and J. M. Cunningham. 2002. Distinct domains of erythroid Krüppel-like factor modulate chromatin remodeling and transactivation at the endogenous β-globin gene promoter. Mol. Cell. Biol. 22:161-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiba, H., M. Muramatsu, A. Nomoto, and H. Kato. 1994. Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 22:1815-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cote, J., J. Quinn, J. L. Workman, and C. L. Peterson. 1994. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 265:53-60. [DOI] [PubMed] [Google Scholar]
- 11.de la Serna, I. L., Y. Ohkawa, and A. N. Imbalzano. 2006. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat. Rev. Genet. 7:461-473. [DOI] [PubMed] [Google Scholar]
- 12.del Mar Pena, L. M., and L. A. Laimins. 2001. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J. Virol. 75:10005-10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demeret, C., C. Desaintes, M. Yaniv, and F. Thierry. 1997. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J. Virol. 71:9343-9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, G., T. R. Broker, and L. T. Chow. 1994. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J. Virol. 68:1115-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dostatni, N., P. F. Lambert, R. Sousa, J. Ham, P. M. Howley, and M. Yaniv. 1991. The functional BPV-1 E2 trans-activating protein can act as a repressor by preventing formation of the initiation complex. Genes Dev. 5:1657-1671. [DOI] [PubMed] [Google Scholar]
- 16.Dostatni, N., F. Thierry, and M. Yaniv. 1988. A dimer of BPV-1 E2 containing a protease resistant core interacts with its DNA target. EMBO J. 7:3807-3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan, H., C. A. Heckman, and L. M. Boxer. 2005. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 25:1608-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan, H. Y., K. W. Trotter, T. K. Archer, and R. E. Kingston. 2005. Swapping function of two chromatin remodeling complexes. Mol. Cell 17:805-815. [DOI] [PubMed] [Google Scholar]
- 19.Favre, M., F. Breitburd, O. Croissant, and G. Orth. 1977. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 21:1205-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fehrmann, F., and L. A. Laimins. 2003. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene 22:5201-5207. [DOI] [PubMed] [Google Scholar]
- 21.Fry, C. J., and C. L. Peterson. 2001. Chromatin remodeling enzymes: who's on first? Curr. Biol. 11:R185-R197. [DOI] [PubMed] [Google Scholar]
- 22.Gallimore, P. H., and A. S. Turnell. 2001. Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene 20:7824-7835. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh, M. K., and M. L. Harter. 2003. A viral mechanism for remodeling chromatin structure in G0 cells. Mol. Cell 12:255-260. [DOI] [PubMed] [Google Scholar]
- 24.Gloss, B., M. Kalantari, and H. U. Bernard. 2005. Analysis of the regulation of viral transcription. Methods Mol. Med. 119:261-278. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin, E. C., and D. DiMaio. 2000. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 97:12513-12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodwin, E. C., L. K. Naeger, D. E. Breiding, E. J. Androphy, and D. DiMaio. 1998. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J. Virol. 72:3925-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawley-Nelson, P., E. J. Androphy, D. R. Lowy, and J. T. Schiller. 1988. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J. 7:525-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou, S. Y., S. Y. Wu, T. Zhou, M. C. Thomas, and C. M. Chiang. 2000. Alleviation of human papillomavirus E2-mediated transcriptional repression via formation of a TATA binding protein (or TFIID)-TFIIB-RNA polymerase II-TFIIF preinitiation complex. Mol. Cell. Biol. 20:113-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, M., F. Qian, Y. Hu, C. Ang, Z. Li, and Z. Wen. 2002. Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nat. Cell Biol. 4:774-781. [DOI] [PubMed] [Google Scholar]
- 30.Ilves, I., S. Kivi, and M. Ustav. 1999. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which is mediated by the viral E2 protein and its binding sites. J. Virol. 73:4404-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ilves, I., K. Maemets, T. Silla, K. Janikson, and M. Ustav. 2006. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 80:3660-3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imbalzano, A. N., H. Kwon, M. R. Green, and R. E. Kingston. 1994. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 370:481-485. [DOI] [PubMed] [Google Scholar]
- 33.Jaskelioff, M., I. M. Gavin, C. L. Peterson, and C. Logie. 2000. SWI-SNF-mediated nucleosome remodeling: role of histone octamer mobility in the persistence of the remodeled state. Mol. Cell. Biol. 20:3058-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kadam, S., and B. M. Emerson. 2003. Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol. Cell 11:377-389. [DOI] [PubMed] [Google Scholar]
- 35.Khavari, P. A., C. L. Peterson, J. W. Tamkun, D. B. Mendel, and G. R. Crabtree. 1993. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature 366:170-174. [DOI] [PubMed] [Google Scholar]
- 36.Kovelman, R., G. K. Bilter, E. Glezer, A. Y. Tsou, and M. S. Barbosa. 1996. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses. J. Virol. 70:7549-7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuzmichev, A., Y. Zhang, H. Erdjument-Bromage, P. Tempst, and D. Reinberg. 2002. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol. Cell. Biol. 22:835-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon, H., A. N. Imbalzano, P. A. Khavari, R. E. Kingston, and M. R. Green. 1994. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 370:477-481. [DOI] [PubMed] [Google Scholar]
- 39.Lambert, P. F., N. Dostatni, A. A. McBride, M. Yaniv, P. M. Howley, and B. Arcangioli. 1989. Functional analysis of the papilloma virus E2 trans-activator in Saccharomyces cerevisiae. Genes Dev. 3:38-48. [DOI] [PubMed] [Google Scholar]
- 40.Lee, D., C. Lim, T. Seo, H. Kwon, H. Min, and J. Choe. 2002. The viral oncogene human papillomavirus E7 deregulates transcriptional silencing by Brm-related gene 1 via molecular interactions. J. Biol. Chem. 277:48842-48848. [DOI] [PubMed] [Google Scholar]
- 41.Lee, D., H. Sohn, G. V. Kalpana, and J. Choe. 1999. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature 399:487-491. [DOI] [PubMed] [Google Scholar]
- 42.Li, R., and M. R. Botchan. 1994. Acidic transcription factors alleviate nucleosome-mediated repression of DNA replication of bovine papillomavirus type 1. Proc. Natl. Acad. Sci. USA 91:7051-7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li, R., and M. R. Botchan. 1993. The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell 73:1207-1221. [DOI] [PubMed] [Google Scholar]
- 44.Li, R., J. Knight, G. Bream, A. Stenlund, and M. Botchan. 1989. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev. 3:510-526. [DOI] [PubMed] [Google Scholar]
- 45.Lieberman, P. M. 2006. Chromatin regulation of virus infection. Trends Microbiol. 14:132-140. [DOI] [PubMed] [Google Scholar]
- 46.Liu, H., H. Kang, R. Liu, X. Chen, and K. Zhao. 2002. Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol. Cell. Biol. 22:6471-6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu, K., Y. Luo, F. T. Lin, and W. C. Lin. 2004. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 18:673-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu, R., H. Liu, X. Chen, M. Kirby, P. O. Brown, and K. Zhao. 2001. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106:309-318. [DOI] [PubMed] [Google Scholar]
- 49.Logie, C., and C. L. Peterson. 1997. Catalytic activity of the yeast SWI/SNF complex on reconstituted nucleosome arrays. EMBO J. 16:6772-6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marzio, G., M. Tyagi, M. I. Gutierrez, and M. Giacca. 1998. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 95:13519-13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McBride, A. A., J. B. Bolen, and P. M. Howley. 1989. Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J. Virol. 63:5076-5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McBride, A. A., J. C. Byrne, and P. M. Howley. 1989. E2 polypeptides encoded by bovine papillomavirus type 1 form dimers through the common carboxyl-terminal domain: transactivation is mediated by the conserved amino-terminal domain. Proc. Natl. Acad. Sci. USA 86:510-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrissey, L. C., J. Barsoum, and E. J. Androphy. 1989. Trans-activation by the bovine papillomavirus E2 protein in Saccharomyces cerevisiae. J. Virol. 63:4422-4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muchardt, C., and M. Yaniv. 1993. A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J. 12:4279-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neely, K. E., A. H. Hassan, C. E. Brown, L. Howe, and J. L. Workman. 2002. Transcription activator interactions with multiple SWI/SNF subunits. Mol. Cell. Biol. 22:1615-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neigeborn, L., and M. Carlson. 1987. Mutations causing constitutive invertase synthesis in yeast: genetic interactions with snf mutations. Genetics 115:247-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pal, S., R. Yun, A. Datta, L. Lacomis, H. Erdjument-Bromage, J. Kumar, P. Tempst, and S. Sif. 2003. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol. Cell. Biol. 23:7475-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peng, Y. C., D. E. Breiding, F. Sverdrup, J. Richard, and E. J. Androphy. 2000. AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J. Virol. 74:5872-5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peterson, C. L. 2002. Chromatin remodeling: nucleosomes bulging at the seams. Curr. Biol. 12:R245-R247. [DOI] [PubMed] [Google Scholar]
- 60.Peterson, C. L., and J. W. Tamkun. 1995. The SWI-SNF complex: a chromatin remodeling machine? Trends Biochem. Sci. 20:143-146. [DOI] [PubMed] [Google Scholar]
- 61.Peterson, C. L., and J. L. Workman. 2000. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin. Genet. Dev. 10:187-192. [DOI] [PubMed] [Google Scholar]
- 62.Phelan, M. L., S. Sif, G. J. Narlikar, and R. E. Kingston. 1999. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 3:247-253. [DOI] [PubMed] [Google Scholar]
- 63.Prakash, S. S., S. R. Grossman, R. B. Pepinsky, L. A. Laimins, and E. J. Androphy. 1992. Amino acids necessary for DNA contact and dimerization imply novel motifs in the papillomavirus E2 trans-activator. Genes Dev. 6:105-116. [DOI] [PubMed] [Google Scholar]
- 64.Rank, N. M., and P. F. Lambert. 1995. Bovine papillomavirus type 1 E2 transcriptional regulators directly bind two cellular transcription factors, TFIID and TFIIB. J. Virol. 69:6323-6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rehtanz, M., H. M. Schmidt, U. Warthorst, and G. Steger. 2004. Direct interaction between nucleosome assembly protein 1 and the papillomavirus E2 proteins involved in activation of transcription. Mol. Cell. Biol. 24:2153-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roberts, C. W., and S. H. Orkin. 2004. The SWI/SNF complex—chromatin and cancer. Nat. Rev. Cancer 4:133-142. [DOI] [PubMed] [Google Scholar]
- 67.Saha, A., J. Wittmeyer, and B. R. Cairns. 2006. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell. Biol. 7:437-447. [DOI] [PubMed] [Google Scholar]
- 68.Salma, N., H. Xiao, E. Mueller, and A. N. Imbalzano. 2004. Temporal recruitment of transcription factors and SWI/SNF chromatin-remodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor γ nuclear hormone receptor. Mol. Cell. Biol. 24:4651-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schweiger, M. R., J. You, and P. M. Howley. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 80:4276-4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sif, S., A. J. Saurin, A. N. Imbalzano, and R. E. Kingston. 2001. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev. 15:603-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sif, S., P. T. Stukenberg, M. W. Kirschner, and R. E. Kingston. 1998. Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev. 12:2842-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith, C. L., R. Horowitz-Scherer, J. F. Flanagan, C. L. Woodcock, and C. L. Peterson. 2003. Structural analysis of the yeast SWI/SNF chromatin remodeling complex. Nat. Struct. Biol. 10:141-145. [DOI] [PubMed] [Google Scholar]
- 73.Spalholz, B. A., Y. C. Yang, and P. M. Howley. 1985. Transactivation of a bovine papilloma virus transcriptional regulatory element by the E2 gene product. Cell 42:183-191. [DOI] [PubMed] [Google Scholar]
- 74.Steger, G., J. Ham, O. Lefebvre, and M. Yaniv. 1995. The bovine papillomavirus 1 E2 protein contains two activation domains: one that interacts with TBP and another that functions after TBP binding. EMBO J. 14:329-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stern, M., R. Jensen, and I. Herskowitz. 1984. Five SWI genes are required for expression of the HO gene in yeast. J. Mol. Biol. 178:853-868. [DOI] [PubMed] [Google Scholar]
- 76.Swindle, C. S., and J. A. Engler. 1998. Association of the human papillomavirus type 11 E1 protein with histone H1. J. Virol. 72:1994-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tamkun, J. W., R. Deuring, M. P. Scott, M. Kissinger, A. M. Pattatucci, T. C. Kaufman, and J. A. Kennison. 1992. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68:561-572. [DOI] [PubMed] [Google Scholar]
- 78.Tan, S. H., L. C. Leong, P. A. Walker, and H. U. Bernard. 1994. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J. Virol. 68:6411-6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thierry, F., and M. Yaniv. 1987. The BPV-1 trans-acting protein can be either an activator or a repressor of the HPV 18 regulatory region. EMBO J. 6:3391-3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ushikai, M., M. J. Lace, Y. Yamakawa, M. Kono, J. Anson, T. Ishiji, S. Parkkinen, N. Wicker, M. E. Valentine, I. Davidson, L. P. Turek, and T. H. Haugen. 1994. trans activation by the full-length E2 proteins of human papillomavirus type 16 and bovine papillomavirus type 1 in vitro and in vivo: cooperation with activation domains of cellular transcription factors. J. Virol. 68:6655-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Utley, R. T., J. Cote, T. Owen-Hughes, and J. L. Workman. 1997. SWI/SNF stimulates the formation of disparate activator-nucleosome complexes but is partially redundant with cooperative binding. J. Biol. Chem. 272:12642-12649. [DOI] [PubMed] [Google Scholar]
- 82.Vignali, M., A. H. Hassan, K. E. Neely, and J. L. Workman. 2000. ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol. 20:1899-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang, W., J. Cote, Y. Xue, S. Zhou, P. A. Khavari, S. R. Biggar, C. Muchardt, G. V. Kalpana, S. P. Goff, M. Yaniv, J. L. Workman, and G. R. Crabtree. 1996. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15:5370-5382. [PMC free article] [PubMed] [Google Scholar]
- 84.Wen, J., S. Huang, H. Rogers, L. A. Dickinson, T. Kohwi-Shigematsu, and C. T. Noguchi. 2005. SATB1 family protein expressed during early erythroid differentiation modifies globin gene expression. Blood 105:3330-3339. [DOI] [PubMed] [Google Scholar]
- 85.Whitehouse, I., A. Flaus, B. R. Cairns, M. F. White, J. L. Workman, and T. Owen-Hughes. 1999. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature 400:784-787. [DOI] [PubMed] [Google Scholar]
- 86.Wu, D. Y., G. V. Kalpana, S. P. Goff, and W. H. Schubach. 1996. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J. Virol. 70:6020-6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu, Z., H. P. Kim, H. H. Xue, H. Liu, K. Zhao, and W. J. Leonard. 2005. Interleukin-21 receptor gene induction in human T cells is mediated by T-cell receptor-induced Sp1 activity. Mol. Cell. Biol. 25:9741-9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang, L., R. Li, I. J. Mohr, R. Clark, and M. R. Botchan. 1991. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature 353:628-632. [DOI] [PubMed] [Google Scholar]
- 89.Yao, J. M., D. E. Breiding, and E. J. Androphy. 1998. Functional interaction of the bovine papillomavirus E2 transactivation domain with TFIIB. J. Virol. 72:1013-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yung, E., M. Sorin, A. Pal, E. Craig, A. Morozov, O. Delattre, J. Kappes, D. Ott, and G. V. Kalpana. 2001. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 7:920-926. [DOI] [PubMed] [Google Scholar]