

Abstract
Although CD8+ T cells play an important role in controlling viral infections, boosting specific CD8+ T cells by prophylactic vaccination with simian immunodeficiency virus (SIV) epitopes fails to provide sterilizing immunity. Viral replication rates and viral contraction rates after the peak viremia hardly depend on the presence of memory CD8+ T cells. To study these paradoxical findings, we parameterize novel mathematical models for acute SIV and human immunodeficiency virus infection. These models explain that failure of vaccination is due to the fact that effector/target ratios are too low during the viral expansion phase. Because CD8+ T cells require cell-to-cell contacts, immune protection requires high effector/target ratios at the primary site of infection. Effector/target ratios become favorable for immune control at the time of the peak in the viral load when the virus becomes limited by other factors, such as the availability of uninfected target cells. At the viral set point, effector/target ratios are much higher, and perturbations of the number of CD8+ effector cells have a large impact on the viral load. Such protective effector/target ratios are difficult to achieve with nucleic acid- or protein-based vaccines.
CD8+ T cells play an important role in controlling viral infection because CD8+ cytotoxic effector T cells (CTLs) clear infected cells displaying viral peptides on their class I HLA molecules. CD8+ T cells are also known to be important in controlling simian immunodeficiency virus (SIV) and human immunodeficiency virus (HIV) infection because (i) the depletion of CD8+ T cells during chronic SIV infection in monkeys increases the viral load (46, 60, 85), (ii) human HIV-positive patients who are heterozygous at class I HLA loci have slower rates of disease progression (18), and (iii) the virus accumulates mutations in CD8+ T-cell epitopes (33, 37, 59). The various epitope-specific CD8+ T-cell responses during chronic viral infection with HIV can amount to up to 10% of the total number of CD8+ T cells (13). Similar percentages have been reported for CD4+ and CD8+ T-cell responses in healthy human carriers of cytomegalovirus (90). Given a total human body count of 1011 CD8+ T cells (20), the total antiviral CD8+ T-cell response amounts to 1010 effector cells. Although it remains difficult to estimate total body numbers of infected target cells (39), such large effector populations probably imply that the human cellular immune response to these chronic viral infections operates at high effector/target ratios. Naturally infected sooty mangabeys that typically remain healthy despite high viral loads also devote more than 1% of their CD4+ and CD8+ T cells to the control of this benign SIV infection (30). Given the estimated naive CD8+ T-cell precursor frequencies of 1:105 cells (14), successful cellular immune control therefore seems to require a 103- to 104-fold expansion of specific T cells.
Prophylactic vaccination is the triumph of immunology, and several childhood diseases are successfully prevented by prophylactic vaccines eliciting humoral antibody responses that provide sterilizing immunity that may last for a lifetime. In striking contrast, cellular immune responses typically fail to prevent subsequent infection (103, 104). The importance of cellular immunity in the control of HIV infection and the poor effectiveness of neutralizing antibodies to this pathogen form a formidable challenge for the development of protective HIV vaccines (17, 65). Live attenuated vaccine candidates provide much better protection than protein-based or nucleic acid-based vaccines (4, 47, 53, 87, 101), and immunogens based upon the more aggressive X4 viruses (5, 12, 82, 86) seem to protect somewhat better than those based on the more natural R5 viruses (2, 44, 73) that provide hardly any protection (65). Attenuated HIV is too dangerous to be used for vaccination in humans (43), and we investigate here why other vaccination approaches fail to provide sterilizing immunity.
SIV infection in macaques is the primary animal model for testing the effects of HIV vaccines, and these experiments seem to be a “best-case scenario” because the epitopes in the vaccines are matched exactly with those in the challenge viruses (53). Nevertheless, in monkeys vaccinated with protein-based or nucleic acid-based vaccines (12, 19, 44, 54, 61, 63, 86) or with single-cycle SIV (32), specific CD8+ T cells are boosted, but the acute phase of the infection develops remarkably similarly to that found in naive monkeys. This similarity has been quantified in a number of studies estimating initial viral replication rates. In both naive and vaccinated animals, the virus grows uncontrolled for a few weeks, with a replication rate of about 1.5 day−1 (21, 22, 57, 71). Such a failure of memory CD8+ T-cell responses to reduce initial viral replication rates is not unique to SIV/HIV and has also been found in other experimental models of rapidly replicating viruses (7, 29, 34). SIV and HIV are special because they replicate in CD4+ T cells that are also boosted by vaccination, leading to increased target cell availability and increased viral replication (88).
During the early expansion phase, massive numbers of CCR5+ CD4+ memory T cells in the gut mucosa become infected and disappear (38, 55, 62, 93), and such a severe depletion of target cells is expected to slow down viral replication (75). Despite equal initial replication rates, the peak viral loads that are obtained in vaccinated monkeys can be 10-fold lower than those in naive monkeys (12, 21, 22, 32, 86). CD8+ T cells are known to be important around the time of the peak, because in their absence, there is hardly any decrease in the viral load after the peak, i.e., a high viral set point is approached (85). When CD8+ T-cell responses are inhibited by blocking costimulation during acute infection in monkeys, one even observes a somewhat lower peak viremia in treated monkeys but a much higher viral set point (80). Because of the enormous damage done by HIV during acute infection, relatively small differences in the peak viral load can have a strong impact on the rate of disease progression afterwards (41). Vaccinated monkeys indeed approach lower viral set points (12, 86) and better preserve central memory CD4+ T cells (54, 61).
After the peak, the viral load contracts, with a half-life of about a day, and surprisingly, this half-life is largely unaffected by the presence of large numbers of specific CD8+ T cells in vaccinated animals (1, 21, 22). This 1-day half-life is similar to the classical half-life of productively infected cells during chronic infection (42, 74, 95). Thus, although the specific CD8+ T-cell response is required for the decline toward a low set point (80, 85), the abundance of specific CD8+ T cells in vaccinated monkeys hardly affects the rate at which the viral load declines.
Several authors have argued that the specific CD8+ T-cell response to SIV (or HIV) is “too late and too little” for effective control during acute infection (1, 21, 22, 81, 93). The late arrival of CD8+ effector cells at the local site of infection is partly explained by the high antigen densities required for the recruitment of large memory CD8+ T-cell populations (22). The main goal of this paper is to explore this further and explain why the CD8+ T-cell response would fail to affect the initial viral replication rate, fail to affect the down slope during the viral decline phase, and nevertheless account for lower peak viral loads. We develop a novel mathematical model with realistic dynamics for viral expansion, contraction, and approach to the viral set point to study the impact of concomitantly expanding CD8+ effector T cells. In finding that large effector populations are required to control rapidly replicating viruses, we provide a better understanding of the lack of sterilizing immunity after boosting cellular immunity.
THEORY
The mathematical model is defined as a set of differential equations:
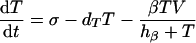 |
(1) |
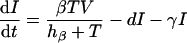 |
(2) |
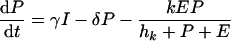 |
(3) |
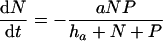 |
(4) |
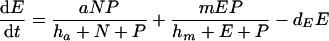 |
(5) |
where V is viral load. Because the dynamics of the virus particles are much faster than that of the infected cells (77), one can write a quasi-steady-state equation for the density of virus particles: V = pP, where p is the virus production per infected cell scaled by the clearance rate of virus particles (27). Uninfected CD4+ target cells, T, are produced at a rate of σ cells day−1, die at rate dT, and can become infected. Virus particles maximally infect β target cells day−1; this infection rate is half-maximal at a target cell density of hβ cells. Productively infected cells appear by infection and are split into an early stage, I, during which little or no viral proteins are expressed, and a late stage, P, during which virus particles are produced. The γ parameter defines an eclipse phase that on average takes γ−1 days, with a length that is exponentially distributed. Because few viral proteins are expressed early in the viral life cycle, it is assumed that early-infected cells, I, are not cleared by the immune response. Infected cells producing virus are cleared by the immune response, E, at a maximum rate, k, day−1, and CD8+ immune effector cells expand at a maximum rate, m, and die at rate dE. Naive CD8+ T cells, N, are activated by productively infected cells, P, and become effector cells at maximum rate a (that is approached when P ≫ ha). The hk and hm parameters are Michaelis-Menten constants that appear by making the total quasi-steady-state assumption (see Appendix) (16, 25, 91, 92). When at least one of the two cell populations in the denominator of the saturation functions is much larger than the Michaelis-Menten constant, their actual values hardly matter for the behavior of the model. The effector/target ratio in the steady state is solved from dE/dt = 0; when the naive T cells are depleted, one obtains E = P(m/de − 1) − hE. For our parameters, m/dE = 2, and because P ≫ hE, we obtain that at steady-state, E ≈ P.
RESULTS
Expansion phase.
For cytotoxic effectors, E, killing an exponentially growing pathogen, one can easily see that effector/target ratios should play an important role. CTLs cannot kill an infinite number of infected target cells per day because it takes time to locate and bind them, to deliver a lethal hit, and to dissociate to find the next target cell (66, 89, 97). This “handling time” creates a saturation effect similar to that of Michaelis-Menten enzyme-substrate kinetics (16, 26, 76). A problem with this limited killing efficiency is that pathogens that expand faster than the CTL response tend to grow uncontrolled (see Fig. A1) (76). At high pathogen densities, the expected killing rate per pathogen becomes proportional to the effector/target ratio, and this ratio continues to decrease when the pathogen load, L, grows faster than the immune response, E.
This “problem” of failing to control the pathogen is an excellent explanation for the similar SIV replication rates in naive and vaccinated monkeys. SIV indeed expands faster than the specific CD8+ T-cell immune response, with estimated growth rates of 1.8 day−1 and 0.9 day−1, respectively (21). During the initial expansion phase, the effector/target ratio at the local site of infection is apparently too low to control the pathogen. For SIV-vaccinated animals, this probably means that the CD8+ effector cells appear too late in the mucosal tissues to control the initial virus growth (22, 81, 93). Whenever the CD8+ T-cell immune response arrives late at the local scene of infection, virus-infected target cells outnumber the initial immune response, and the initial effector/target ratio will be too low for immediate immune control (see Appendix). These results imply that under such conditions, the immune response fails to catch up, and the virus will grow uncontrolled with ever-declining effector/target ratios (see Fig. A1a). Thus, for viruses that replicate faster than the immune response, “too late” implies “too little” (76).
Peak viremia.
Why, then, would the virus be controlled after 2 to 3 weeks? These results suggest that the CD8+ T-cell immune response can control the virus only when viral replication has been slowed down by other limiting factors (see Appendix). Because there is a massive depletion of CCR5+ CD4+ T cells in the gut during the first weeks of infection (55, 62, 93), it is very likely that viral replication rates slow down due to the limited availability of uninfected target cells (75). When viral replication is slowing down, the immune response can catch up and achieve effector/target ratios that are sufficient to control the virus (see Fig. A1b). Because cellular immune control can start only when the virus is limited by something else, the model offers a natural explanation for the very similar viral replication rates and the fairly similar peak viral loads in naive monkeys and monkeys boosted with DNA- or protein-based vaccines (12, 19, 21, 22, 44, 54, 61, 63, 86). In this scenario, the CD8 immune response becomes important only around the peak viral load, which is in good agreement with the observation that the peak viral load is similar in monkeys in which CD8+ T cells were depleted or inhibited (80, 85).
Decline phase.
Although monkeys lacking a CD8+ T-cell response have similar peak viral loads, their set-point viral levels fail to decline after the peak (80, 85). CD8+ T cells are therefore important around the peak and during the subsequent decline phase. However, if CD8+ T cells are so important during the approach to a low set point, it seems strange that (i) the down slope itself is hardly affected by the presence of specific CD8+ T cells in vaccinated animals (1, 21, 22) and (ii) HIV-positive patients with markedly different immune responses show little variation in the death rates of productively infected cells during chronic HIV infection (15, 35, 68).
An important factor that is expected to contribute to an upper limit of the down slope is the “eclipse phase,” i.e., the time it takes before a productively infected cell begins to transcribe proviral DNA and to produce viral proteins in large quantities. This eclipse phase has been estimated to be approximately 1 day (28, 69, 79, 84). It therefore seems likely that there is a delay of about 1 day before infected cells become good targets for CTL killing (84). As described above (see THEORY), we develop a novel mathematical model that aims to have realistic dynamics for the expansion and decline phases. This model allows for an eclipse phase with an average length of 1 day (γ = 1 day−1), has a killing term that is limited by the CTLs when there are more infected cells than CTLs, and is limited by the infected cells when there are more CTLs than infected cells (see Appendix) (16, 25, 91, 92). An eclipse phase of approximately 1 day introduces a new timescale that corresponds very well with the invariant down slope of 0.5 day−1. The combination of the eclipse phase with the improved killing term provides a robust explanation for the observed invariant down slopes (compare Fig. 1 with Fig. A2).
FIG. 1.

Acute infection dynamics in the model described in THEORY. The lines depict the target cells (T) (light solid line), the viral load (V) (heavy solid line), the immune response (E) (dashed line), and the naive T cells (N) (dashed-dotted line). From left to right, the production rate varies from p = 1 to p = 10 to p = 100 virions day−1 (and hence, β = 6.3, 0.63, and 0.063, respectively). From top to bottom, the killing rate varies from k = 1 to k = 10 to k = 100 day−1. Other parameters are as follows: γ = δ = 1 day−1, σ = 108 cells day−1, dT = 0.01 day−1, d = 0.02 day−1, dE = 0.5 day−1, m = 1.5 day−1, ha = hm = 1,000 cells, hk = 105 cells, hβ = 108 cells, and a = 0.1 day−1.
Explaining paradoxical observations.
The model presented above (see THEORY) studies replication rates observed in SIV-infected monkeys; i.e., the maximum replication rate of the virus is 1.5 day−1, which is considerably faster than the growth rate of the immune response of approximately 1 day−1 (21, 22). The model includes naive (or memory) CD8+ T cells that are activated at a sufficiently high antigen density, which is weighted by intraclonal competition (24, 40, 50, 51, 52, 98) to account for the late recruitment of large memory populations (22). During the first few weeks of acute infection, there are many target cells in the gut mucosa, and viral growth is exponential at the predefined rate of 1.5 day−1 (see Appendix). Little is known about the number of viral particles produced per productively infected cell (39). Therefore, the scaled production parameter, p, is varied 100-fold from left to right in Fig. 1 (constraining the initial viral replication rate to 1.5 day−1) (see Appendix). The same uncertainty holds for the killing rate. Asquith et al. (6) estimated that CTLs specific for human T-lymphotropic virus type 1 (HTLV-1)-infected cells kill no more than five target cells per day. Stinchcombe et al. (89) and Wiedemann et al. (97) showed that CTLs kill the target cells that they are associated with in a few minutes. Two-photon microscopy studies suggest that CTL killing takes about half an hour (66). Part of the differences between these studies could depend on the time searching for new target cells rather than killing them. We therefore vary k from 1 day−1 to 100 day−1 from top to bottom in Fig. 1. All simulations started with 1,000 naive CD8+ T cells that are specific for one (or a few) immunodominant epitope(s).
For most of the wide parameter domain studied in Fig. 1, the model has the typical expansion phase, immune control that starts around the peak, i.e., after viral growth has been slowed down by target cell limitation, a down slope that is gradual and not far from the invariant 0.5 day−1, and a slow approach to the viral set point. In the top row, the killing rate is comparable to the normal death rate of infected cells, and the viral load at the set point is due to a combination of target cell limitation and immune control. In the middle row, effector cells kill maximally 10 target cells day−1, and the viral load decreases several orders of magnitude after the peak, with a down slope approaching 0.5 day−1 (note that the down slope of the effector cells is also dE = 0.5 day−1). Although much lower viral set points are obtained, the peak viral load has hardly decreased by increasing the killing rate 10-fold (Fig. 1, compare a to c with d to f). Increasing the killing rate to the probably unrealistic 100 target cells day−1 does lead to markedly lower peak viral loads (Fig. 1g to i), and immune control starts before the virus is limited by target cells. At very high killing rates, immune control requires only a limited clonal expansion of the CD8+ T cells, which enables the CTL to catch up with the more replicating virus by the recruitment of naive cells. Overall, the viral set point increases with the virus production rate, p, and decreases with the CTL killing rate, k (Fig. 1) (68).
Prophylactic vaccination.
Because immune control starts only after viral growth has been slowed down by other mechanisms, the impact of prophylactic vaccination with DNA or protein is expected to be relatively minor. Viral outgrowth can be prevented only if the immune response starts with a sufficiently high effector/target ratio at the initial site of infection. Otherwise, the virus will outgrow the slower CD8+ effectors, and immune control will start only around the normal peak values, i.e., “too late implies too little.” Vaccinated monkeys are simulated in Fig. 2, with an increased activation rate of the CD8+ T cells, N, now representing memory cells induced by previous vaccination. Because there is no evidence for reactivation of the CD8+ memory cells during the first 10 days of the expansion phase in vaccinated monkeys (1, 12, 21, 22), the recruitment of memory cells is made only 10-fold faster than that of the naive T cells. Because effector cells arrive late and cannot catch up with the virus, increasing the number of memory cells hardly affects the initial replication rate, reduces the peak viral load somewhat, and leads to the same viral set point (Fig. 2).
FIG. 2.

SIV dynamics in vaccinated monkeys. The lines depict the target cells (T) (light solid line), the viral load (V) (heavy solid line), the immune response (E) (dashed line), and the memory T cells (N) (dashed-dotted line). By increasing the activation rate 10-fold to a = 1 day−1, the naive T cells of equation 4 now represent memory T cells. In panels a to d, simulations start with various levels of memory cells, i.e., N1 = 102, 103, 104, and 105 cells, respectively. Other parameters are described in the legend of Fig. 1e, i.e., p = k = 10. Vaccination changes the initial infection dynamics only at high numbers of memory cells; otherwise, the response is too late and too slow and becomes important after the peak. Note that the peak viremia decreases somewhat from panel a to panel c and that the peak has disappeared in panel d. Because the model has only one attractor for these parameter values, the same viral set point is approached in all four panels. In reality, vaccinated animals approach lower set points.
Since vaccination is no more than a change in initial conditions, the ultimate viral set point levels in the model are not altered by vaccination (Fig. 2). (Mathematical models can account for lower set points after vaccination only if they have multiple attractors [53a, 99], and for the parameter values presented here, our model has a single attracting set point; in rather unrealistic parameter regimens, i.e., when the target cell-limited steady state is too low to sustain the immune response, bistability is possible.) Repeated vaccinations of macaques with single-cycle SIV can decrease the peak viral load during subsequent challenge with SIVmac238 by 1 to 3 logs (32), while the initial replication rate seems very similar to that in unvaccinated control monkeys. For the largest initial effector/target ratio in Fig. 2d, the peak viral load has indeed blunted markedly and resembles the viral set-point level. At such high memory cell densities, the immune response can catch up with the virus before it is limited by target cells (Fig. 2d).
To summarize, the modeling confirms that prophylactic vaccinations boosting the CD8+ T-cell response need affect neither the viral replication rate nor the down slope after the peak. The explanation is that CD8+ T cells are too slow; i.e., they expand slower than the virus. As a result, immune control can become important only when viral replication is slowing down for other reasons (like the availability of uninfected target cells). The only way to control a rapidly replicating virus by slow CD8+ T-cell immunity is to prevent the initial viral outgrowth (see Appendix) (76). In the absence of a persisting infection that is maintaining an active local effector population at the site of infection, CD8+ T-cell recruitment into the local tissues is too slow to allow for swift initial control (1, 21, 22, 45), and CD8+ T cells are expected to fail on rapidly growing viruses.
Perturbations during the chronic phase.
During the chronic phase, immune control is important, and small changes in the number of effector cells can have a large effect in the model. Increasing the number of CD8+ T cells by therapeutic vaccination (58, 94) or depleting them with a monoclonal antibody (46, 67, 85) during the chronic phase of the infection indeed markedly affects the viral load. The former is surprising because one would expect a minor effect of therapeutic vaccination, since there is enough antigen available to stimulate the CD8+ T cells. In our model, there is a major effect of a minor increase in CD8+ T-cell numbers by therapeutic vaccination (Fig. 3a). The explanation is that at equilibrium effector/target ratios, the effector cells determine the viral load and vise versa. Artificially increasing the effector cells therefore decreases the viral load. When the therapeutic stimulation of the effectors peters out, the system returns to its original steady state (Fig. 3a). Because effector/target ratios are much more favorable during the chronic stage of HIV infection, these results also predict that it should be beneficial to combine antiretroviral therapy (ART) with therapeutic vaccination: lowering the viral load and increasing the immune response could tip the balance in favor of good immune control (65). The opposite perturbation of the chronic steady state is to deplete the CD8+ T cells with monoclonal antibody (46, 67, 85). After a 10-fold depletion of the specific CD8+ T-cell immune response in the model, there is a rapid increase of the viral load followed by a rapid recovery of the CD8+ T-cell response, which is in good agreement with experimental data (46, 67, 85). Afterward, the viral load approaches the original set-point level (Fig. 3b).
FIG. 3.

Therapeutic vaccination (a) and CD8+ T-cell depletion (b). Considering the chronic phase, we ignore the naive T cells. In panel a, P in equation 5 is replaced with P + D, where D is antigen-loaded dendritic cells that are introduced at day zero and that disappear exponentially [D(t) = 105e−0.02t]. Note that a small increase in the immune response (dashed line) markedly decreases the viral load (solid line). In panel b, the specific CD8+ T-cell response is depleted to 10% of its steady-state value at day zero. Parameters are described in the legend of Fig. 1e, i.e., p = k = 10.
DISCUSSION
Because CD8+ T cells exert their effects by cell-to-cell contacts, large numbers of effectors per infected target cell are required to control an infection (8). In humans, this is confirmed by the enormous magnitude (i.e., 10%, or 1010 CD8+ T cells) of the cellular immune responses observed in individuals chronically infected with HIV and cytomegalovirus (13, 90). Such huge densities of effector cells are apparently required to control chronic viral infection. Since the initial naive lymphocyte precursor densities are small (14), an acute infection has to have significant clonal expansion. Because this takes time, CD8+ T-cell immune responses are expected to be too slow to catch up with rapidly expanding infections like SIV and HIV. Therefore, we have argued that CD8+ T-cell immune control starts to become important only after viral growth has slowed down by other control mechanisms, like target cell limitation.
Monkeys vaccinated with SIV antigens carry expanded antigen-specific CD8+ T cells and nevertheless fail to control initial viral replication when challenged with SIV (12, 19, 21, 22, 44, 54, 61, 63, 81, 86). Because high effector/target ratios are required to control SIV infection, we have explained these observations with the time it takes these memory cells to be reactivated and recruited to the primary site of infection and/or to be expanded into a sufficiently large clone of effector cells. The situation is very different for vaccines boosting B-cell immunity, and it has indeed been argued that the better protection that is achieved with live attenuated SIV vaccines is due to the induction of neutralizing antibodies (53). Each B cell produces millions of antibody molecules that collectively can clear large numbers of virus particles. The humoral response can therefore catch up with a rapidly growing virus because its effector molecules easily outnumber their targets.
Whether the failure of prophylactic vaccination is due to the evolution of immune escape mutations during acute infection has been discussed (10, 11, 63). Although immune escape is rampant and is an important hurdle for HIV vaccine development (37), it is an insufficient explanation for the failure of the vaccinations discussed here (63). Upon challenge with live virus, the specific CD8+ T-cell responses induced by the vaccines expand successfully (12, 21, 22, 63), suggesting that the virus has not yet escaped. Immune escape mutations are not expected before CD8+ T cells exert a sufficient selection pressure (10), which should occur only when the effector/target ratios are sufficiently high, i.e., after the peak of the viremia.
The best evidence that the initial replication rate of the virus plays a crucial role in the eventual control of the infection comes from monkeys treated with ART during the first weeks of infection (56, 83). After withdrawing ART, the viral load typically remains low, and monkeys appear to be protected against rechallenge with homologous, and sometimes heterologous, SIV. These monkeys mount CD8+ T-cell responses to the virus, and depletion of CD8+ T cells by monoclonal antibodies increases the viral load (56). Slowing down viral replication during the very early stage is expected to allow the CD8+ T-cell response to catch up early (see Fig. A1), which may preserve specific CD4+ T-cell responses and lead to excellent immune control when ART is withdrawn. Infections with attenuated viruses that are replicating slower than normal virus are also expected to lead to better immune control (which is indeed observed) (53). Additionally, chronic infection with attenuated virus is expected to maintain an activated effector population in precisely those tissues that are most affected by the infection. These effector cells would be able to provide immediate local immune control upon rechallenge with virus. Killing a virus very early in infection, i.e., before it has outgrown the effector cells by its more rapid replication, is the best strategy to limit viral expansion, according to our models.
The conjecture that localized populations of activated CD8+ effector cells play a crucial role in protective immunity to virus infection is supported by circumstantial evidence. First, in a cohort of African sex workers that remain seronegative despite high exposure to HIV, HIV-specific CD8+ T cells have been found in the cervix (48). These populations seem to be maintained by frequent exposure to HIV because several of the women have seroconverted after a prolonged period of low exposure to HIV, e.g., a break from sex work or increased condom usage (49). Second, repeated immunizations with a multiprotein DNA/modified vaccinia virus Ankara HIV-1 vaccine inoculated in the rectal cavity of monkeys provided much better vaccine efficacy than conventional procedures (31). However, it was suggested that this localized vaccination works via innate immunity and nonspecific barriers because protection could not be correlated with cellular or humoral immunity (31). Other evidence suggesting that CD8+ effector populations localized in the tissues provide short-lived protection comes from the immunity to heterologous infection with influenza virus, where CD8+ effector T cells residing the lung and airways typically provide protection for about 1 month (78, 100, 102). In summary, CD8+ T cells seem to play a crucial role in controlling chronic infections, become important at a relatively late stage of acute infection, and provide protection to new viral infections if sufficiently large effector T-cell populations are maintained at the local site of infection.
FIG. A1.

Cytotoxic immune response to an acute infection. Consider the model with density-dependent pathogen growth:
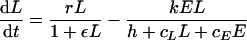 |
of a pathogen, L, growing exponentially at a rate, r, as long as ɛL ≪ 1 (depicted by the solid line), and an immune response, E, growing exponentially at a rate of dE/dt = mE (depicted by the dashed line). For ɛ = cL = cE = 0, one obtains the mass-action model dL/dt = (r − k′E)L, where k′ = k/h. The heavy solid line in panel a shows that the pathogen growth drops to zero at the critical immune response E = r/k′. We take expansion rates that are realistic for SIV infection of macaques, i.e., r = 1.5 > m = 1 day−1 (21), and set k equal to 2 and h equal to 103 cells. The light solid line in panel a depicts uncontrolled pathogen growth that is obtained for the same parameters when we allow for saturation in the number of targets by setting cL equal to 1 (16, 26, 76). In panel b, the replication rate of the pathogen is reduced at high pathogen densities (by setting ɛ equal to 10−7 cells). The saturated immune response (with cL = 1) can eradicate the pathogen after its growth has slowed down. This model accounts for the observed limited effects of prophylactic vaccination, because the initial replication rate of the pathogen remained unaffected by vaccination, and similar peak values were observed; see panel c, where we start with E = 0.01, 0.1, 1, or 10 effector cells at time point zero. After the peak dL/dt ≈ rL − kE, and because the effector population is very large, the rate at which the pathogen is cleared is unrealistically fast. This is unrealistic because there are many effector cells per target cell, each killing target cells at rate k. We solve this problem by setting cE equal to 1, which makes the killing also limited by the target cells (d). By setting the killing rate, k, equal to 2 day−1, the down slope in panel d approaches the observed invariant, r − k = −0.5 day−1.
FIG. A2.

Acute infection dynamics without an eclipse phase. The effect of the eclipse phase on the down slope after the peak in viral load can be seen by comparing this figure with Fig. 1. The eclipse phase is eliminated from the model by increasing the maturation parameter, γ, by 100-fold and decreasing the death rate, δ, of virus-producing cells to keep the same expected life span of 2 days for productively infected cells. For low killing rates, k = 1 day−1, the immune response hardly affects the viral load, and there is hardly any down slope. For intermediate killing rates, k = 10 day−1, the down slope is much steeper than in the model with an eclipse phase, and the virus is eliminated after 3 weeks. Such a steep down slope and the clearance of the virus are unrealistic. The lines depict the target cells (T) (light solid line), the viral load (V) (heavy solid line), the immune response (E) (dashed line), and naive T cells (N) (dash-dotted line). Parameters are described in the legend of Fig. 1 but with γ = 100 day−1 and δ = 0.5 day−1.
Acknowledgments
I thank Alan Perelson, Ruy Ribeiro, and Miles Davenport for extensive discussions and Christian Althaus, Joost Beltman, José Borghans, Vitaly Ganusov, and Can Keşmir for critical readings of earlier versions of this paper and for helpful comments.
I thank the Dutch NWO (VICI grant 016.048.603) and the HFSP (grant RGP0010/2004) for financial support.
APPENDIX
Current mathematical models for acute HIV infection tend to ignore saturation effects and have simple “mass-action” terms for the killing of infected target cells by immune effector cells (23, 64, 68, 70, 72, 75). In mass-action terms, the killing rate is determined by the product of the concentrations, and the actual effector/target ratio of the cellular interaction does not play a role. Consider the mass-action model for the pathogen load: dL/dt = (r − kE)L, where r is the exponential growth rate of the pathogen, L, and k is the killing rate by cytotoxic effectors (CTLs). This pathogen will be controlled whenever the immune response exceeds the critical level, E > r/k (Fig. A1a), which is independent of the size of the pathogen population. Control of a pathogen consisting of 10 or 10 million infected cells requires the same population size of cytotoxic effectors, E, which is entirely unrealistic.
In models with mass-action killing terms (and an exponentially increasing immune response) (Fig. A1), prophylactic vaccination is expected to have a major effect because vaccination should markedly decrease the time required to control the pathogen. An exponentially expanding immune response, dE/dt = mE, will breach the critical level, E = r/k, at day t = ln{r/[kE(0)]}/m. Increasing the initial number of effector cells, E(0), therefore decreases the time to control. This is evidently not the case after vaccination with SIV antigens (1, 21, 22).
Moreover, CTL cannot kill an infinite number of infected target cells per day (6). It takes time to locate and bind them and deliver a lethal hit (66, 89, 97). This “handling time” creates a saturation effect similar to Michaelis-Menten enzyme-substrate kinetics (16, 26, 76); i.e., the killing term should take the form −kEL/(h + L), where the Michaelis-Menten constant, h, is the pathogen density at which the killing rate per CTL is half-maximal. An implication of this much more realistic model is that pathogens that expand faster than the immune response tend to grow uncontrolled (76). At large pathogen densities, pathogen growth approaches dL/dt = rL − kE, which continues to increase when L grows faster than E (Fig. A1b). Importantly, in this model, the per-capita killing rate approaches kE/L, and the pathogen is controlled when the effector/target ratio exceeds the same critical level, i.e., when E/L > r/k. Because SIV expands faster than the specific CD8+ T-cell immune response (with estimated growth rates of 1.8 day−1 and 0.9 day−1, respectively) (21), virus-infected target cells will rapidly outnumber the immune response and grow uncontrolled because the global effector/target ratio is declining (Fig. A1a).
This model suggests that the CD8+ T-cell immune response can control the virus only when viral replication has been slowed down by other limiting factors. Because there is massive depletion of CCR5+ CD4+ T cells in the gut during the first weeks of infection (55, 62, 93), it is very likely that viral replication rates slow down due to a limited availability of uninfected target cells. Additionally, innate immune responses may slow down viral replication. Both can be included by density-dependent viral replication:
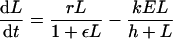 |
where the replication rate is the same initially but slows down with increasing pathogen densities (i.e., at L = 1/ɛ, the replication rate has halved). When viral replication is slowing down, the immune response can catch up and achieve effector/target ratios that are sufficient to control the virus (Fig. A1b). Because immune control can start only when the virus is limited by something else, the model naturally explains why vaccinated monkeys have peak viral loads similar to those of unvaccinated monkeys (12, 21, 22).
Accepting the notion that the immune response is important during the decline phase, one has to face the problem that current mathematical models fail to account for a moderate down slope that is largely independent of the magnitude of the immune response. As long as infected target cells are not limiting, the total killing rate in the model is approximately kE, and because the immune response has to be large, the down slope is expected to be very steep (Fig. A1b and c and A2). This is partly an artifact of the simplicity of the model, because the killing rate per infected cell should approach some maximum value when the effector/target ratio is large. One can improve the model by considering a scheme where the CTL first has to interact with a target cell for some time to bind viral peptides presented by major histocompatibility complex molecules, after which it will deliver its lethal hit and dissociate to possibly bind other target cells.
From the general scheme where an unbound CD8+ effector cell, Eu, binds an unbound target cell, Pu, to form a complex, C, that after a time dissociates into an unbound effector cell and a dying target cell, Pd, i.e., Eu + Pu ↔ C → Eu + Pd, with the conservation equations E = Eu + C and P = Pu + C, one can make the “total quasi-steady-state assumption,” dC/dt = 0, and obtain the following:
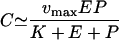 |
where vmax is the maximum reaction rate and K is the Michaelis-Menten constant (16, 91, 92). The killing rate per infected cell then approaches its maximum, vmax cells day−1, when E ≫ K + P, i.e., when there are enough effector cells per target cell. When there are many target cells and few effector cells, i.e., when P ≫ K + E, the killing rate of an infected cell is proportional to the effector/target ratio, i.e., approaches vmaxE/P. Such a killing term can account for a down slope of 0.5 day−1 if the killing rate is set to k = r + 0.5 (Fig. A1d). This seems a rather strong parameter constraint, however, given that we know so little about the rate at which CTLs kill their infected target cells (6, 8, 9, 36, 96).
Maximal replication rate.
Because the infected cells in the model presented above (see THEORY) consist of two subpopulations, we compute here the maximum replication rate of the virus. During the initial phase of the infection in the gut, the availability of target cells should not yet be limiting, and the CD8+ T-cell response should be negligible. The initial replication rate of the viral infection is therefore realized by the following simplification of the model:
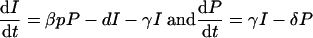 |
To compute the replication rate, we simplify by assuming that the ratio of producing cells to infected cells (R = P/I) will rapidly approach steady state:
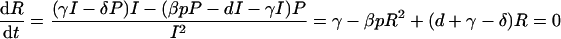 |
which has only one positive root for parameters where d + γ − δ > 0,
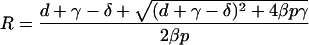 |
Adding dI/dt to dP/dt to compute the total number, L (for load), of productively infected cells gives dL/dt = (βp − δ)P − dI, where L = P + I, P = RI, and I = L/(1 + R). This gives the following exponential growth function:
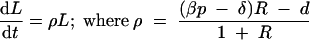 |
is the replication rate. For every parameter setting studied here, we enforced a replication rate of 1.5 per day by solving β from ρ = 3/2 in dL/dt, with R defined as described above:
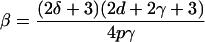 |
By this procedure, which establishes an inverse relation between β and p, we indeed obtained a replication rate of 1.5 day−1 for various values of p (Fig. 1). Note that assuming that dR/dt = 0 is not the same as a quasi-steady-state assumption, dP/dt = 0, which turns out to be a poor approximation during the expansion phase (not shown).
Footnotes
Published ahead of print on 3 January 2007.
REFERENCES
- 1.Abdel-Motal, U. M., J. Gillis, K. Manson, M. Wyand, D. Montefiori, K. Stefano-Cole, R. C. Montelaro, J. D. Altman, and R. P. Johnson. 2005. Kinetics of expansion of SIV Gag-specific CD8+ T lymphocytes following challenge of vaccinated macaques. Virology 333:226-238. [DOI] [PubMed] [Google Scholar]
- 2.Allen, T. M., L. Mortara, B. R. Mothé, M. Liebl, P. Jing, B. Calore, M. Piekarczyk, R. Ruddersdorf, D. H. O'Connor, X. Wang, C. Wang, D. B. Allison, J. D. Altman, A. Sette, R. C. Desrosiers, G. Sutter, and D. I. Watkins. 2002. Tat-vaccinated macaques do not control simian immunodeficiency virus SIVmac239 replication. J. Virol. 76:4108-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reference deleted.
- 4.Amara, R. R., K. Patel, G. Niedziela, P. Nigam, S. Sharma, S. I. Staprans, D. C. Montefiori, L. Chenareddi, J. G. Herndon, H. L. Robinson, H. M. McClure, and F. J. Novembre. 2005. A combination DNA and attenuated simian immunodeficiency virus vaccine strategy provides enhanced protection from simian/human immunodeficiency virus-induced disease. J. Virol. 79:15356-15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amara, R. R., F. Villinger, J. D. Altman, S. L. Lydy, S. P. O'Neil, S. I. Staprans, D. C. Montefiori, Y. Xu, J. G. Herndon, L. S. Wyatt, M. A. Candido, N. L. Kozyr, P. L. Earl, J. M. Smith, H. L. Ma, B. D. Grimm, M. L. Hulsey, J. Miller, H. M. McClure, J. M. McNicholl, B. Moss, and H. L. Robinson. 2001. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292:69-74. [DOI] [PubMed] [Google Scholar]
- 6.Asquith, B., A. J. Mosley, A. Barfield, S. E. Marshall, A. Heaps, P. Goon, E. Hanon, Y. Tanaka, G. P. Taylor, and C. R. Bangham. 2005. A functional CD8+ cell assay reveals individual variation in CD8+ cell antiviral efficacy and explains differences in human T-lymphotropic virus type 1 proviral load. J. Gen. Virol. 86:1515-1523. [DOI] [PubMed] [Google Scholar]
- 7.Bachmann, M. F., T. M. Kundig, H. Hengartner, and R. M. Zinkernagel. 1997. Protection against immunopathological consequences of a viral infection by activated but not resting cytotoxic T cells: T cell memory without “memory T cells”? Proc. Natl. Acad. Sci. USA 94:640-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barber, D. L., E. J. Wherry, and R. Ahmed. 2003. Cutting edge: rapid in vivo killing by memory CD8 T cells. J. Immunol. 171:27-31. [DOI] [PubMed] [Google Scholar]
- 9.Barchet, W., S. Oehen, P. Klenerman, D. Wodarz, G. Bocharov, A. L. Lloyd, M. A. Nowak, H. Hengartner, R. M. Zinkernagel, and S. Ehl. 2000. Direct quantitation of rapid elimination of viral antigen-positive lymphocytes by antiviral CD8+ T cells in vivo. Eur. J. Immunol. 30:1356-1363. [DOI] [PubMed] [Google Scholar]
- 10.Barouch, D. H., J. Kunstman, J. Glowczwskie, K. J. Kunstman, M. A. Egan, F. W. Peyerl, S. Santra, M. J. Kuroda, J. E. Schmitz, K. Beaudry, G. R. Krivulka, M. A. Lifton, D. A. Gorgone, S. M. Wolinsky, and N. L. Letvin. 2003. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 77:7367-7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barouch, D. H., J. Powers, D. M. Truitt, M. G. Kishko, J. C. Arthur, F. W. Peyerl, M. J. Kuroda, D. A. Gorgone, M. A. Lifton, C. I. Lord, V. M. Hirsch, D. C. Montefiori, A. Carville, K. G. Mansfield, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin. 2005. Dynamic immune responses maintain cytotoxic T lymphocyte epitope mutations in transmitted simian immunodeficiency virus variants. Nat. Immunol. 6:247-252. [DOI] [PubMed] [Google Scholar]
- 12.Barouch, D. H., S. Santra, J. E. Schmitz, M. J. Kuroda, T. M. Fu, W. Wagner, M. Bilska, A. Craiu, X. X. Zheng, G. R. Krivulka, K. Beaudry, M. A. Lifton, C. E. Nickerson, W. L. Trigona, K. Punt, D. C. Freed, L. Guan, S. Dubey, D. Casimiro, A. Simon, M. E. Davies, M. Chastain, T. B. Strom, R. S. Gelman, D. C. Montefiori, M. G. Lewis, E. A. Emini, J. W. Shiver, and N. L. Letvin. 2000. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290:486-492. [DOI] [PubMed] [Google Scholar]
- 13.Betts, M. R., D. R. Ambrozak, D. C. Douek, S. Bonhoeffer, J. M. Brenchley, J. P. Casazza, R. A. Koup, and L. J. Picker. 2001. Analysis of total human immunodeficiency virus (HIV)-specific CD4+ and CD8+ T-cell responses: relationship to viral load in untreated HIV infection. J. Virol. 75:11983-11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blattman, J. N., R. Antia, D. J. Sourdive, X. Wang, S. M. Kaech, K. Murali-Krishna, J. D. Altman, and R. Ahmed. 2002. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J. Exp. Med. 195:657-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonhoeffer, S., G. A. Funk, H. F. Gunthard, M. Fischer, and V. Muller. 2003. Glancing behind virus load variation in HIV-1 infection. Trends Microbiol. 11:499-504. [DOI] [PubMed] [Google Scholar]
- 16.Borghans, J. A. M., R. J. De Boer, and L. A. Segel. 1996. Extending the quasi-steady state approximation by changing variables. Bull. Math. Biol. 58:43-63. [DOI] [PubMed] [Google Scholar]
- 17.Burton, D. R., R. C. Desrosiers, R. W. Doms, W. C. Koff, P. D. Kwong, J. P. Moore, G. J. Nabel, J. Sodroski, I. A. Wilson, and R. T. Wyatt. 2004. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 5:233-236. [DOI] [PubMed] [Google Scholar]
- 18.Carrington, M., G. W. Nelson, M. P. Martin, T. Kissner, D. Vlahov, J. J. Goedert, R. Kaslow, S. Buchbinder, K. Hoots, and S. J. O'Brien. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283:1748-1752. [DOI] [PubMed] [Google Scholar]
- 19.Casimiro, D. R., F. Wang, W. A. Schleif, X. Liang, Z. Q. Zhang, T. W. Tobery, M. E. Davies, A. B. McDermott, D. H. O'Connor, A. Fridman, A. Bagchi, L. G. Tussey, A. J. Bett, A. C. Finnefrock, T. M. Fu, A. Tang, K. A. Wilson, M. Chen, H. C. Perry, G. J. Heidecker, D. C. Freed, A. Carella, K. S. Punt, K. J. Sykes, L. Huang, V. I. Ausensi, M. Bachinsky, U. Sadasivan-Nair, D. I. Watkins, E. A. Emini, and J. W. Shiver. 2005. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 79:15547-15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark, D. R., R. J. De Boer, K. C. Wolthers, and F. Miedema. 1999. T cell dynamics in HIV-1 infection. Adv. Immunol. 73:301-327. [DOI] [PubMed] [Google Scholar]
- 21.Davenport, M. P., R. M. Ribeiro, D. L. Chao, and A. S. Perelson. 2004. Predicting the impact of a nonsterilizing vaccine against human immunodeficiency virus. J. Virol. 78:11340-11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenport, M. P., L. Zhang, A. Bagchi, A. Fridman, T. M. Fu, W. Schleif, J. W. Shiver, R. M. Ribeiro, and A. S. Perelson. 2005. High-potency human immunodeficiency virus vaccination leads to delayed and reduced CD8+ T-cell expansion but improved virus control. J. Virol. 79:10059-10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Boer, R. J., and C. A. Boucher. 1996. Anti-CD4 therapy for AIDS suggested by mathematical models. Proc. R. Soc. Lond. B Biol. Sci. 263:899-905. [DOI] [PubMed] [Google Scholar]
- 24.De Boer, R. J., A. A. Freitas, and A. S. Perelson. 2001. Resource competition determines selection of B cell repertoires. J. Theor. Biol. 212:333-343. [DOI] [PubMed] [Google Scholar]
- 25.De Boer, R. J., and A. S. Perelson. 1994. T cell repertoires and competitive exclusion. J. Theor. Biol. 169:375-390. [DOI] [PubMed] [Google Scholar]
- 26.De Boer, R. J., and A. S. Perelson. 1995. Towards a general function describing T cell proliferation. J. Theor. Biol. 175:567-576. [DOI] [PubMed] [Google Scholar]
- 27.De Boer, R. J., and A. S. Perelson. 1998. Target cell limited and immune control models of HIV infection: a comparison. J. Theor. Biol. 190:201-214. [DOI] [PubMed] [Google Scholar]
- 28.Dixit, N. M., M. Markowitz, D. D. Ho, and A. S. Perelson. 2004. Estimates of intracellular delay and average drug efficacy from viral load data of HIV-infected individuals under antiretroviral therapy. Antivir. Ther. 9:237-246. [PubMed] [Google Scholar]
- 29.Doherty, P. C., and J. P. Christensen. 2000. Accessing complexity: the dynamics of virus-specific T cell responses. Annu. Rev. Immunol. 18:561-592. [DOI] [PubMed] [Google Scholar]
- 30.Dunham, R., P. Pagliardini, S. Gordon, B. Sumpter, J. Engram, A. Moanna, M. Paiardini, J. N. Mandl, B. Lawson, S. Garg, H. M. McClure, Y. X. Xu, C. Ibegbu, K. Easley, N. Katz, I. Pandrea, C. Apetrei, D. L. Sodora, S. I. Staprans, M. B. Feinberg, and G. Silvestri. 2006. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 108:209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellenberger, D., R. A. Otten, B. Li, M. Aidoo, I. V. Rodriguez, C. A. Sariol, M. Martinez, M. Monsour, L. Wyatt, M. G. Hudgens, E. Kraiselburd, B. Moss, H. Robinson, T. Folks, and S. Butera. 2006. HIV-1 DNA/MVA vaccination reduces the per exposure probability of infection during repeated mucosal SHIV challenges. Virology 352:216-225. [DOI] [PubMed] [Google Scholar]
- 32.Evans, D. T., J. E. Bricker, H. B. Sanford, S. Lang, A. Carville, B. A. Richardson, M. Piatak, Jr., J. D. Lifson, K. G. Mansfield, and R. C. Desrosiers. 2005. Immunization of macaques with single-cycle simian immunodeficiency virus (SIV) stimulates diverse virus-specific immune responses and reduces viral loads after challenge with SIVmac239. J. Virol. 79:7707-7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez, C. S., I. Stratov, R. De Rose, K. Walsh, C. J. Dale, M. Z. Smith, M. B. Agy, S. L. Hu, K. Krebs, D. I. Watkins, D. H. O'Connor, M. P. Davenport, and S. J. Kent. 2005. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J. Virol. 79:5721-5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flynn, K. J., G. T. Belz, J. D. Altman, R. Ahmed, D. L. Woodland, and P. C. Doherty. June. 1998. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity 8:683-691. [DOI] [PubMed] [Google Scholar]
- 35.Funk, G. A., A. Oxenius, M. Fischer, M. Opravil, B. Joos, M. Flepp, R. Weber, H. F. Gunthard, and S. Bonhoeffer. 2006. HIV replication elicits little cytopathic effects in vivo: analysis of surrogate markers for virus production, cytotoxic T cell response and infected cell death. J. Med. Virol. 78:1141-1146. [DOI] [PubMed] [Google Scholar]
- 36.Ganusov, V. V., and R. J. De Boer. 2006. Estimating costs and benefits of CTL escape mutations in SIV/HIV infection. PLoS Comput. Biol. 2:182-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goulder, P. J., and D. I. Watkins. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat. Rev. Immunol. 4:630-640. [DOI] [PubMed] [Google Scholar]
- 38.Guadalupe, M., E. Reay, S. Sankaran, T. Prindiville, J. Flamm, A. McNeil, and S. Dandekar. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 77:11708-11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haase, A. T., K. Henry, M. Zupancic, G. Sedgewick, R. A. Faust, H. Melroe, W. Cavert, K. Gebhard, K. Staskus, Z. Q. Zhang, P. J. Dailey, H. H. Balfour, Jr., A. Erice, and A. S. Perelson. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985-989. [DOI] [PubMed] [Google Scholar]
- 40.Hataye, J., J. J. Moon, A. Khoruts, C. Reilly, and M. K. Jenkins. 2006. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science 312:114-116. [DOI] [PubMed] [Google Scholar]
- 41.Hel, Z., J. R. McGhee, and J. Mestecky. 2006. HIV infection: first battle decides the war. Trends Immunol. 27:274-281. [DOI] [PubMed] [Google Scholar]
- 42.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123-126. [DOI] [PubMed] [Google Scholar]
- 43.Hofmann-Lehmann, R., J. Vlasak, A. L. Williams, A. L. Chenine, H. M. McClure, D. C. Anderson, S. O'Neil, and R. M. Ruprecht. 2003. Live attenuated, nef-deleted SIV is pathogenic in most adult macaques after prolonged observation. AIDS 17:157-166. [DOI] [PubMed] [Google Scholar]
- 44.Horton, H., T. U. Vogel, D. K. Carter, K. Vielhuber, D. H. Fuller, T. Shipley, J. T. Fuller, K. J. Kunstman, G. Sutter, D. C. Montefiori, V. Erfle, R. C. Desrosiers, N. Wilson, L. J. Picker, S. M. Wolinsky, C. Wang, D. B. Allison, and D. I. Watkins. 2002. Immunization of rhesus macaques with a DNA prime/modified vaccinia virus Ankara boost regimen induces broad simian immunodeficiency virus (SIV)-specific T-cell responses and reduces initial viral replication but does not prevent disease progression following challenge with pathogenic SIVmac239. J. Virol. 76:7187-7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jansen, V. A., H. K. Altes, G. A. Funk, and D. Wodarz. 2005. Contrasting B cell- and T cell-based protective vaccines. J. Theor. Biol. 234:39-48. [DOI] [PubMed] [Google Scholar]
- 46.Jin, X., D. E. Bauer, S. E. Tuttleton, S. Lewin, A. Gettie, J. Blanchard, C. E. Irwin, J. T. Safrit, J. Mittler, L. Weinberger, L. G. Kostrikis, L. Zhang, A. S. Perelson, and D. D. Ho. 1999. dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189:991-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson, R. P., and R. C. Desrosiers. 1998. Protective immunity induced by live attenuated simian immunodeficiency virus. Curr. Opin. Immunol. 10:436-443. [DOI] [PubMed] [Google Scholar]
- 48.Kaul, R., F. A. Plummer, J. Kimani, T. Dong, P. Kiama, T. Rostron, E. Njagi, K. S. MacDonald, J. J. Bwayo, A. J. McMichael, and S. L. Rowland-Jones. 2000. HIV-1-specific mucosal CD8+ lymphocyte responses in the cervix of HIV-1-resistant prostitutes in Nairobi. J. Immunol. 164:1602-1611. [DOI] [PubMed] [Google Scholar]
- 49.Kaul, R., S. L. Rowland-Jones, J. Kimani, T. Dong, H. B. Yang, P. Kiama, T. Rostron, E. Njagi, J. J. Bwayo, K. S. MacDonald, A. J. McMichael, and F. A. Plummer. 2001. Late seroconversion in HIV-resistant Nairobi prostitutes despite pre-existing HIV-specific CD8+ responses. J. Clin. Investig. 107:341-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kedl, R. M., J. W. Kappler, and P. Marrack. 2003. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 15:120-127. [DOI] [PubMed] [Google Scholar]
- 51.Kedl, R. M., W. A. Rees, D. A. Hildeman, B. Schaefer, T. Mitchell, J. Kappler, and P. Marrack. 2000. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 192:1105-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kedl, R. M., B. C. Schaefer, J. W. Kappler, and P. Marrack. 2002. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat. Immunol. 3:27-32. [DOI] [PubMed] [Google Scholar]
- 53.Koff, W. C., P. R. Johnson, D. I. Watkins, D. R. Burton, J. D. Lifson, K. J. Hasenkrug, A. B. McDermott, A. Schultz, T. J. Zamb, R. Boyle, and R. C. Desrosiers. 2006. HIV vaccine design: insights from live attenuated SIV vaccines. Nat. Immunol. 7:19-23. [DOI] [PubMed] [Google Scholar]
- 53a.Korthals Altes, H. K., R. M. Ribeiro, and R. J. De Boer. 2003. The race between initial T helper expansion and virus growth upon HIV infection influences polyclonality of the response and viral setpoint. Proc. R. Soc. Lond. B Biol. Sci. 270:1349-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Letvin, N. L., J. R. Mascola, Y. Sun, D. A. Gorgone, A. P. Buzby, L. Xu, Z. Y. Yang, B. Chakrabarti, S. S. Rao, J. E. Schmitz, D. C. Montefiori, B. R. Barker, F. L. Bookstein, and G. J. Nabel. 2006. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science 312:1530-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li, Q., L. Duan, J. D. Estes, Z. M. Ma, T. Rourke, Y. Wang, C. Reilly, J. Carlis, C. J. Miller, and A. T. Haase. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434:1148-1152. [DOI] [PubMed] [Google Scholar]
- 56.Lifson, J. D., J. L. Rossio, M. Piatak, Jr., T. Parks, L. Li, R. Kiser, V. Coalter, B. Fisher, B. M. Flynn, S. Czajak, V. M. Hirsch, K. A. Reimann, J. E. Schmitz, J. Ghrayeb, N. Bischofberger, M. A. Nowak, R. C. Desrosiers, and D. Wodarz. 2001. Role of CD8+ lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J. Virol. 75:10187-10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Little, S. J., A. R. McLean, C. A. Spina, D. D. Richman, and D. V. Havlir. 1999. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu, W., L. C. Arraes, W. T. Ferreira, and J. M. Andrieu. 2004. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat. Med. 10:1359-1365. [DOI] [PubMed] [Google Scholar]
- 59.Matano, T., M. Kobayashi, H. Igarashi, A. Takeda, H. Nakamura, M. Kano, C. Sugimoto, K. Mori, A. Iida, T. Hirata, M. Hasegawa, T. Yuasa, M. Miyazawa, Y. Takahashi, M. Yasunami, A. Kimura, D. H. O'Connor, D. I. Watkins, and Y. Nagai. 2004. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 199:1709-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matano, T., R. Shibata, C. Siemon, M. Connors, H. C. Lane, and M. A. Martin. 1998. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 72:164-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mattapallil, J. J., D. C. Douek, A. Buckler-White, D. Montefiori, N. L. Letvin, G. J. Nabel, and M. Roederer. 2006. Vaccination preserves CD4 memory T cells during acute simian immunodeficiency virus challenge. J. Exp. Med. 203:1533-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mattapallil, J. J., D. C. Douek, B. Hill, Y. Nishimura, M. Martin, and M. Roederer. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093-1097. [DOI] [PubMed] [Google Scholar]
- 63.McDermott, A. B., D. H. O'Connor, S. Fuenger, S. Piaskowski, S. Martin, J. Loffredo, M. Reynolds, J. Reed, J. Furlott, T. Jacoby, C. Riek, E. Dodds, K. Krebs, M. E. Davies, W. A. Schleif, D. R. Casimiro, J. W. Shiver, and D. I. Watkins. 2005. Cytotoxic T-lymphocyte escape does not always explain the transient control of simian immunodeficiency virus SIVmac239 viremia in adenovirus-boosted and DNA-primed Mamu-A*01-positive rhesus macaques. J. Virol. 79:15556-15566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McLean, A. R., V. C. Emery, A. Webster, and P. D. Griffiths. 1991. Population dynamics of HIV within an individual after treatment with zidovudine. AIDS 5:485-489. [DOI] [PubMed] [Google Scholar]
- 65.McMichael, A. J. 2006. HIV vaccines. Annu. Rev. Immunol. 24:227-255. [DOI] [PubMed] [Google Scholar]
- 66.Mempel, T. R., M. J. Pittet, K. Khazaie, W. Weninger, R. Weissleder, H. Von Boehmer, and U. H. Von Andrian. 2006. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity 25:129-141. [DOI] [PubMed] [Google Scholar]
- 67.Metzner, K. J., X. Jin, F. V. Lee, A. Gettie, D. E. Bauer, M. Di Mascio, A. S. Perelson, P. A. Marx, D. D. Ho, L. G. Kostrikis, and R. I. Connor. 2000. Effects of in vivo CD8+ T cell depletion on virus replication in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J. Exp. Med. 191:1921-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Müller, V., A. F. Marée, and R. J. De Boer. 2001. Small variations in multiple parameters account for wide variations in HIV-1 set-points: a novel modelling approach. Proc. R. Soc. Lond. B Biol. Sci. 268:235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nelson, P. W., J. E. Mittler, and A. S. Perelson. 2001. Effect of drug efficacy and the eclipse phase of the viral life cycle on estimates of HIV viral dynamic parameters. J. Acquir. Immune Defic. Syndr. 26:405-412. [DOI] [PubMed] [Google Scholar]
- 70.Nowak, M. A., and C. R. Bangham. 1996. Population dynamics of immune responses to persistent viruses. Science 272:74-79. [DOI] [PubMed] [Google Scholar]
- 71.Nowak, M. A., A. L. Lloyd, G. M. Vasquez, T. A. Wiltrout, L. M. Wahl, N. Bischofberger, J. Williams, A. Kinter, A. S. Fauci, V. M. Hirsch, and J. D. Lifson. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71:7518-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nowak, M. A., and R. M. May. 2000. Virus dynamics. Mathematical principles of immunology and virology. Oxford University Press, Oxford, United Kingdom.
- 73.Pal, R., D. Venzon, N. L. Letvin, S. Santra, D. C. Montefiori, N. R. Miller, E. Tryniszewska, M. G. Lewis, T. C. VanCott, V. Hirsch, R. Woodward, A. Gibson, M. Grace, E. Dobratz, P. D. Markham, Z. Hel, J. Nacsa, M. Klein, J. Tartaglia, and G. Franchini. 2002. ALVAC-SIV-gag-pol-env-based vaccination and macaque major histocompatibility complex class I (A*01) delay simian immunodeficiency virus SIVmac-induced immunodeficiency. J. Virol. 76:292-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. [DOI] [PubMed] [Google Scholar]
- 75.Phillips, A. N. 1996. Reduction of HIV concentration during acute infection: independence from a specific immune response. Science 271:497-499. [DOI] [PubMed] [Google Scholar]
- 76.Pilyugin, S. S., and R. Antia. 2000. Modeling immune responses with handling time. Bull. Math. Biol. 62:869-890. [DOI] [PubMed] [Google Scholar]
- 77.Ramratnam, B., S. Bonhoeffer, J. Binley, A. Hurley, L. Zhang, J. E. Mittler, M. Markowitz, J. P. Moore, A. S. Perelson, and D. D. Ho. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 354:1782-1785. [DOI] [PubMed] [Google Scholar]
- 78.Ray, S. J., S. N. Franki, R. H. Pierce, S. Dimitrova, V. Koteliansky, A. G. Sprague, P. C. Doherty, A. R. De Fougerolles, and D. J. Topham. 2004. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity 20:167-179. [DOI] [PubMed] [Google Scholar]
- 79.Reddy, B., and J. Yin. 1999. Quantitative intracellular kinetics of HIV type 1. AIDS Res. Hum. Retrovir. 15:273-283. [DOI] [PubMed] [Google Scholar]
- 80.Regoes, R. R., R. Antia, D. A. Garber, G. Silvestri, M. B. Feinberg, and S. I. Staprans. 2004. Roles of target cells and virus-specific cellular immunity in primary simian immunodeficiency virus infection. J. Virol. 78:4866-4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reynolds, M. R., E. Rakasz, P. J. Skinner, C. White, K. Abel, Z. M. Ma, L. Compton, G. Napoe, N. Wilson, C. J. Miller, A. Haase, and D. I. Watkins. 2005. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: too late and too little. J. Virol. 79:9228-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rose, N. F., P. A. Marx, A. Luckay, D. F. Nixon, W. J. Moretto, S. M. Donahoe, D. Montefiori, A. Roberts, L. Buonocore, and J. K. Rose. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539-549. [DOI] [PubMed] [Google Scholar]
- 83.Rosenberg, E. S., M. Altfeld, S. H. Poon, M. N. Phillips, B. M. Wilkes, R. L. Eldridge, G. K. Robbins, R. T. D'Aquila, P. J. Goulder, and B. D. Walker. 2000. Immune control of HIV-1 after early treatment of acute infection. Nature 407:523-526. [DOI] [PubMed] [Google Scholar]
- 84.Rouzine, I. M., R. A. Sergeev, and A. I. Glushtsov. 2006. Two types of cytotoxic lymphocyte regulation explain kinetics of immune response to human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 103:666-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmitz, J. E., M. J. Kuroda, S. Santra, V. G. Sasseville, M. A. Simon, M. A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B. J. Scallon, J. Ghrayeb, M. A. Forman, D. C. Montefiori, E. P. Rieber, N. L. Letvin, and K. A. Reimann. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857-860. [DOI] [PubMed] [Google Scholar]
- 86.Shiver, J. W., T. M. Fu, L. Chen, D. R. Casimiro, M. E. Davies, R. K. Evans, Z. Q. Zhang, A. J. Simon, W. L. Trigona, S. A. Dubey, L. Huang, V. A. Harris, R. S. Long, X. Liang, L. Handt, W. A. Schleif, L. Zhu, D. C. Freed, N. V. Persaud, L. Guan, K. S. Punt, A. Tang, M. Chen, K. A. Wilson, K. B. Collins, G. J. Heidecker, V. R. Fernandez, H. C. Perry, J. G. Joyce, K. M. Grimm, J. C. Cook, P. M. Keller, D. S. Kresock, H. Mach, R. D. Troutman, L. A. Isopi, D. M. Williams, Z. Xu, K. E. Bohannon, D. B. Volkin, D. C. Montefiori, A. Miura, G. R. Krivulka, M. A. Lifton, M. J. Kuroda, J. E. Schmitz, N. L. Letvin, M. J. Caulfield, A. J. Bett, R. Youil, D. C. Kaslow, and E. A. Emini. 2002. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415:331-335. [DOI] [PubMed] [Google Scholar]
- 87.Stahl-Hennig, C., M. Eisenblatter, M. Franz, H. Stoiber, K. Tenner-Racz, Y. S. Suh, E. Jasny, B. Falkensammer, M. Ugucchioni, G. Georgsson, C. Baroni, M. P. Dierich, J. D. Lifson, R. M. Steinman, K. Uberla, P. Racz, and R. Ignatius. 2007. A single vaccination with attenuated SIVmac 239 via the tonsillar route confers partial protection against challenge with SIVmac 251 at a distant mucosal site, the rectum. Front. Biosci. 12:2107-2123. [DOI] [PubMed] [Google Scholar]
- 88.Staprans, S. I., A. P. Barry, G. Silvestri, J. T. Safrit, N. Kozyr, B. Sumpter, H. Nguyen, H. McClure, D. Montefiori, J. I. Cohen, and M. B. Feinberg. 2004. Enhanced SIV replication and accelerated progression to AIDS in macaques primed to mount a CD4 T cell response to the SIV envelope protein. Proc. Natl. Acad. Sci. USA 101:13026-13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stinchcombe, J. C., G. Bossi, S. Booth, and G. M. Griffiths. 2001. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15:751-761. [DOI] [PubMed] [Google Scholar]
- 90.Sylwester, A. W., B. L. Mitchell, J. B. Edgar, C. Taormina, C. Pelte, F. Ruchti, P. R. Sleath, K. H. Grabstein, N. A. Hosken, F. Kern, J. A. Nelson, and L. J. Picker. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202:673-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tzafriri, A. R., and E. R. Edelman. 2004. The total quasi-steady-state approximation is valid for reversible enzyme kinetics. J. Theor. Biol. 226:303-313. [DOI] [PubMed] [Google Scholar]
- 92.Tzafriri, A. R., and E. R. Edelman. 2005. On the validity of the quasi-steady state approximation of bimolecular reactions in solution. J. Theor. Biol. 233:343-350. [DOI] [PubMed] [Google Scholar]
- 93.Veazey, R. S., and A. A. Lackner. 2005. HIV swiftly guts the immune system. Nat. Med. 11:469-470. [DOI] [PubMed] [Google Scholar]
- 94.Von Gegerfelt, A. S., M. Rosati, C. Alicea, A. Valentin, P. Roth, J. Bear, G. Franchini, P. S. Albert, N. Bischofberger, J. D. Boyer, D. B. Weiner, P. Markham, Z. R. Israel, J. H. Eldridge, G. N. Pavlakis, and B. K. Felber. 29 November 2006. Long-lasting decrease of viremia in chronically SIVmac251-infected macaques after therapeutic DNA immunization. J. Virol. doi: 10.1128/JVI.01990-06. [DOI] [PMC free article] [PubMed]
- 95.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117-122. [DOI] [PubMed] [Google Scholar]
- 96.Wick, W. D., O. O. Yang, L. Corey, and S. G. Self. 2005. How many human immunodeficiency virus type 1-infected target cells can a cytotoxic T-lymphocyte kill? J. Virol. 79:13579-13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wiedemann, A., D. Depoil, M. Faroudi, and S. Valitutti. 2006. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA 103:10985-10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Willis, R. A., J. W. Kappler, and P. C. Marrack. 2006. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc. Natl. Acad. Sci. USA 103:12063-12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wodarz, D., and M. A. Nowak. 1999. Specific therapy regimes could lead to long-term immunological control of HIV. Proc. Natl. Acad. Sci. USA 96:14464-14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woodland, D. L., K. H. Ely, S. R. Crowe, M. Tighe, J. W. Brennan, A. G. Harmsen, and L. S. Cauley. 2002. Antiviral memory T-cell responses in the lung. Microbes Infect. 4:1091-1098. [DOI] [PubMed] [Google Scholar]
- 101.Wyand, M. S., K. Manson, D. C. Montefiori, J. D. Lifson, R. P. Johnson, and R. C. Desrosiers. 1999. Protection by live, attenuated simian immunodeficiency virus against heterologous challenge. J. Virol. 73:8356-8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zammit, D. J., D. L. Turner, K. D. Klonowski, L. Lefrancois, and L. S. Cauley. 2006. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity 24:439-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zinkernagel, R. M. 2000. What is missing in immunology to understand immunity? Nat. Immunol. 1:181-185. [DOI] [PubMed] [Google Scholar]
- 104.Zinkernagel, R. M. 2002. Immunity, immunopathology and vaccines against HIV? Vaccine 20:1913-1917. [DOI] [PubMed] [Google Scholar]