

Abstract
Short RNA interference is more and more widely recognized as an effective method to specifically suppress viral functions in eukaryotic cells. Here, we used an experimental system that allows suppression of the Sendai virus (SeV) M protein by using a target sequence, derived from the green fluorescent protein gene, that was introduced in the 3′ untranslated region of the M protein mRNA. Silencing of the M protein gene was eventually achieved by a small interfering RNA (siRNA) directed against this target sequence. This siRNA was constitutively expressed in a cell line constructed by transduction with an appropriate lentivirus vector. Suppression of the M protein was sufficient to diminish virus production by 50- to 100-fold. This level of suppression had no apparent effect on viral replication and transcription, supporting the lack of M involvement in SeV transcription or replication control.
Enveloped viruses derive their envelope from cellular membranes after the viral components have assembled at the lipid bilayer. The assembly process brings together the glycoproteins spanning the lipid bilayer with the inner core of the virus particle. The inner layer of the membrane generally contains a viral protein that bridges the glycoproteins and the inner core, dubbed the matrix or M protein. M is generally considered an essential protein, without which the production of virus particle production is highly impaired if not impossible.
The M protein of Sendai virus (SeV-M), a member of the Paramyxovirinae subfamily, Paramyxoviridae family, is no exception to the rule. It is synthesized in the cytoplasm and self-associates to form a leaflet at the inner face of the plasma membrane (for a recent review, see reference 45). In the virus particle, it similarly carpets the inner part of the viral envelope, interacting with the two surface glycoproteins, HN and F, on the one hand and with the viral ribonucleoprotein complex (N protein plus viral RNA) associated with the L and P proteins on the other hand (for a review, see reference 29). In addition to its role in virus particle formation, paramyxovirus M has been reported to participate in the regulation of RNA synthesis (19, 27, 38, 40, 44). Such a role for M in viral transcription control has been described for other negative-stranded RNA viruses, such as vesicular stomatitis virus (VSV) and rabies virus (both members of the Rhabdoviridae family) (9, 11, 26, 31, 49) as well as for the influenza viruses (Orthomyxovirus family) (32, 48). In addition, VSV-M has been implicated in the shutoff of cellular transcription (3, 4), and rabies virus-M has been implicated in the stimulation of viral replication in vivo (14, 15).
Our laboratory has long been interested in the SeV-M protein and, in particular, in its function in virus particle formation (13, 35, 36, 43). To perform a structure-function analysis, one would ideally like to silence expression of the resident SeV-M gene and replace it with M mutants in a search for residues or domains that can modulate its functions. One approach would consist of deleting the M gene and in producing SeV infectious particles with the use of helper cell lines expressing M. Then, through M mutants expressed in the helper cells, or following expression of M mutants from plasmids in regular cell lines, characterization of M domains essential for its functions could be envisaged. This approach has indeed been developed for SeV (25). However, it turns out to be impractical for structure-function studies, as cell lines constitutively expressing SeV-M are difficult to produce, partly because of the possible toxicity of the protein. Similarly, transient expression of M proteins to complement SeV particle formation is inefficient, likely because of a too-low level of expression.
We have therefore investigated another approach based on short RNA interference (siRNA) technology. We targeted the resident M gene for suppression by inserting an siRNA target nucleotide sequence in the 3′ untranslated sequence of its mRNA. We derived the target sequence from the green fluorescent protein (GFP) gene, creating a recombinant Sendai virus (rSeV-M-gfpt) that grows normally in regular cells. We developed in parallel a cell line constitutively expressing siRNAs targeted to the GFP sequence (siGFP-RNAs). In the end, by growing SeV-M-gfpt in a cell line expressing the siGFP-RNAs, we could achieve a suppression of M sufficient to provoke about a 100-fold diminution of SeV particles production. Under these conditions, we found no alteration in the accumulation of viral RNAs produced by transcription or replication. In the end, this observation supports the lack of M involvement in the control of SeV RNA synthesis, a conclusion contrasting with the previously published data (38).
MATERIALS AND METHODS
Cells.
BSR-T7 cells (a gift from K.-K. Conzelmann) were grown in BHK-21 medium (Glasgow minimal essential medium; Gibco). HeLa cells were grown in regular minimal essential medium (Gibco) supplemented with 5% fetal calf serum (FCS) in a 5% CO2 atmosphere. To prepare the A549-LV-NGFR and A549-LV-siGFP cell lines, A549 cells were transduced with the appropriate lentiviral vectors (LV-siGFP and LV-NGFR, as described by Wiznerowicz and Trono [50]). Following transduction, the cells were selected twice in a cell sorter for expression of the nerve growth factor receptor (NGFR; Becton-Dickinson FACSCan2). About 50,000 cells were incubated for 30 min at 4°C, in the dark, in 100 μl of phosphate-buffered saline (PBS) containing an anti-NGFR monoclonal antibody coupled to phycoerythrin (1:50). The cells were then washed twice with PBS and finally resuspended in 300 μl PBS containing 2% FCS. After selection, the cells were concentrated and reseeded in minimal essential medium containing 10% FCS. The cells resulting from a second selection, found to express NGFR in >95% of the cases, were expanded and frozen in liquid nitrogen as a source of culture renewal.
Viruses.
Wild-type Sendai virus (rSeV), of the Z strain, was recovered from the full-length clone as described by Fouillot-Coriou and Roux (16). rSeV-GFP was rescued from the plasmid pFL5 harboring a GFP gene in between the M and F genes (see below). rSeV-M-gfpt was rescued from the plasmid pFL4-M-gfpt (see below). Rescue of the recombinant Sendai viruses, all of the Z strain type, was done as described before (18), except that the vaccinia virus T7-based method was replaced with that using BSR-T7 cells. In this method, BSR-T7 cells were transfected with the full-length genome cDNA and with pTM1-based plasmids (harboring an encephalomyocarditis virus internal ribosomal entry site [33]) expressing the N, P/Cstop, and L functions (47). Twenty-four hours posttransfection, the cells were collected and injected into 9-day-old embryonated chicken eggs. Three days later, the allantoic fluids were collected, 1 ml of which was pelleted through a 25% glycerol cushion, resuspended in polyacrylamide gel electrophoresis (PAGE) loading buffer, analyzed by PAGE, and Coomassie blue stained to evaluate rescue (presence of the viral proteins). When needed, further egg-to-egg passages were performed. Infectious viral stocks were finally prepared in 9-day-old embryonated chicken eggs and reached titers ranging from 5 × 108 to 5 × 109 PFU/ml. Infections of cells were performed as described elsewhere (34) at the multiplicities of infection (MOIs) indicated in the figure legends. LV-siGFP and LV-NGFR retrovirus vectors were prepared as described previously (50). Virus purification from cell supernatants was performed as described before (16). Briefly, cell supernatants were clarified and the virus particles, sent to the pellet through a 25% glycerol cushion by a 2-h centrifugation at 50,000 rpm, 4°C (Beckmann rotor SW 55), were directly resuspended in sodium dodecyl sulfate (SDS)-PAGE sample buffer. When the infected cells are metabolically radioactively labeled, this method yields in vast majority a set of viral proteins corresponding to that found in bona fide virus particles.
Plasmids and sequences.
pEBS-GFP, a plasmid expressing GFP under the control of the polymerase II promoter, was a kind gift of the laboratory of Michel Strubin, University of Geneva Medical School, Geneva, Switzerland (5). The pSuper construct was described previously (6). Briefly, this plasmid expresses under the control of the polymerase III H1 promoter target gene-specific transcripts predicted to fold back on itself to form 19-bp stem-loop structures representing precursors of siRNAs. To prepare pSuper-siGFP, 200 pmol of the following oligonucleotides were used: 5′-GATCCCCGAACGGCATCAAGGTGAACTTCAAGAGAGTTCACCTTGATGCCGTTCTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAGAACGGCATCAAGGTGAACTCTCTTGAAGTTCACCTTG-ATGCCGTTCGGG-3′, carrying the antisense and sense GFP sequence (5′-GAACGGCAUCAAGGUGAA-3′), were annealed at 95°C for 5 min in 100 mM Na acetate, 30 mM HEPES pH 7.4, and 2 mM Mg-acetate in a total volume of 50 μl and cooled down to 4°C. Two μl of the annealed oligonucleotides was phosphorylated with T4 phospho-nucleotide kinase (PNK) for 30 min at 37°C, followed by 10 min of incubation at 70°C to inactivate the PNK. Annealing and phosphorylation of the two oligonucleotides created compatible BglII and HindIII sites. Height picomoles of the annealed oligonucleotides were finally ligated into BglII/HindIII-digested pSuper. The plasmid pFl5 containing the SeV full-length sequence is a derivative of the original pFl3 plasmid (18). pFl3 was engineered in the first place to remove ATG (G-to-C substitution) at position 69, creating the pFL4 plasmid. The latter was subsequently modified by Machiko Nishio (Daniel Kolakofsky's laboratory, University of Geneva Medical School) to give pFl5. pFl5 contains four new unique restriction sites. In addition, in one of the unique restriction sites (MluI), an insert of 30 nucleotides containing an extra cistron was introduced at position 4832 (accession no. AB105968) in the 3′ untranslated region (UTR) of the F gene. To generate the pFL5-GFP clone, the GFP gene sequence was introduced in the MluI site. pFL4-M-gfpt was constructed from pFL4 by insertion of the GFP target sequence (gfpt; see the underlined GFP sequence, above) in the BspM1 site (position 4743) present in the 5′ UTR of the M gene.
Cell transfection.
For transfection, the cells were seeded at low density (∼3 × 105 cells in Costar six-well plates). The next day, the medium was replaced drop by drop with a preprepared mix containing the plasmid(s) and Fugene (Roche) at 3 μl/μg of plasmid in a total of 200 μl of basic salt solution. Six hours later, 3 ml of Dulbecco's modified Eagle's medium plus 10% FCS replaced the transfection mix. The next day, fresh FCS-free Dulbecco's modified Eagle's medium was added. When applied along with infection, transfection started at the end of the virus incubation time (1 h).
RNA sample preparations.
Nucleocapsid RNAs from infected cells were prepared as described before (8). Infected cells were disrupted in lysis buffer 1 (0.6% NP-40, 50 mM Tris-HCl [pH 8.0], 10 mM NaCl) (37). Postnuclei supernatants were made 5 mM in EDTA and loaded onto linear 20 to 40% (wt/wt) CsCl gradients (Beckman SW60). After centrifugation (40,000 rpm, 12°C, overnight), the nucleocapsids banding in the CsCl gradient were collected and treated as described. Total cellular RNAs were isolated using the TRIzol (Life Technologies) extraction method following the supplier's instructions.
Northern blot analysis.
Northern blot assays were performed as described before (8). For nucleocapsid RNA analysis, 32P-labeled 5′ ex-riboprobes of the plus and minus polarity were used (37). For N and M mRNA analysis, 5′-32P-labeled oligonucleotides of negative polarity specific for the N (5′-CGTCTGTCGTCCTCTTAGGG-3′) and the M (5′-GAAAATCAGCCGCATTCTCTCAATCTTGCT-3′) genes were used to probe 20 μg of total cellular RNA purified from infected or mock-infected cells. Twenty pmoles of oligonucleotides was phosphorylated with T4 polynucleotide kinase in the presence of 50 μCi of [γ-32P]ATP according to a standard procedure (42). The blotted membranes were revealed by phosphorimaging and quantified using ImageQuant version 5.0.
Primer extension analysis.
Two or 10 μg of total cellular RNA purified from infected or mock-infected cells was reacted with 0.8 pmol of the appropriate 5′-32P-labeled oligonucleotide in primer extension reactions according to a standard procedure (42). The products of the reaction were electrophoresed on 6% denaturing polyacrylamide gels. After fixation, the gels were revealed and the images were quantified as above. For the N and M gene analysis the oligonucleotides used were as above. For the P, F, and HN genes, the oligonucleotides were as follows: P (5′-GCTGGACAATCTGAGACAGAGC-3′), F (5′-CGGGTCTGATAGCAATTGCAGG-3′), and HN (5′-CCCTTGCGATAACCTCTTGC-3′).
Western blot analysis.
Western blot methodology has been described previously (35). The protein concentration of each cell sample was determined with the Bio-Rad protein microassay according to the manufacturer's protocol, and equal amounts of total proteins were analyzed within an experiment (between 3 and 15 μg, depending on the experiment; see figure legends). The anti-M, anti-N, and anti-P monoclonal antibodies, obtained from Claes Orvell, Laboratory of Clinical Virology, Huddinge Hospital, Huddinge, Sweden, were used at, respectively, 1:2,000, 1:5,000, and 1:1,000 dilutions. The anti-F0 (FSDS), the anti-HN (HNSDS), and N (NSDS) sera were prepared against the PAGE-purified proteins. These were prepared by subcutaneous injections of rabbits with proteins extracted from a Coomassie blue-stained gel. The anti-HA tag monoclonal antibody, purchased from BAbco (HA.11 16B12) and the anti-GFP antibody protein, purchased from Clontec (8372-1), were used at a 1:5,000 dilution.
RESULTS
Setup of a “universal” siRNA target.
Efficient use of siRNAs depends on the identification of suitable target sequences. Although there are more and more rules to identify these targets (for a recent report, see reference 20), this step still bears some degree of uncertainty. We therefore designed a unique target sequence that we hoped would be effective for any SeV gene silencing. We picked a sequence of the GFP gene, mainly because the GFP gene is not part of the gene pool of animal cells. Besides, GFP expression and suppression can be easily monitored. Figure 1 shows the target efficiency of this sequence. When GFP was expressed from a transfected plasmid in HeLa cells, cotransfection of a pSuper-siGFP plasmid expressing the anti-GFP siRNAs reduced GFP levels by 10-fold (9.0% remaining) (Fig. 1A). GFP was also expressed upon infection of cells with a recombinant SeV carrying a GFP transgene (rSeV-GFP) (Fig. 1C, top).
FIG. 1.
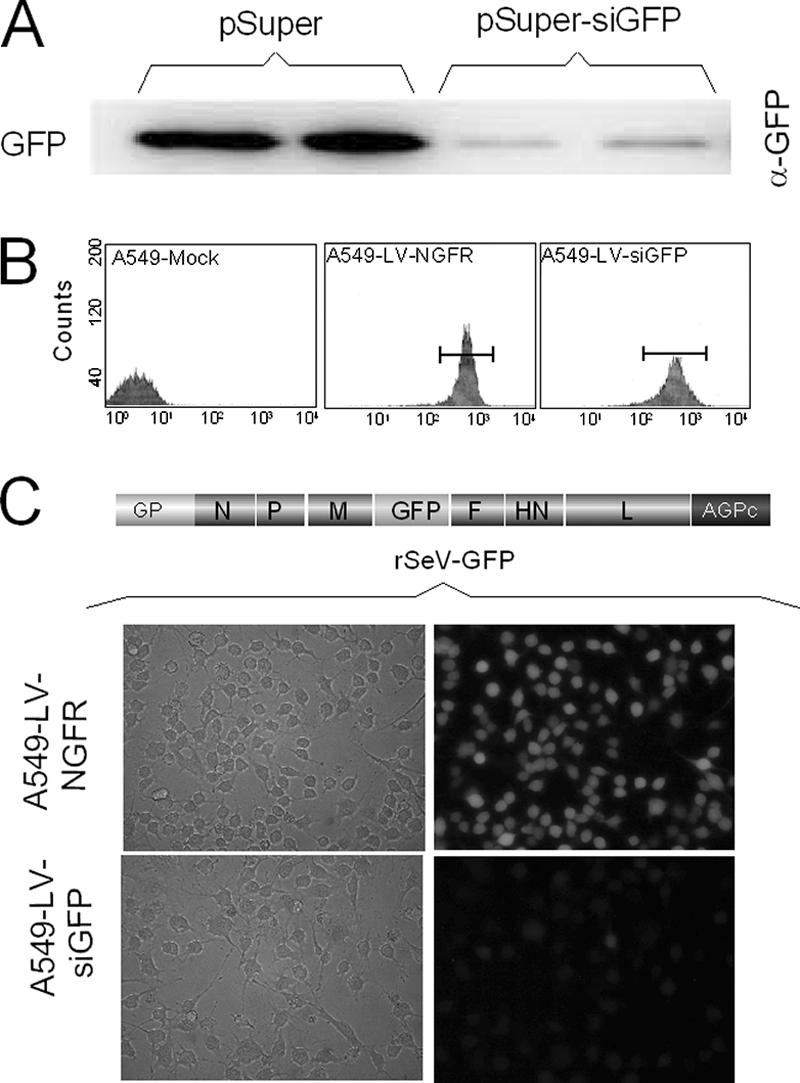
GFP target sequence amenable to silencing by specific siRNAs. A. HeLa cells were transfected with 0.2 μg of pEBS-GFP with either 1 μg of pSuper or pSuper-siGFP. Sixty hours posttransfection the cells were collected and washed with PBS. Cellular extracts were prepared for Western blot analysis using anti-GFP antibody. Fifteen micrograms of total protein (Bradford) was loaded per lane (duplicate samples). B. A549 cells were transduced with the lentivirus vectors expressing anti-GFP-siRNAs and NGFR or NGFR alone as indicated in Materials and Methods. Transduced cells were selected twice in a row by flow cytometry using anti-NGFR antibodies coupled to phycoerythrin (see Materials and Methods). The panels show the results of the second selection process, in which more than 95% of the cells were found positive for the NGFR marker. C. A549-LV-NGFR and A549-LV-siGFP were infected with a recombinant SeV-GFP (MOI = 20). Forty hours later, GFP expression was observed by microscopy under regular (left) or UV light (right; microscope Nikon Eclipse TE 300), and pictures were taken with a Nikon Coolpix digital camera 950.
In this case, its suppression by the anti-GFP siRNAs expressed from pSuper-siGFP was relatively inefficient (less than 50% reduction) (data not shown). This result was likely due to a poor transfection efficiency of pSuper-siGFP in a case where all the cells expressed GFP. To avoid the possible drawback of low transfection efficiency, we built a lentivirus vector (50) to generate a cell line constitutively producing siGFPs. A549 cells were transduced with this lentivirus expressing the anti-GFP siRNAs under the control of the H1 promoter (A549-LV-siGFP). The NGFR gene was also expressed from the vector, under the control of the EF-1a promoter, and served as a selection marker for the transduced cells. A549 cells were also transduced with a lentivirus expressing NGFR alone to derive a control cell line (A549-LV-NGFR). Two rounds of selection by flow cytometry readily led to cell lines in which close to 100% of the cells were positive for NGFR surface expression (Fig. 1B).
Upon infection of A549-LV-siGFP cells with rSeV-GFP, GFP silencing was now effective in all the cells (Fig. 1C). Western blot analysis with anti-GFP antibody confirmed this result by showing the specific loss of GFP (Fig. 2B; 20-fold reduction), whereas there was no significant change in the levels of two other viral proteins, N and M, used as internal markers. The availability of cells constitutively expressing effective siGFPs prompted us to prepare a recombinant SeV harboring the GFP target sequence in the 3′ UTR of the M mRNA (rSeV-M-gfpt) (Fig. 2A). The GFP insertion (gfpt) had no detectable effect on virus growth or on M protein expression. A549-LV-siGFP and A549-LV-NGFR cells were then infected in parallel with rSeV-M-gfpt or rSeV-GFP. Western blot assays of cellular extracts of these infections (Fig. 2B) showed that the M protein originating from rSeV-M-gfpt infection was efficiently reduced (10% remaining) in the A549-LV-siGFP cells (compare the siRNA + and − lanes). In contrast, the M protein from rSeV-GFP, expressed from a viral gene lacking the target sequence, was not affected (lane rSeV-GFP/siRNA+).
FIG. 2.

Silencing of the M gene carrying a GFP target sequence. A. Schematic outline of the recombinant SeV harboring a GFP target sequence in the M gene 5′ UTR (SeV-M-gfpt). B. A549-LV-NGFR and A549-LV-siGFP cells were infected with rSeV-GFP or rSeV-M-gfpt (MOI = 10). Forty hours postinfection, cellular extracts were prepared and analyzed by Western blotting using anti-N, anti-M, and anti-GFP monoclonal antibodies. C. A549-LV-NGFR and A549-LV-siGFP cells were infected with rSeV-M-gfpt (MOI = 10). At 16, 36, and 48 h postinfection, the cells and the clarified cell supernatants were collected. Cell extracts and purified virus particles were analyzed by Western blotting using anti-N and anti-M antibodies. Fifteen micrograms of total cellular protein (Bradford) corresponding to 1/30 of the cellular samples and 1/3 of the virus particle samples was analyzed. D. Total cellular RNAs were purified at 48 h postinfection from A549-LV-NGFR and A549-LV-siGFP cells infected with rSeV-GFP or rSeV-M-gfpt as in panel A. Twenty μg was analyzed by agarose gel electrophoresis and Northern blotting, using specific M or N oligonucleotides of negative polarity. NC(+), viral nucleocapsid RNA of positive polarity; siRNA - or +, A549-LV-NGFR or A549-LV-siGFP cells, respectively. *, migration of an M mRNA size marker.
Suppression of M significantly reduces virus particle production.
The availability of an M suppression system, effective in all the infected cells, allowed us to analyze the effects of this suppression on the viral multiplication cycle. We first examined the production of virus particles. A549-LV-siGFP and A549-LV-NGFR cells were therefore infected with rSeV-M-gfpt, and at increasing times after infection, cell samples were collected to monitor the efficiency of M suppression. In parallel, the virus particles produced in the cell supernatants were collected. Figure 2C (cell extracts) shows that M suppression was as effective early after infection, during the peak of viral protein synthesis (16 h, 2% remaining), as at later times (36 and 48 h, 5% remaining). Correspondingly, and in accordance with the role of M in virus particle assembly and budding, virus particle production was markedly decreased at all time points, although the reduction was particularly spectacular (∼50-fold reduction in M produced in the supernatant, 2.5% and 1.3% remaining for 36 and 48 h postinfection, respectively) at later times when virus particle accumulation was the highest. It is important to remember that the samples in the analysis of Fig. 2C do not represent equivalent fractions of the total. As indicated in the figure legend, 1/3 of the virus particle (VP), but 1/30 of the cellular extracts, was actually loaded on the polyacrylamide gel for Western blot analysis. Under these conditions, it is noteworthy that the N/M ratios in virus particles were modified, changing from about 1/10 to 1/0.5, under normal and M suppression conditions, respectively. This observation suggests that the amount of M involved in budding is somehow flexible (see Discussion, below).
Suppression of M protein parallels M message disappearance, but N message remains normal.
Under conditions of M suppression, the amount of the N protein was not found affected, either positively or negatively (Fig. 2B and C). This reproducible observation was made under conditions where equal amounts of total cellular proteins (15 μg) were analyzed by Western blotting. This argued for the lack of effect of M on viral transcription, as found for other Mononegavirales (see introduction). Total cell RNA was then analyzed with Northern blots probed with negative-sense RNAs specific for the M or the N genes (Fig. 2D, upper and lower panels, respectively). While under conditions of M suppression (rSeV-M-gfpt, siRNA+) the M message was clearly ablated, the N message level showed no variation. Note that the upper bands in both panels refer to the antigenomic RNAs. These were equally not affected by M suppression.
Quantitative estimation of the viral proteins upon M suppression.
To better document the effect of M suppression on the level of the other viral proteins, quantitative Western blot assays were set up for all the structural viral proteins, at the exclusion of L, for which a suitable antibody was missing. The quantitative Western blot assays involved preliminary experiments to establish the amounts of cellular extracts analyzed as well as the dilution of each antibody, so that the signals obtained were in a linear range with the amount of protein loaded. Figure 3A shows part of this effort, with Western blots loaded with increasing amounts of total cellular proteins (see legend to Fig. 3) and displaying signals that increased accordingly. Figure 3B presents a graphic quantification with error bars, with the proteins present in the control cells [siRNA(-)] taken as the reference (arbitrarily set at 100). Suppression of M without alteration of the N protein amount was confirmed. The amount of P was equally shown not to be affected. In contrast, the amounts of F and HN were increased by ∼2-fold.
FIG. 3.

Effect of M suppression on viral protein intracellular accumulation. A. A549-LV-NGFR or A549-LV-siGFP cells (siRNA − and +, respectively) were infected with rSeV or rSeV-M-gfpt (MOI = 10) or mock infected. Forty hours postinfection, cells were collected and disrupted in lysis buffer I (see Materials and Methods). Equal amounts, 3 (1×) or 15 (5×) μg of proteins was analyzed by Western blotting using specific anti-N, -P, -M, -F, and -HN antibodies. B. The amount of each protein was quantified (three independent experiments), and their values were expressed relative to that measured in the A549-LV-NGFR cells, arbitrarily taken as 100. wt (-), wild-type SeV in A549-LV-NGFR; wt (+), wild-type SeV in A549-LV-siGFP; gfpt (-), rSeV-M-gfpt in A549-LV-NGFR; gfpt (+), rSeV-M-gfpt in A549-LV-siGFP.
Quantitative measures of viral RNAs upon M suppression.
The same approach was applied to the measure of viral mRNAs. Quantitative primer extensions were run using two gel loads (Fig. 4A), and the signals generated were plotted taking the values reached in the control cells [siRNA(-)] as reference (Fig. 4B). Under conditions of M suppression [gfpt (+siRNAs)], the accumulation of mRNAs was systematically lower than that found in cells not expressing the siRNAs [gfpt (-siRNAs)]. This decrease, however, was equally observed upon infection with the wild-type (wt) virus [compare wt (-siRNAs) with wt (+siRNAs), i.e., in the absence of M suppression. When this decrease in the control infection was taken into account to correct for the values measured in the cells infected with rSeV-M-gfpt, then no evidence of significant changes in viral mRNA accumulation could be monitored under conditions of M suppression (Fig. 4C).
FIG. 4.

Effect of M suppression on viral messenger accumulation. A. A549-LV-NGFR and A549-LV-siGFP cells were infected with rSeV or SeV-M-gfpt (MOI = 10) or kept mock infected. Forty hours postinfection, cells were collected and total cell RNA purification was performed. Two (1×) and 10 (5×) μg of total RNA was used in primer extension assays with gene-specific primers of negative polarity as described in Materials and Methods. For the N gene, upper signals (NC+) correspond to primer amplification templated by the antigenome RNA. Numbers at right correspond to primer extension product theoretical sizes (nucleotides) whose relative gel migrations have been practically verified (not shown). B. Primer extension signals for panel A were quantified and expressed standardized to the values obtained in A549-LV-NGFR cells taken arbitrarily as 100. N and M genes, three experiments; P, HN, and F0, two experiments. Bars, error from the means. C. The values presented in B for the infections by rSeV-M-gfpt [gfpt(-/+)] were corrected for the decrease observed in the rSeV-infected A549-LV-siGFP cells. Bars are as for panel B.
Finally, Northern blot assays were performed on viral nucleocapsid RNAs to estimate the level of genome and antigenome replication (Fig. 5). Once more, no change in the amount of accumulated genomes [NC(-)] or antigenomes [NC(+)] could be detected under conditions of M suppression. This result corroborates the observations made in Fig. 2D and 4A ([NC(+)]) and shows that viral replication was not affected by M suppression.
FIG. 5.
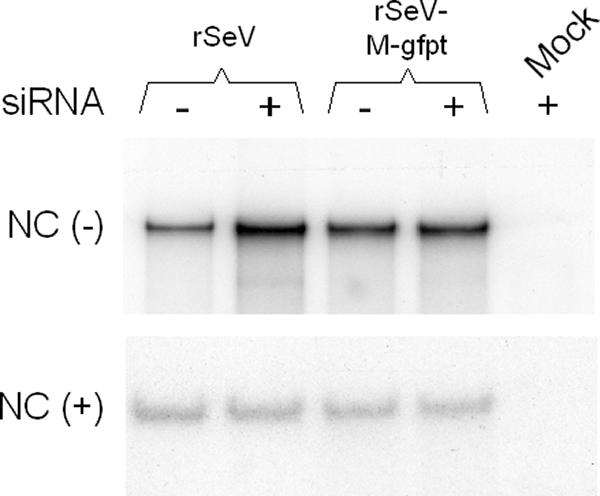
Effect of M suppression on genome and antigenome accumulation. Nucleocapsid RNAs were purified from infected cells at 36 h postinfection as described in Materials and Methods and analyzed by Northern blotting using plus-sense (upper gel) and minus-sense (lower gel) riboprobes (see Materials and Methods), scoring for genomic [NC(-)] and antigenomic [NC(+)] RNA, respectively. siRNA - or + refers to A549-LV-NGFR- or A549-LV-siGFP-infected cells. Mock, RNA samples obtained from mock-infected cells.
Interferon system induction in A549-LV-siGFP.
The estimation of the viral mRNAs showed a general deficit in A549-LV-siGFP cells compared to the A549-LV-NGFR control cells (Fig. 4). This deficit (of about 40% to 60%) was not related to the M protein suppression, since it occurred upon wild-type SeV infection as well (Fig. 4B). We considered, then, the possibility that the α-GFP-siRNA expression could induce the interferon (IFN) system. One standard test to check for IFN system activation is to measure the level of the STAT1 protein, known to be upregulated in response to IFN (22). In regular A549 cells and A549-LV-NGFR control cells, as well as in A549-LV-siGFP cells, mock IFN treated and mock infected, STAT1 levels remained at the background level (Fig. 6, -/-). This contrasted with its marked presence following the IFN addition (-/+), indicating that the cells responded to the treatment. Upon SeV infection, however, the level of STAT1 returned to the background level. Note that the levels of the N protein, which served as measures of the extent of the infections, were never affected, even after IFN treatment, in accordance with the ability of SeV to counteract the antiviral effect of the IFN system (17). In conclusion, it was not possible to document a difference in the status of the cells expressing the siRNAs regarding the IFN system induction in the context of SeV infections.
FIG. 6.
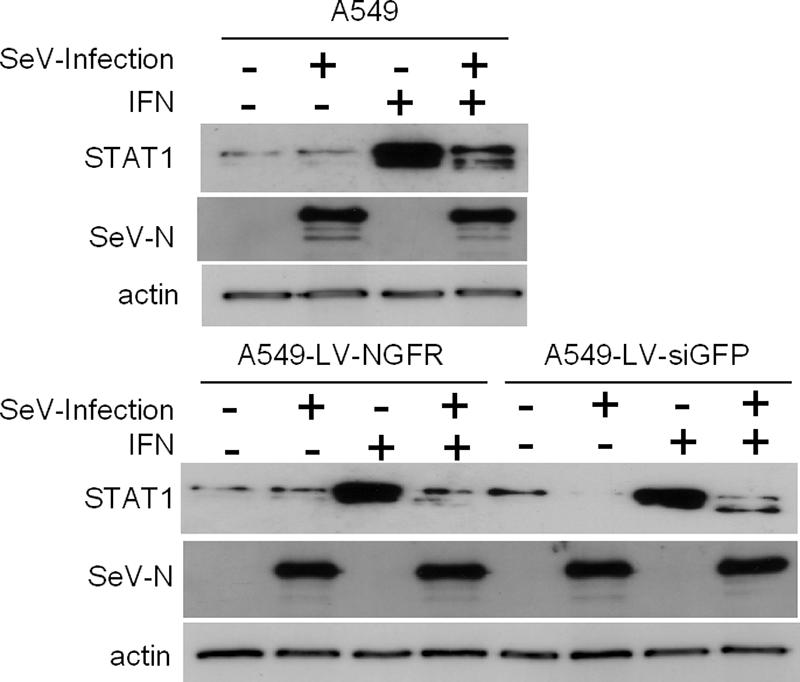
Interferon system induction in A549 cells expressing anti-GFP siRNA. A549, A549-LV-NGFR, and A549-LVsiGFP were treated or not with type I IFN-α (1,000 U/ml) for 24 h. They were then infected or not with wild-type SeV (MOI = 10). Thirty-six hours postinfection, the cells were collected and the cellular extracts were prepared as described in Materials and Methods. Aliquots corresponding to 15 μg of total proteins were analyzed by Western blotting using anti-NSDS (1/5,000 dilution), anti-STAT1 (1/1,000 dilution; S21120; Transduction Laboratories), and antiactin (rabbit antiserum; provided by C. Chaponnier, University of Geneva Medical School, Switzerland).
DISCUSSION
The extent to which siRNAs can suppress protein accumulation depends, in part, on the relative abundance of its mRNA. SeV multiplies efficiently in cells in culture, making it one of the most productive viruses of the family. The number of SeV-M messengers per infected cell has been estimated to amount to ∼30,000 (23), a number that exceeds by almost 10-fold that measured in measles virus-infected cells (10, 39). It was therefore comforting to realize that efficient suppression of SeV-M was feasible. This achievement was likely due to the experimental approach. The setup of a cell line transduced with a lentivirus vector constitutively expressing the siRNA targeted to the M gene provided (i) the assurance of getting close to 100% of the cells expressing the siRNAs and (ii) the conditions for their accumulation at a high enough concentration so as to suppress even abundant messages.
The efficient suppression of M paralleled highly significant decreases in viral particle production. This was expected for a protein known to play a central role in virus particle assembly and budding and confirms the observation made before with an rSeV carrying a deleted M gene (25). More surprising was the apparent change of N/M ratio in the residual particles produced (Fig. 2D), possibly reflecting a flexible representation of this protein in the virus particle. Such flexibility was not expected for a virion component considered essential for particle formation. Whether this reflects the production of complete particles with reduced (about 20- to 50-fold) loads of M, the production of defective particles lacking nucleocapsids or the budding of M protein by itself cannot be resolved from the data obtained in this study. The issue, however, could bear some relevance for understanding the mechanism of virus particle formation. It is noteworthy to remember, for example, that HN, one of the two viral glycoproteins, was found dispensable in the process (16).
In contrast to the important effect of M suppression on virus particle production, no significant changes were observed in the accumulation of viral RNAs in the cells. These results put SeV apart from other members of the Mononegavirales family (VSV, rabies virus, human respiratory syncytial virus, and measles virus), as far as the matrix protein function is concerned (see the references cited in the introduction), and diverge from the conclusions reached before, as far as SeV-M itself is concerned (38). As for the divergence with the other members of the family, no clear explanation can be proposed, except to admit that each family member may have evolved along similar, but not identical, paths. The discrepancy with the results of Ogino and colleagues (38), on the other hand, may flow from different experimental approaches. Ogino and colleagues purified SeV nucleocapsids from virus particles to perform in vitro transcription assays. They observed an increase in transcription after incubation with tubulin, a treatment that detaches SeV-M from the nucleocapsids. This increase in transcription, then, argues for the inhibitory role of M in transcription. Accordingly, in the infected cells, M would similarly bind to transcribing nucleocapsids to carry them to the viral assembly complex and similarly exert its inhibitory effect. Our data do not support this view. We would then propose that SeV-M is not active at this step, but the C protein, instead, could be. SeV-C has been shown to inhibit viral RNA synthesis (7, 30, 46), it is known to interact with the RNA polymerase L protein (21, 24), and it harbors a membrane-targeting signal (D. Garcin and D. Kolakofsky, unpublished data) that could serve to bring the nucleocapsids to the site of assembly, where M would come into play. This scenario is consistent with a clear overtranscription phenotype associated with SeV-C gene mutants (12, 28, 30) and with the observation that an rSeV deficient in the four C proteins was claimed to exhibit a defect in infectious virus production (28).
Regarding the viral protein levels in cells upon M protein suppression, these findings parallel the results of the RNA levels, even if the HN and F amounts were found to be increased. This increase, however, can probably be accounted for by budding inhibition leading to accumulation of these structural proteins for which the natural cellular pool is the lowest.
To end with comments regarding the data obtained, one should mention that M suppression was not complete. This opens the possibility that some remaining M still exerts its function. We consider this unlikely, since previous findings suggest a mass action effect for M.
siRNA silencing technology has been effective in suppressing proteins of viruses belonging to different families (hepatitis C virus, hepatitis B virus, human immunodeficiency virus, and human papillomavirus), including non-retro-RNA viruses of positive (poliovirus) or negative (influenza virus) polarity. siRNA is equally well suited for viruses with a life cycle restricted to the cytoplasm (respiratory syncytial virus [for a review, see reference 41]). For the negative-stranded RNA viruses, the target of the siRNA silencing was shown to be restricted to the mRNAs and did not involve the encapsidated RNAs (1, 2). This latter observation is confirmed in the present study. As for the general decrease (∼2-fold) of the viral messages in the A549-LV-siGFP cells, we have no clear explanation. The effect is seen upon infection with the wt as well with the GFP-targeted virus and, therefore, cannot be accounted for by M suppression. Whether it reflects a different permissivity of the cells to viral infection (not apparent at the replication level) or a faster turnover of the messengers in these cells is open to question. The attempt to demonstrate a difference in the IFN system status of the A549-LV-siGFP cells failed. This failure is consistent with the previous demonstration of a lack of interferon induction upon siRNA expression in the course of negative-stranded RNA virus infections (2).
In addition to addressing the role of M in SeV multiplication, the present study set up an experimental system that bears some broader features. This integrated silencing system can be used to suppress the product of any individual SeV gene, provided that the gene carries the GFP target sequence in its 5′ UTR. We see two clear advantages to this approach. (i) Viruses carrying an siRNA target (like SeV-M-gfpt) are expected to be expressing wild-type functions until they are grown on siGFP-producing cells. (ii) These SeV-targeted viruses can themselves be used to drive expression of complementing genes, from efficiently replicating minigenomes, as described previously (35). This approach is different from those using mutated viruses or viruses with one gene deleted. These approaches need helper cell lines to provide the complementing functions. The difference in how the mRNAs are provided could bias some steps of the multiplication cycle if it affects the intracellular distribution of the complementing protein. This approach can be extended to other viral systems for which expression systems through minigenomes are available.
Acknowledgments
We are indebted to Daniel Kolakofsky for critical comments and participation in writing the manuscript and to Didier Trono for general support.
This work was supported by grants from the Swiss National Foundation for Scientific Research, the Société Académique de Genève, and the Swisslife Stiftung.
Footnotes
Published ahead of print on 27 December 2006.
REFERENCES
- 1.Barik, S. 2004. Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Res. 102:27-35. [DOI] [PubMed] [Google Scholar]
- 2.Bitko, V., and S. Barik. 2001. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. 1:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black, B. L., and D. S. Lyles. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol. 66:4058-4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black, B. L., R. B. Rhodes, M. McKenzie, and D. S. Lyles. 1993. The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. J. Virol. 67:4814-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bontron, S., N. Lin-Marq, and M. Strubin. 2002. Hepatitis B virus X protein associated with UV-DDB1 induces cell death in the nucleus and is functionally antagonized by UV-DDB2. J Biol. Chem. 277:38847-38854. [DOI] [PubMed] [Google Scholar]
- 6.Brummelkamp, T. R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550-553. [DOI] [PubMed] [Google Scholar]
- 7.Cadd, T., D. Garcin, C. Tapparel, M. Itoh, M. Homma, L. Roux, J. Curran, and D. Kolakofsky. 1996. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J. Virol. 70:5067-5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calain, P., and L. Roux. 1995. Functional characterisation of the genomic and antigenomic promoter of the Sendai virus. Virology 211:163-173. [DOI] [PubMed] [Google Scholar]
- 9.Carroll, A. R., and R. R. Wagner. 1979. Role of the membrane (M) protein in endogenous inhibition of in vitro transcription by vesicular stomatitis virus. J. Virol. 29:134-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cattaneo, R., G. Rebmann, A. Schmid, K. Baczko, V. ter Meulen, and M. A. Billeter. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 6:681-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clinton, G. M., S. P. Little, F. S. Hagen, and A. S. Huang. 1978. The matrix (M) protein of vesicular stomatitis virus regulates transcription. Cell 15:1455-1462. [DOI] [PubMed] [Google Scholar]
- 12.Curran, J., J.-B. Marq, and D. Kolakofsky. 1992. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology 189:647-656. [DOI] [PubMed] [Google Scholar]
- 13.de Melo, M., G. Mottet, C. Orvell, and L. Roux. 1992. Sendai virus M protein is found in two distinct isoforms defined by monoclonal antibodies. Virus Res. 24:47-64. [DOI] [PubMed] [Google Scholar]
- 14.Finke, S., and K. K. Conzelmann. 2003. Dissociation of rabies virus matrix protein functions in regulation of viral RNA synthesis and virus assembly. J. Virol. 77:12074-12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finke, S., R. Mueller-Waldeck, and K. K. Conzelmann. 2003. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 84:1613-1621. [DOI] [PubMed] [Google Scholar]
- 16.Fouillot-Coriou, N., and L. Roux. 2000. Structure-function analysis of the Sendai virus F and HN cytoplasmic domain: different role for the two proteins in the production of virus particle. Virology 270:464-475. [DOI] [PubMed] [Google Scholar]
- 17.Garcin, D., P. Latorre, and D. Kolakofsky. 1999. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J. Virol. 73:6559-6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcin, D., T. Pelet, P. Calain, L. Roux, J. Curran, and D. Kolakofsky. 1995. A highly recombinogenic system for the recovery of infectious Sendai paramyxovirus from cDNA: generation of a novel copy-back nondefective interfering virus. EMBO J. 14:6087-6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghildyal, R., C. Baulch-Brown, J. Mills, and J. Meanger. 2003. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch. Virol. 148:1419-1429. [DOI] [PubMed] [Google Scholar]
- 20.Gong, D., and J. Ferrell. 2004. Picking a winner: new mechanistic insights into the design of effective siRNAs. Trends Biotechnol. 22:451-454. [DOI] [PubMed] [Google Scholar]
- 21.Grogan, C. C., and S. A. Moyer. 2001. Sendai virus wild-type and mutant C proteins show a direct correlation between L polymerase binding and inhibition of viral RNA synthesis. Virology 288:96-108. [DOI] [PubMed] [Google Scholar]
- 22.Haspel, R. L., M. Salditt-Georgieff, and J. E. Darnell, Jr. 1996. The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO J. 15:6262-6268. [PMC free article] [PubMed] [Google Scholar]
- 23.Homann, H. E., P. H. Hofschneider, and W. J. Neubert. 1990. Sendai virus gene expression in lytically and persistently infected cells. Virology 177:131-140. [DOI] [PubMed] [Google Scholar]
- 24.Horikami, S. M., R. E. Hector, S. Smallwood, and S. A. Moyer. 1997. The Sendai virus C protein binds the L polymerase protein to inhibit viral RNA synthesis. Virology 235:261-270. [DOI] [PubMed] [Google Scholar]
- 25.Inoue, M., Y. Tokusumi, H. Ban, T. Kanaya, M. Shirakura, T. Tokusumi, T. Hirata, Y. Nagai, A. Iida, and M. Hasegawa. 2003. A new Sendai virus vector deficient in the matrix gene does not form virus particles and shows extensive cell-to-cell spreading. J. Virol. 77:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito, Y., A. Nishizono, K. Mannen, K. Hiramatsu, and K. Mifune. 1996. Rabies virus M protein expressed in Escherichia coli and its regulatory role in virion-associated transcriptase activity. Arch. Virol. 141:671-683. [DOI] [PubMed] [Google Scholar]
- 27.Kras'ko, A. G., and A. G. Bukrinskaia. 1986. In vitro transcription of human influenza and parainfluenza viruses and its regulation. Vopr. Virusol. 31:528-532. [PubMed] [Google Scholar]
- 28.Kurotani, A., K. Kiyotani, A. Kato, T. Shioda, Y. Sakai, K. Mizumoto, T. Yoshida, and Y. Nagai. 1998. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells 3:111-124. [DOI] [PubMed] [Google Scholar]
- 29.Lamb, R. A., and D. Kolakofsky. 2001. Paramyxoviridae: the viruses and their replication, p. 1305-1340. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 1. Lippincott, Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 30.Latorre, P., T. Cadd, M. Itoh, J. Curran, and D. Kolakofsky. 1998. The various Sendai virus C proteins are not functionally equivalent and exert both positive and negative effects on viral RNA accumulation during the course of infection. J. Virol. 72:5984-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, Y., L. Luo, and R. R. Wagner. 1989. Transcription inhibition site on the M protein of vesicular stomatitis virus located by marker rescue of mutant tsO23(III) with M-gene expression vectors. J. Virol. 63:2841-2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melnikov, S. Y., A. V. Mikheeva, I. A. Leneva, and Y. Z. Ghendon. 1985. Interaction of M protein and RNP of fowl plague virus in vitro. Virus Res. 3:353-365. [DOI] [PubMed] [Google Scholar]
- 33.Moss, B., O. Elroy-Stein, T. Mizukami, W. A. Alexander, and T. R. Fuerst. 1990. New mammalian expression vectors. Nature 348:91-92. [DOI] [PubMed] [Google Scholar]
- 34.Mottet, G., J. Curran, and L. Roux. 1990. Intracellular stability of nonreplicating paramyxovirus nucleocapsids. Virology 176:1-7. [DOI] [PubMed] [Google Scholar]
- 35.Mottet, G., A. Mühlemann, C. Tapparel, F. Hoffmann, and L. Roux. 1996. A Sendai virus vector leading to the efficient expression of mutant M proteins interfering with virus particle budding. Virology 221:159-171. [DOI] [PubMed] [Google Scholar]
- 36.Mottet, G., V. Muller, and L. Roux. 1999. Characterization of Sendai virus M protein mutants that can partially interfere with virus particle production. J. Gen. Virol. 80:2977-2986. [DOI] [PubMed] [Google Scholar]
- 37.Mottet, G., and L. Roux. 1989. Budding efficiency of Sendai virus nucleocapsids: influence of size and ends of the RNA. Virus Res. 14:175-187. [DOI] [PubMed] [Google Scholar]
- 38.Ogino, T., M. Iwama, Y. Ohsawa, and K. Mizumoto. 2003. Interaction of cellular tubulin with Sendai virus M protein regulates transcription of viral genome. Biochem. Biophys. Res. Commun. 311:283-293. [DOI] [PubMed] [Google Scholar]
- 39.Plumet, S., W. P. Duprex, and D. Gerlier. 2005. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 79:6900-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reuter, T., B. Weissbrich, S. Schneider-Schaulies, and J. Schneider-Schaulies. 2006. RNA interference with measles virus N, P, and L mRNAs efficiently prevents and with matrix protein mRNA enhances viral transcription. J. Virol. 80:5951-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saksela, K. 2003. Human viruses under attack by small inhibitory RNA. Trends Microbiol. 11:345-347. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 43.Stricker, R., G. Mottet, and L. Roux. 1994. The Sendai virus matrix protein appears to be recruited in the cytoplasm by the viral nucleocapsid to function in viral assembly and budding. J. Gen. Virol. 75:1031-1042. [DOI] [PubMed] [Google Scholar]
- 44.Suryanarayana, K., K. Baczko, V. ter Meulen, and R. R. Wagner. 1994. Transcription inhibition and other properties of matrix proteins expressed by M genes cloned from measles viruses and diseased human brain tissue. J. Virol. 68:1532-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takimoto, T., and A. Portner. 2004. Molecular mechanism of paramyxovirus budding. Virus Res. 106:133-145. [DOI] [PubMed] [Google Scholar]
- 46.Tapparel, C., S. Hausmann, T. Pelet, J. Curran, D. Kolakofsky, and L. Roux. 1997. Inhibition of Sendai virus genome replication due to promoter-increased selectivity: a possible role for the accessory C proteins. J. Virol. 71:9588-9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vulliemoz, D., S. Cordey, G. Mottet-Osman, and L. Roux. 2005. Nature of a paramyxovirus replication promoter influences a nearby transcription signal. J. Gen. Virol. 86:171-180. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe, K., H. Handa, K. Mizumoto, and K. Nagata. 1996. Mechanism for inhibition of influenza virus RNA polymerase activity by matrix protein. J. Virol. 70:241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson, T., and J. Lenard. 1981. Interaction of wild-type and mutant M protein of vesicular stomatitis virus with nucleocapsids in vitro. Biochemistry 20:1349-1354. [DOI] [PubMed] [Google Scholar]
- 50.Wiznerowicz, M., and D. Trono. 2003. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 77:8957-8951. [DOI] [PMC free article] [PubMed] [Google Scholar]