

Abstract
Mutation is the basis of adaptation. Yet, most mutations are detrimental, and elevating mutation rates will impair a population's fitness in the short term. The latter realization has led to the concept of lethal mutagenesis for curing viral infections, and work with drugs such as ribavirin has supported this perspective. As yet, there is no formal theory of lethal mutagenesis, although reference is commonly made to Eigen's error catastrophe theory. Here, we propose a theory of lethal mutagenesis. With an obvious parallel to the epidemiological threshold for eradication of a disease, a sufficient condition for lethal mutagenesis is that each viral genotype produces, on average, less than one progeny virus that goes on to infect a new cell. The extinction threshold involves an evolutionary component based on the mutation rate, but it also includes an ecological component, so the threshold cannot be calculated from the mutation rate alone. The genetic evolution of a large population undergoing mutagenesis is independent of whether the population is declining or stable, so there is no runaway accumulation of mutations or genetic signature for lethal mutagenesis that distinguishes it from a level of mutagenesis under which the population is maintained. To detect lethal mutagenesis, accurate measurements of the genome-wide mutation rate and the number of progeny per infected cell that go on to infect new cells are needed. We discuss three methods for estimating the former. Estimating the latter is more challenging, but broad limits to this estimate may be feasible.
Some viral infections for which vaccines are unavailable or ineffective can be treated with antiviral drugs. One of the more interesting mechanisms suspected for some antiviral drugs is lethal mutagenesis, pushing a within-host population of viruses to extinction by overwhelming it with an elevated mutation rate. Lethal mutagenesis has emerged as a concept and practice without much theoretical underpinning. Consequently, it is not even clear how to make the measurements to ascertain whether lethal mutagenesis is operating. Much of the viral literature equates lethal mutagenesis with the error catastrophe originally proposed by Eigen in the context of quasispecies (1, 13, 27). Indeed, the very idea of lethal mutagenesis seems to have been inspired by theories of the error catastrophe. Ironically, the two concepts are not the same: an error catastrophe is an evolutionary shift in genotype space, whereas extinction is a demographic process, a drop in the absolute abundance of individuals in the population. An error catastrophe can delay or even prevent extinction by shifting the population to genotypes that are robust to mutation (see below), while lethal mutagenesis is by definition a process that pushes the population to extinction.
Despite the confusion over error catastrophes and their relation to extinction, empirical evidence broadly supports the principle of lethal mutagenesis. Chemical mutagens have been used to artificially increase error rates in a variety of RNA viruses, including vesicular stomatitis virus (VSV) (33, 39), human immunodeficiency virus type 1 (HIV-1) (40), poliovirus type 1 (12, 33), foot-and-mouth disease virus (55), lymphocytic choriomeningitis virus (30), Hantaan virus (54), and hepatitis C virus (66). The drugs severely reduced viral titers and in some cases achieved extinction. Thus, lethal mutagenesis appears to have merit in principle and also to be biochemically feasible with various drugs.
Whereas the theory of error catastrophe has been developed and expounded for decades, the theory of lethal mutagenesis remains to be developed, which is our purpose here. More specifically, our intent is to synthesize existing empirical and theoretical work to explain the quantities relevant to lethal mutagenesis. None of the theory offered here is specifically original; rather, it is the application of simple models and the interpretation of those results in the context of empirical methods that makes this paper original.
Understanding the genetic and demographic bases of population extinction has been the goal of many papers in the evolutionary and the ecological literature. The ecological literature has addressed population size and inability to adapt as key features of extinction (21, 37, 38). In the evolutionary literature, a major focus has been to discover the reason why parthogenetic plants and animals do not persist (4, 45). Processes such as mutational meltdown via Muller's ratchet and fixation of deleterious genes in small populations have been entertained as mechanisms of extinction (31, 42, 47). Muller's ratchet is the progressive accumulation of deleterious mutations in finite, asexual populations. If back mutations cannot occur, then any finite asexual population will eventually reach the point at which each genome carries at least one deleterious mutation. The mutation-free wild-type genome is forever lost from the population at this point. By the same mechanism, eventually each genome in the population will carry at least two mutations and then at least three, and so on, and all genomes with fewer mutations are forever lost and cannot be reconstructed without recombination.
Lethal mutagenesis is distinct from those processes because the latter require small populations. Lethal mutagenesis is a deterministic process that will overwhelm the largest of populations. Furthermore, the time scale over which lethal mutagenesis operates is potentially much shorter than the time scale usually attributed to processes such as Muller's ratchet, one of the few other extinction mechanisms that can operate in relatively large populations (50). Viral extinction may occur at two levels: (i) a clearance of the infection within one host or (ii) extinction of the virus across the entire population of hosts. There are mathematical similarities between the two cases but profound biological differences. Historically, the domain of lethal mutagenesis has been extinction within a host, which is what we consider here. To eradicate a virus by lethal mutagenesis across the entire population of hosts would require treatment of virtually every infected host throughout the time of its infection, which is not practical. Below, therefore, the use of the word “population” in reference to viruses refers to the viruses within one host.
THEORY
Approach and models.
Lethal mutagenesis is a form of extinction. Most basically, it requires that deleterious mutations are happening often enough that the population cannot maintain itself, but it is otherwise no different from any other extinction process in which fitness is not great enough for one generation of individuals to fully replace themselves in the next. (We consider viral fitness to be the average number of progeny capable of infecting new cells produced by a specific viral genome. Thus, the more viable progeny a virus produces, the higher its fitness.) Although lethal mutagenesis is a genetic (evolutionary) phenomenon because it is driven by mutations, it is not an exclusively genetic one because it also depends on absolute reproductive rates. Consequently, there will be no universal mutation rate that signals extinction for all viruses or even for the same virus under different conditions. Consider the following example. For a virus to establish an infection in the body, it must produce an excess number of progeny, whereby one infected cell gives rise, on average, to more than one new infected cell; otherwise, the infection will not spread. Lethal mutagenesis is a mechanism by which that excess is curtailed and rendered negative. If the excess is 49, whereby one infected cell creates 50 new infected cells, mutagenesis will need to be high enough to harm 98% of the progeny; if the excess is only one, mutagenesis will need to be high enough to harm only 50% of the progeny. Thus, the ecology or natural history of the infection will determine how much mutagenesis is required for extinction, and that dependence on ecology means that there is no universal genetic law for lethal mutagenesis.
The dependence of lethal mutagenesis on fitness means that it indeed contains an evolutionary component in addition to the ecological one. This evolutionary component requires a detailed understanding of the relationship between mutation rate and fitness, and that relationship is affected by how fitness changes with increasing numbers of mutations in the genome. These evolutionary properties defy intuition, so we resort to mathematical models. The models will proceed in three steps. First, we specify how fitness declines with increasing numbers of mutations in the genome. Here, we consider three simple models. Second, we consider the relationship between mutation rate and average fitness when the population has reached genetic equilibrium. As has already been established in the population genetics literature, this relationship is a simple one that applies across broad classes of models. It is also an important part of the lethal mutagenesis threshold. Third, we add the ecological component to the lethal mutagenesis model to achieve the threshold.
General assumptions.
Our models make several simplifying assumptions about the viral mutation process and mutational effects on viral fitness. Our approach best suits an infection that, except for treatment, is maintained indefinitely. Foremost, the viral population size is very large and the target cell population is even larger. We assume discrete generations in an ongoing infection process in which virus in each infected cell is subjected to a genomic-mutation rate of U mutations per genome per replication. Viral progeny are released and go on to infect new cells, where they are again subjected to a mutation rate of U per genome.
For all models, mutations occur at random and are equally likely to affect any site in the genome. Under this assumption, the number of mutations in a genome follows a Poisson distribution. The Poisson distribution assigns a probability of occurrence to each possible number of mutations that may arise in a genome (i.e., 0, 1, 2,…, ∞) and is characterized by a single parameter, U, which gives the mean number of mutations per genome. In what follows below, we neglect finite population effects on mutation frequencies and assume that recombination is absent and that all mutations are unique and either deleterious or neutral; the possibilities of beneficial mutations, compensatory evolution, parallel evolution, and reversion are consequently absent. Since we assume that all mutations are either neutral or deleterious, we can subdivide the mutation rate U into component Un, comprising purely neutral mutations, and component Ud, comprising mutations with a (deleterious) fitness effect, and write U = Un + Ud.
All models assume that the fitness of individuals with j deleterious mutations is independent of the identity of those mutations. For convenience and without loss of generality, the relative fitness of mutation-free genotypes is set at unity (w0 = 1); models that require absolute fitnesses are indicated where needed and parameterized accordingly.
Three models.
We consider three simple models (Fig. 1). None of these models is considered biologically realistic, but they collectively span the spectrum of possibilities usually addressed. (i) In the multiplicative fitness landscape, each additional deleterious mutation reduces viral fitness by a fraction s, independently of the number of mutations already present, and the fitness of a genotype carrying j nonneutral mutations is wj = (1 − s)j. (ii) In the Eigen two-class fitness landscape, the wild-type genotype has one fitness (arbitrarily set to 1) and all genotypes with one or more nonneutral mutations have the same (lower) fitness (18, 19). We denote the fitness of the non-wild-type sequences by wj > 0 = 1 − s. Note that mutations in the Eigen model are conditionally neutral: each mutation individually has a fitness effect of 1 − s, but multiple nonneutral mutations have the same fitness effect as a single nonneutral mutation. (iii) In the truncation landscape, a small number of mutations can be tolerated without effect, but any genotype carrying too many mutations is inviable. More specifically, genotypes carrying 0 to k nonneutral mutations have fitness levels of 1, and genotypes with k + 1 or more mutations are dead (wj = 1 for j ≤ k and wj = 0 for j > k). Note that mutations in the truncation landscape are conditionally deleterious: each mutation individually has no fitness effect, but k + 1 or more mutations are lethal.
FIG. 1.

Fitness models considered in this work. The multiplicative model [wj = (1 − s)j, shown for s = 0.5], the Eigen model (w0 = 1, wj > 0 = 1 − s, shown for s = 0.5), and the truncation model (wj = 1 for j ≤ k, wj = 0 for j > k, shown for k = 2) are shown. The mutation number j counts nonneutral mutations only.
The multiplicative model assumes that mutations have independent effects; hence, there is no genetic interaction (epistasis). The Eigen model represents an extreme case of antagonistic epistasis (in which mutations have reduced impact levels as they accumulate), and the truncation model represents an extreme case of synergistic epistasis (in which mutations have increased impact levels as they accumulate). The Eigen model is well known because it exhibits an error threshold, as discussed below.
RESULTS
Mean fitness level at equilibrium declines exponentially with the deleterious-mutation rate.
When a population is first subjected to a sustained, increased level of mutation, fitness will typically decline over several generations, until it eventually levels off and reaches a new equilibrium. The reason for this gradual approach to the new equilibrium is that mutations accumulate with each succeeding generation. The ultimate drop in fitness is due to the total burden of mutations from many preceding generations. At equilibrium, further accumulation of deleterious mutations is fully counteracted by selection against those mutations already present. Whether mutagenesis is lethal (ultimately causes population extinction) will depend in part on this equilibrium fitness.
For each of the three models considered here, with an exception noted below, the mean fitness at equilibrium (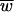 ) is simply the Poisson fraction of mutation-free genotypes (not counting neutral mutations),
) is simply the Poisson fraction of mutation-free genotypes (not counting neutral mutations), 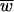 = e−Ud (35). It is remarkable that this equilibrium is independent of the selective effects of those mutations and independent of epistasis (in the absence of recombination). It is important to use the deleterious-mutation rate Ud rather than the overall genome-wide mutation rate U in this expression (61). Recall that beneficial mutations are not allowed in our models.
= e−Ud (35). It is remarkable that this equilibrium is independent of the selective effects of those mutations and independent of epistasis (in the absence of recombination). It is important to use the deleterious-mutation rate Ud rather than the overall genome-wide mutation rate U in this expression (61). Recall that beneficial mutations are not allowed in our models.
This relationship between mutation rate and mean fitness level does not hold at high mutation rates in the Eigen error catastrophe model: an error catastrophe actually maintains fitness above e−Ud (Fig. 2). An error catastrophe is an evolutionary phenomenon in which high-fitness genotypes are lost from the population because they are sensitive to mutations, and the population evolves to genotypes that are low in fitness but robust to the effects of mutations. Thus, mean fitness in the Eigen model behaves as a hybrid of two processes. At low mutation rates, fitness declines exponentially with increasing mutation rate, as in the other models. This relationship applies up to the error threshold. At mutation rates above the error threshold, fitness stops declining: there is no change in mean fitness level because all genotypes are insensitive to mutation. Therefore, contrary to common perceptions, in the Eigen two-class fitness landscape an error catastrophe actually retards the extinction of the population.
FIG. 2.

Equilibrium mean fitness level as a function of deleterious-mutation rate Ud. The solid curve is 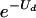 , which is the equilibrium for all models in which an error catastrophe is absent or has not occurred. The dashed line is the mean fitness level for the simple Eigen model beyond the error catastrophe, in which the best genotype has a fitness level of 1.0 and all mutants have fitness levels of 0.1 (no back mutations are allowed). In models without error thresholds, the mean fitness levels decay to arbitrarily small values for high mutation rates, whereas an error catastrophe slows down this decay and, in the simple Eigen model, sets a lower bound on the mean fitness level of the population.
, which is the equilibrium for all models in which an error catastrophe is absent or has not occurred. The dashed line is the mean fitness level for the simple Eigen model beyond the error catastrophe, in which the best genotype has a fitness level of 1.0 and all mutants have fitness levels of 0.1 (no back mutations are allowed). In models without error thresholds, the mean fitness levels decay to arbitrarily small values for high mutation rates, whereas an error catastrophe slows down this decay and, in the simple Eigen model, sets a lower bound on the mean fitness level of the population.
In conclusion, except at the error catastrophe, mean fitness level depends entirely on the deleterious-mutation rate, not on the fitness effects of mutations or on the way mutations interact. The higher the mutation rate, the lower the population's fitness. This result holds for infinite, asexual populations; for sexual populations, it holds only if there is no epistasis (35). This result also relies on the assumption of no back mutation. When this assumption is relaxed, mean fitness is determined by fitness effects (53, 61, 63), and low population sizes amplify this effect (36). Nevertheless, 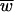 = e−Ud is a good first-order approximation even in these cases (8). Note also that none of the results that we present in the following sections are strictly dependent on the assumption of no back mutations. We employ this assumption mainly for mathematical simplicity and clarity of presentation.
= e−Ud is a good first-order approximation even in these cases (8). Note also that none of the results that we present in the following sections are strictly dependent on the assumption of no back mutations. We employ this assumption mainly for mathematical simplicity and clarity of presentation.
An extinction criterion.
The results described above provide the mean fitness level on a relative scale, in which the best (wild-type) genotype has a fitness level of 1. Unfortunately, models of relative fitness (which include all of Eigen's error threshold models) do not enable direct calculation of extinction rates, because actual numbers of offspring cancel out in those types of models (see the supplemental material). The difference between extinction and survival depends on actual birth rates or numbers of offspring and thus depends on absolute fitness (6, 60, 62). Extinction is a demographic phenomenon. Although the relationship between mutation rate and population mean fitness level is central to the calculation of extinction conditions, knowledge of only mean relative fitness level is insufficient to determine extinction, as we show now.
Deterministically, a decline in the population size will occur when the average number of offspring per parent is less than 1 for all genotypes:
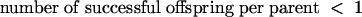 |
(1) |
If the inability of parents to replace themselves continues indefinitely, population extinction will ultimately result (neglecting limiting cases of infinitesimal declines). To extend this result to lethal mutagenesis, it is necessary to separate the evolutionary (mutational) and demographic components. If we define R as the number of successful viral offspring released per cell in the absence of mutation, the general condition for a decline in the population size is then
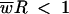 |
(2) |
(see the supplemental material). This formula multiplies relative fitness level (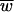 ) by the maximum number of offspring per parent to create a measure of absolute fitness, i.e., number of progeny. The
) by the maximum number of offspring per parent to create a measure of absolute fitness, i.e., number of progeny. The 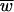 term represents the evolutionary component, and the R term represents the demographic or ecological component; our use of R is in fact borrowed from the demography literature.
term represents the evolutionary component, and the R term represents the demographic or ecological component; our use of R is in fact borrowed from the demography literature.
Formally, R applies to the wild-type, mutation-free genotype and is the number of progeny released from one infected cell that go on to establish infections in other cells. The reason for basing R on the best or mutation-free genotype is so that all effects of mutation may be subsumed into  . This dependence on the best genotype poses some empirical challenges when these quantities are being measured. These difficulties will be addressed below, although one of those dependencies will be overcome by a modification introduced next.
. This dependence on the best genotype poses some empirical challenges when these quantities are being measured. These difficulties will be addressed below, although one of those dependencies will be overcome by a modification introduced next.
If both terms in equation 2 are constant over time, then condition 2 ensures population extinction. However, R may not be constant. As is recognized in the literature on demography (2), the value of R will often be density dependent, largest when the population is at its lowest density. If, by the same argument, R increases as the viral population nears extinction, inequality 2 may be satisfied initially but later reverse and lead to a stable population reduced in size. A more stringent condition is thus the replacement of R with Rmax, representing the maximum reproductive rate of the mutation-free genotype across all viral population densities. Using the results for the equilibrium mean fitness level at mutation rate Ud, equation 2 is easily modified to provide a sufficient condition for lethal mutagenesis in the absence of an error catastrophe:
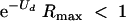 |
(3) |
This extinction criterion can be justified from two perspectives. As suggested above, this condition merely states that the average number of successful offspring per infected cell is less than 1 once the mutation-selection equilibrium has been attained (successful offspring are those that go on to infect new cells). The population size must then decline. A different justification for this result comes from the theory of branching processes (32). e−Ud is both the mean fitness level and also the fraction of offspring with no nonneutral mutations. If this number is so low that the best genotype produces (on average) less than 1 viable offspring that successfully initiates a new infection, then the population will go extinct eventually.
Recall that e−Ud is the mean fitness level in all models lacking an error threshold and in the Eigen model before the error catastrophe. It may be impossible to satisfy condition 3 in the Eigen model if the error threshold occurs before the mean fitness level drops to the requisite value e−Ud. Extinction is impossible in this specific case because all mutant genotypes have absolute fitness levels high enough to replace themselves, and fitness cannot drop lower. Of course, this extreme case is unrealistic, but it serves to illustrate the possibility that some realistic models may not obey criterion 3.
Dynamics of extinction: population decline may not happen immediately.
It is instructive to consider some numerical examples to develop a sense of the overall process of lethal mutagenesis. Figure 3 shows the decline in fitness over the first 10 generations of exposure to mutation rate Ud = 2 for each of our three models. The equilibrium mean fitness level is 0.135 for this mutation rate in all three models. There are five curves in each graph because two of the models are illustrated with two values each for the selection coefficient s. The calculations assumed a starting genotype lacking mutations, and the curves represent the initial changes in fitness as the viral population evolves toward equilibrium. The data shown in Fig. 3A are the same as those in Fig. 3B, except that the mean absolute fitness level on the vertical scale in Fig. 3B is exactly twice what it is in Fig. 3A: the mutation-free genotype was assumed to have four offspring in Fig. 3A and eight offspring in Fig. 3B. Gene frequency evolution is thus the same for the corresponding curves between Fig. 3A and B, but the two graphs differ in whether the outcome is ultimately extinction or survival. In both graphs, the extinction threshold is an absolute fitness level of 1, indicated with the horizontal black line. Extinction (lethal mutagenesis) would never occur for any of the curves in Fig. 3B but will eventually occur for all the curves in Fig. 3A. The reason for the difference in extinction is evident from our criterion, e−UdRmax < 1. Thus, 0.135 × 4 = 0.54 < 1 implies extinction in Fig. 3A but 0.135 × 8 = 1.08 > 1 implies survival in Fig. 3B. All but one of the curves in Fig. 3A have crossed the extinction threshold by generation 10; hence, their populations would be declining in generation 10; one curve in Fig. 3A has not crossed the threshold by generation 10 but would by generation 12. Even after crossing the extinction threshold, the population may persist for hundreds or thousands of generations, depending on its initial size and how close to 1.0 the absolute fitness level remains at the mutation-selection equilibrium.
FIG. 3.

Decay in average fitness level over the initial 10 generations of mutagenesis with mutation rate Ud = 2. All models illustrated have the same equilibrium mean relative fitness level of e−2 = 0.135. Multiplicative models are indicated with circles (filled, s = 0.1; open, s = 0.5), truncation models by squares (open, k = 1; filled, k = 3), and the Eigen model by filled diamonds (s = 0.9). Graphs A and B represent the same changes in relative fitness level but different absolute fitness levels (relative fitness levels have been multiplied by R = 4.0 in A and by R = 8.0 in B for conversion to absolute fitness levels). The extinction threshold is shown as a thick black line at the absolute fitness level of 1. All populations in panel A will eventually go extinct, although one of the multiplicative models does not cross the extinction threshold until generation 12. None of the populations in panel B will go extinct, because their intrinsic fecundities (R) are high enough to offset the deleterious effects of a mutation rate of Ud = 2. Once a curve drops below the extinction threshold, the population size begins declining, but the time until complete loss of the population depends on initial population size and may take many generations. Gene frequency dynamics are the same in both graphs despite the different outcomes in extinction.
These graphs emphasize two points. First, the same gene frequency dynamics operate regardless of whether the outcome is extinction or survival (while population size remains large). Thus, there is no genetic signature of lethal mutagenesis to distinguish it from the same mutagenesis and relative fitness effects observed when the population survives. In particular, lethal mutagenesis operating in large populations is not a runaway mutation accumulation in which mutations increase indefinitely (although stochastic effects will augment mutation accumulation once the population becomes small). The ecology of the infection determines whether the outcome is extinction or survival via its effect on offspring number, Rmax, but the genetics behaves the same. Second, lethal mutagenesis is usually a progressive decline. Unless mutation rates are elevated to extreme levels, absolute fitness may stay above the extinction threshold for several generations, until mutations have accumulated. Even after the threshold is breached, reproduction continues, just not enough to maintain population size. This prediction is compatible with results from mutagenesis experiments indicating that viral extinction is not sudden. For example, the presence of the base analog 5-hydroxydeoxycytidine resulted in the loss of HIV-1 infectivity after 9 to 24 serial passages in human cells (40). Similarly, 5-fluorouracil or 5-azacytidine caused occasional extinction of foot-and-mouth disease virus populations after 11 to 21 serial passages (55).
Estimating parameters of lethal mutagenesis.
The extinction threshold for lethal mutagenesis involves two components. One is evolutionary and depends only on the deleterious-mutation rate. The other is demographic, an absolute fecundity specific to the infection, and applies to the best genotype. Both present difficulties in estimation, especially in vivo. However, the deleterious-mutation rate is potentially the most important and most empirically tractable of the two.
Measuring the deleterious-mutation rate, Ud.
The meaning of Ud is straightforward: it is the genomic rate of deleterious mutations per generation. Using the tools of molecular biology, this value is perhaps most easily sought as the product of two numbers, the genome-wide mutation rate U times the fraction of mutations that are deleterious, α = Ud/U. The proportion of mutations that are deleterious, α, has been estimated as 70% in VSV for randomly generated point mutations (40% lethal, 30% viable but deleterious [51]). Direct estimates of α for other viruses are not available. Interestingly, however, our models can provide an indirect estimation of a component of α, the fraction of mutations that are nonviable mutations. We obtain this estimate by reanalyzing data from a study that addressed the impact of the mutagen ribavirin on poliovirus type 1 infectivity (12). Mutations were counted by sequencing of biological clones obtained from isolated PFU, and infectivity was measured as the number of PFU per standard amount of genomic RNA. These two variables are plotted in Fig. 4b of reference 12. To carry out our analysis, we have to assume that the observed numbers of mutations equal the mutation rates (U). Exact equality exists only in the first generation after mutation (Table 1), but equating numbers with rates seems a reasonable approximation in this case because viruses were sequenced only a few replication rounds after mutagenesis. Since the infectivity assay distinguishes only viable and nonviable mutants, we must take into account only neutral (s = 0) and lethal (s = 1) mutations. Equation S8 in the supplemental material takes the following form here: infectivity = e−γm, where γ is the fraction of lethal mutations and m is the mutation count. (Note that α measures the fraction of deleterious mutations, which is a superset of the lethal mutations. We always have γ ≤ α.) A least-squares regression yields an estimate of γ = 0.33 ± 0.13, which means that approximately one-third of the mutations produce noninfectious virions. This result is similar to that obtained for VSV in the absence of drugs. Hence, even though the effects of individual mutations depend on the environment, the overall fraction of lethal mutations might be roughly constant for different RNA viruses in different environments. It would obviously be most useful to lethal mutagenesis applications if the value of α were relatively constant.
FIG. 4.

Lethal mutagenesis threshold according to mutation rate Ud and maximum fecundity Rmax, from inequality 3. The relationship is log linear, so that changes in mutation rate have a much larger effect on extinction than changes in fecundity. In turn, modest increases in mutation rate, especially for RNA viruses, may be especially amenable to achievement of extinction.
TABLE 1.
Number of mutations per genome equals the mutation rate only in the first generation before selection
| Generation | Avg no. of mutations/genome in modela:
|
||
|---|---|---|---|
| Multiplicative | Threshold (k = 2) | Eigen | |
| 1 (before selection) | U | U | U |
| 1 (after selection) | U − sUd | 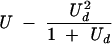 |
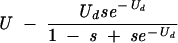 |
| 2 (before selection) | 2U − sUd |  |
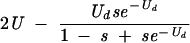 |
| Equilibriumb (before selection) | Ud/s | 2 + Ud | 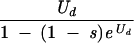 |
Table cells give the average numbers of mutations per genome as functions of overall mutation rate U, deleterious mutation rate Ud, generation number, and model. The models assume that mutations occur as the genome is replicated and packaged, with selection ensuing later, when that genome infects a new cell. Thus, lethal mutations would be observed in the genomes of individual virus particles (hence before selection) but not in a consensus sequence obtained from a plaque (after selection). The process of mutation alone increases average mutation number with each generation, whereas selection reduces it.
Equilibrium number of deleterious mutations only. The number given for the Eigen model assumes that an error catastrophe has not occurred; after the error catastrophe, the equilibrium number of mutations is infinite if unlimited mutations are allowed. The equilibrium for the multiplicative model was given in reference 31 and that for the Eigen model in reference 59.
Whereas α, the fraction of all mutations that are deleterious, might be independent of context and thus be estimable from experiments done outside the context of specific drugs, mutation rate estimations must be carried out in the presence of the mutagenic drug. Three approaches to mutation rate estimation have been commonly used. Each has its own strengths, but none is free of difficulties, as summarized in Table 2.
TABLE 2.
Summary of methods for estimating mutation rates and their respective advantages and drawbacks
| Method | What is measured | Advantage(s) | Drawbacks |
|---|---|---|---|
| Luria-Delbrück | Rate of mutation to a (selectable) phenotype | Easily assayed for appropriate phenotypes | Mutation rate must be converted to entire genome; does not estimate fraction of mutations that are deleterious; few phenotypes can be assayed; not feasible in vivo |
| Mutations in genomic sequences | Accumulated no. of mutations | Mutations can be observed even in the absence of detectable phenotypes; counts can be genome-wide | Accumulated total is reduced by selection and increased by no. of generations; deleterious fraction is unknown |
| Mutation accumulation | Declines in rate and variance of fitness with population bottlenecks | Provides a mutation rate for deleterious effects, omitting neutral mutations; genome-wide; easily assayed | Mutations with large deleterious effects are omitted; protocol is prone to recovery of beneficial mutations that bias the estimates; not feasible in vivo |
The classic method for estimating mutation rates is the Luria-Delbrück fluctuation test (41; see reference 65 for a review): an easily selected phenotype is scored in replicate cultures inoculated with phenotype-negative genotypes. For example, a small inoculum of an antibody-sensitive virus would be used to infect a number of identical cultures without antibody, grown for a period of time and then plated in the presence of the antibody to test for the appearance of resistant mutants. In its simplest version, the proportion of phenotype-negative cultures is used to estimate the mutation rate from the formula P0 = e−μΔN; P0 is the proportion of mutant-free cultures, ΔN is the change in population size during the growth of the cultures, and μ is the rate per replication event at which the phenotype converts into the selectable state. Under some additional assumptions, it is possible to estimate μ from the entire observed distribution of the number of phenotype-positive individuals per culture (24, 56). To extrapolate phenotypic mutation rates to genome-wide mutation rates (U), it is further necessary to know the number of different mutations that give rise to the selectable phenotype. This method has been used to estimate mutation rates for influenza A virus (57), measles virus (52), bacteriophage Φ6 (11), and VSV (24). Extrapolation to genomic error rates was done for the last three viruses, yielding U = 1.4, U = 0.03, and U = 0.07, respectively. Note that this extrapolation may be subject to a potentially large and difficult-to-quantify error. If there are few ways of mutating to the phenotype, then the estimates obtained from the Luria-Delbrück assay may deviate considerably from the genomic average because site-to-site variation can be large. One should also consider that biases with Luria-Delbrück estimates may exist when a substantial fraction of mutations are lethal, but we have not explored this possibility.
A second approach to estimating the mutation rate is to measure the number of mutations in sequences (mutation count), as has been done in several mutagenesis experiments (12, 29, 30, 33, 40, 54, 55, 66). The difficulty is that there is no feasible method for converting numbers of mutations into a mutation rate. In particular, when mutation-free templates are used initially, the observed numbers of mutations increase with each succeeding generation, counteracted by selection against the mutations. The net accumulation thus depends on the fitness effects of the mutations, mutation rate, and time; the observed accumulation will not generally allow a unique determination of mutation rate without independent knowledge of fitness effects and number of generations. If all mutations are neutral, the number of mutations increases by the neutral mutation rate each generation, whereas if all mutations are lethal, then mutations do not accumulate, because mutated genomes die. In between these two extremes, any observed rate of mutation accumulation could stem from a high rate of highly deleterious mutations or a low rate of weakly deleterious mutations. Table 1 shows that the number of mutations per genome equals U only in the first generation. After this point, the number of mutations per genome depends on the deleterious-mutation rate, the generation number, and the selective effect of mutations. We are unaware of any empirical study in which these considerations have been applied when estimating mutation rates from mutation frequencies.
Although there are many complications in estimating mutation rates from counts, we see from Table 1 that one simple method may be feasible: expose viral genomes to a single generation of mutagenesis and measure the counts before selection. For example, cells could be infected at a multiplicity near 1 in the presence of a drug. The drug may prolong the infectious cycle, but as long as nearly all cells are infected initially, there will be few cells to be infected in second cycles. Either the virus should kill the cell or the infected cell should resist superinfection for this method to give meaningful results. Individual virions from the resulting culture are then sequenced directly, in the absence of any subsequent infection or other biological amplification process that would cause a bias against deleterious mutations.
Yet, even this simple protocol, which is technically feasible with many viruses, gives a direct estimate of mutation rate only if viral replication within a cell does not select against mutations that arise within that cell (as when the infecting genome is the template for all copies). If the nature of replication is unknown or differs from the single-template mechanism, one solution may be to measure mutation rates in parts of the genome that would not be subject to selection within the cell. Another limitation of this method is that mutations may have accumulated prior to the beginning of the experiment or may be introduced by reverse transcription during the synthesis of the cDNA. Sophisticated technology has been developed for measuring mutation rates during a single infection cycle and in the absence of selection in retroviruses (see reference 44 for a review). This technology, based on the use of genetic constructions that carry nonviral genes, offers a valuable tool for lethal mutagenesis experiments. These genes are partially released from selection and hence can be used to estimate mutation rates more accurately. A similar approach was undertaken using a cognate mutational target in a study with tobacco mosaic virus in which the viral gene that encodes the movement protein was complemented by a plant transgene (43).
The two methods described above provide estimates of mutation rates but require independent estimates of the fraction of deleterious mutations. A third method, mutation accumulation, partly overcomes this problem. Mutation accumulation experiments offer a method for directly estimating deleterious-mutation rates, but only for viable mutations. For VSV (20) and bacteriophage φ6 (7), this method has yielded estimates of Ud = 1.2 and Ud = 0.07, respectively. (Note that these values are not directly comparable to the Luria-Delbrück estimates of U for the same viruses because of differences in the methodologies.) Starting from a single clone, several lineages are founded and propagated at the lowest possible population size, which is typically done as plaque-to-plaque transfers. Small population size facilitates the accumulation of all nonlethal mutations, even deleterious ones, through genetic drift. Together, the average rate of fitness decline and the variance between lines enable estimates of the deleterious-mutation rate and the average deleterious effect; the Bateman-Mukai (3, 46), maximum-likelihood (34), and minimum-distance (25) methods are different statistical approaches to these estimates. A benefit of estimating mutation rates by this method is that neutral mutations do not affect the estimates, and of course, neutral mutations are also not relevant to lethal mutagenesis. On the other hand, unlike for experiments with higher organisms, it is impossible to maintain population size to a single individual, so some degree of selection is inevitable (e.g., during plaque outgrowth), introducing a bias against strongly deleterious mutations. As a consequence, lethal mutations will be completely missed. Another limitation of this design for viruses is that plaques may become undetectable at very low fitness levels, a situation that is particularly likely in mutagenized populations.
Measuring maximum fecundity, Rmax.
The second parameter in the extinction threshold is a type of fecundity, Rmax. R is the average number of offspring per cell infected by the mutation-free genotype that would go on to establish new infected cells. This parameter specifically applies in vivo, so its measurement is not trivial. Some histological observations on plants inoculated with tobacco mosaic virus suggest that R may be as low as 3 to 6 particles per cell (43).
It is easiest to contemplate the fecundity value R as the product of two parameters, S and b (R = Sb). The parameter S is the success rate, corresponding to the survival of mutation-free progeny in establishing infections in new cells, and b is the burst size, i.e., the number of viable viral offspring released from a cell infected by the mutation-free genome. In general, b is a number much larger than 1, but S is always strictly smaller than 1, being reduced by senescent decay, immune clearance, and other properties specific to the infection in vivo. Thus, R may be much less than the number of offspring (burst size) per infected cell, because the success rate, or survival of progeny virus, may be low. Additionally, the values of S (and possibly b) may be density dependent, larger when the viral load is low and smaller when the viral load is high. Indeed, for a viral infection to reach an equilibrium density, one or both of these quantities must decline as the infection grows. Therefore, as mentioned previously, the R value sufficient to satisfy our extinction criterion must be the maximum across all stages of the infection, Rmax. Otherwise, mutagenesis might reduce viral load down to the point that inequality 3 is reversed.
The mutagenic drug may impair b or S directly, contributing to achievement of the lethal threshold by nonmutagenic processes. For example, ribavirin negatively impacts viral fitness by possibly four molecular mechanisms besides mutagenesis (28). Those effects facilitate satisfaction of the extinction criterion of equation 3 by reducing Rmax. Indeed, if a mutagenic drug is so harmful to viral reproduction that Rmax is <1, extinction will occur regardless of the mutagenic effect. (In general, Fig. 4 can be used to calculate the combination of effects on Ud and Rmax that will cause extinction together.)
There are several difficulties in estimating Rmax and thus b and S. First, the model assigns these values to the wild-type, mutation-free genotype. It may not be possible to confine assays to mutation-free genotypes or even to know which genomes are mutation free. This difficulty could lead to underestimation of both parameters, as genomes with accumulated mutations will likely have lower values of b and S than those that are mutation free. Another complication is that mutagenesis may confound the estimates of b and S. For example, the estimate of b, the number of viral progeny produced per infected cell, might seem to be obtained easily, but mutations arising in progeny that kill or otherwise harm them may interfere with progeny counts (which are typically done by plaque assays) and thus lead to underestimation of b. Finally, the estimate of b will likely depend on whether b was measured at a low or high multiplicity of infection.
The difficulties in obtaining direct estimates of Rmax may require working with crude upper limits and indirect estimates. Fortunately, great accuracy in the estimate of Rmax is not essential, because the mutation rate satisfying equation 3 appears as an exponent, so small changes in Ud can overwhelm large differences in Rmax (Fig. 4). A gross upper limit to Rmax might be obtained by setting S to 1 and measuring b in cell cultures. For example, for bacteriophage Φ6, the burst size was estimated as b = 76 PFU per cell (11), whereas in an animal virus, such as VSV, a single infected cell can often produce several thousand particles (22, 23). These estimates for b are not fully independent of the mutation rate, as mentioned above, but they give an idea of the order of magnitude of b for cellular cultures in the absence of mutagens. If we replace Rmax by b in equation 3, we have
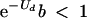 |
(4) |
This is a crude but, most probably, conservative condition for lethal mutagenesis. For example, for b = 100, a deleterious-mutation rate of Ud ≈ 4.6 would suffice to fulfill this condition, whereas Ud ≈ 6.9 would be necessary for b = 1,000. If basal mutation rates in RNA viruses were around 1 (14, 15), given that the majority of mutations are deleterious (17, 51), an approximately five- to sevenfold increase in mutation rates would be sufficient to achieve lethal mutagenesis. Since some mutagens are known to reduce the replicative capacities of viruses by mechanisms independent of mutagenesis (28) and b is a crude upper limit to Rmax, more-modest increases in Ud will probably suffice to induce lethal mutagenesis. A series of experiments with RNA viruses or retroviruses replicating in the presence of base analogs have shown that reductions of several orders of magnitude in viral titers and even extinction can be achieved with modest increases in mutation counts, ranging from less than twofold to sixfold relative to those for the untreated controls (12, 30, 33, 40, 55).
DISCUSSION
Lethal mutagenesis is an elevation of mutation rate to the point that a population is so overwhelmed by deleterious mutations that it cannot maintain itself. This method has been suggested as the basis of successful treatments of viral infections by use of drugs known to elevate mutation rates. This paper has developed a simple theoretical condition for the operation of lethal mutagenesis in the viral infection of a culture or host:  , where Ud is the genomic rate of deleterious mutation and Rmax is the maximum average number of viral progeny per cell infected with wild-type virus that go on to establish new infected cells. Although the Eigen error catastrophe theory is often invoked as the theoretical basis of lethal mutagenesis, that process is different from lethal mutagenesis and may actually retard lethal mutagenesis.
, where Ud is the genomic rate of deleterious mutation and Rmax is the maximum average number of viral progeny per cell infected with wild-type virus that go on to establish new infected cells. Although the Eigen error catastrophe theory is often invoked as the theoretical basis of lethal mutagenesis, that process is different from lethal mutagenesis and may actually retard lethal mutagenesis.
The goal of treatment could be to reduce viremia during an acute infection or to end a persistent infection. With an acute infection, it is likely that any decrease in mean fitness due to mutagenesis will slow the ascent of the viremia and thereby augment recovery by the immune system. For this case, mutagenesis need not surpass the extinction threshold to have a beneficial effect: any reduction in viral fitness will reduce the rate at which the within-host viral population expands, potentially enabling the immune system to clear the infection earlier. Our theory applies also to cases of persistent infection, which is associated with the usual application of lethal mutagenesis.
Extinction threshold equation 3 is sufficient to cause viral decline but is possibly conservative and may specify a higher mutation rate than necessary. For example, the within-host growth rate of the viral population could be important to the outcome of the infection, and a slowing of viral growth rate will be achieved even if 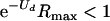 is not satisfied. The exact mutation-extinction threshold lies in the ecology of each type of infection, and a specific model of these dynamics, as well as of the impact of mutation on the different infection parameters, is required. What generalities will be found by studying specific models remains to be seen, however.
is not satisfied. The exact mutation-extinction threshold lies in the ecology of each type of infection, and a specific model of these dynamics, as well as of the impact of mutation on the different infection parameters, is required. What generalities will be found by studying specific models remains to be seen, however.
As a second example of how our extinction criterion is possibly conservative, stochastic effects may contribute to the fixation of deleterious mutations in finite populations through processes such as mutational meltdown mediated by Muller's ratchet (42). For a population of size N with deleterious-mutation rate Ud, the expected number of individuals without any nonneutral mutations at equilibrium is 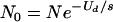 . For N0 < 1, there is a high probability that these mutation-free individuals are lost by chance or never exist, and the loss will be irreversible without back mutation (9, 26, 31). This stochastic mutation accumulation process, or Muller's ratchet, can ultimately lead to the extinction of the population (42). Small populations are prone to fixing deleterious mutations, as has been amply demonstrated in several experiments (10, 16, 64). The emphasis in those studies has been small population size, but as the important quantity in the above formula is N0, increasing the mutation rate through the use of chemical mutagens is an alternative to reducing the total population size. For the multiplicative model, beyond a mutation rate of Ud = s ln N, the population risks extinction even without deterministic lethal mutagenesis. Not surprisingly, the combination of population bottlenecking and chemical mutagenesis has proven to be the most efficient way to achieve viral extinction (55).
. For N0 < 1, there is a high probability that these mutation-free individuals are lost by chance or never exist, and the loss will be irreversible without back mutation (9, 26, 31). This stochastic mutation accumulation process, or Muller's ratchet, can ultimately lead to the extinction of the population (42). Small populations are prone to fixing deleterious mutations, as has been amply demonstrated in several experiments (10, 16, 64). The emphasis in those studies has been small population size, but as the important quantity in the above formula is N0, increasing the mutation rate through the use of chemical mutagens is an alternative to reducing the total population size. For the multiplicative model, beyond a mutation rate of Ud = s ln N, the population risks extinction even without deterministic lethal mutagenesis. Not surprisingly, the combination of population bottlenecking and chemical mutagenesis has proven to be the most efficient way to achieve viral extinction (55).
An interesting outcome of the theory presented here is that there is no genetic signature of lethal mutagenesis that distinguishes it from nonlethal mutagenesis. Mutagenesis itself obviously has a genetic signature, but whether extinction will result does not. The same elevated mutation rate may or may not cause population extinction, and at least while the population is still large, the genetic evolution of deleterious mutations is the same whether the population is stable or declining. There is no mutational runaway accumulation of mutations accompanying lethal mutagenesis. The reason for this genetic independence of population survival versus extinction is that genetic evolution depends on relative fitness, whereas population survival depends on absolute fitness, i.e., total numbers of offspring.
There are demonstrations, both in vivo and in vitro, that the addition of mutagens can lead to the extinction of the viral population (29, 30, 40, 55). Whether these results constitute a clear demonstration of lethal mutagenesis depends on the other potential effects of the mutagen. The most thorough empirical study of this problem measured mutation counts (as approximations of rates) in poliovirus subjected to ribavirin treatment in vitro (12). Even if those estimates of mutation counts are accepted as rates, it is further necessary to estimate the number of viruses produced by one cell that go on to infect other cells. At the highest dose in that study, mutation counts were indeed quite high per genome (15.5, a high rate even if due to an accumulation over a few generations), and it seems likely a priori that mutagenesis would have been high enough to ensure extinction (Fig. 4). However, it is also possible that other effects of the drug would have been high enough to eradicate the virus without mutagenesis.
Every viral infection that could potentially be treated with a mutagen falls into one of three categories: (i) 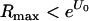 , where U0 is the deleterious-mutation rate in the absence of mutagenesis, in which the virus cannot successfully establish an infection, and mutagenesis is not necessary for extinction but might shorten the total duration of infection; (ii)
, where U0 is the deleterious-mutation rate in the absence of mutagenesis, in which the virus cannot successfully establish an infection, and mutagenesis is not necessary for extinction but might shorten the total duration of infection; (ii) 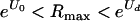 , in which mutagenesis is necessary for extinction; and (iii)
, in which mutagenesis is necessary for extinction; and (iii)  , in which extinction does not occur despite mutagenesis. The last case is potentially a worry because the elevated mutation rate might facilitate evolution to a part of the fitness landscape that was otherwise not likely to be accessed. In general, if mutagenesis increases the mutation rate closer to the mutation rate optimum for the virus, then mutagenesis will presumably be counterproductive for treatment. This possibility seems unlikely for RNA viruses, as their intrinsic mutation rates are so high. However, the relevant parameters are not adequately known to exclude this possibility, so the caution seems warranted.
, in which extinction does not occur despite mutagenesis. The last case is potentially a worry because the elevated mutation rate might facilitate evolution to a part of the fitness landscape that was otherwise not likely to be accessed. In general, if mutagenesis increases the mutation rate closer to the mutation rate optimum for the virus, then mutagenesis will presumably be counterproductive for treatment. This possibility seems unlikely for RNA viruses, as their intrinsic mutation rates are so high. However, the relevant parameters are not adequately known to exclude this possibility, so the caution seems warranted.
The assumption of a constant mutation rate across all infected cells is possibly valid for in vitro systems but may be violated in vivo. In a multicellular host, refugia might exist with low drug concentrations, as has been observed for HIV-1 patients under antiretroviral therapy. Mutagenesis levels may rise and fall with drug concentrations and cause genetic differentiation of viruses replicating in different compartments (5). Any decrease in mutation rate, whether spatially or temporally, will obviously work against lethal mutagenesis.
Our assumption that all mutations are deleterious or neutral is unrealistic. Beneficial mutations invariably exist. Furthermore, the spectrum of beneficial effects may vary during the course of mutagenesis, such that more beneficial mutations become available as mean fitness level declines (49, 58). A low rate of beneficial mutations should not preclude lethal mutagenesis per se, although it will raise the threshold for extinction, so that a higher dose of mutagen will be required to achieve the same effect. There are two mechanisms by which beneficial mutations can work. First, some genuine beneficial mutations may increase the intrinsic replicatory ability of the virus, increasing b and thus R. Second, mutations can confer partial or complete resistance to the mutagen (48). While partial resistance might be possible to overcome with an increased mutagen dosage, complete resistance will prevent lethal mutagenesis. A treatment strategy for preventing the evolution of significant or complete resistance could be combination therapy with several mutagens or with a mutagen in combination with other antiviral drugs.
Supplementary Material
Acknowledgments
J.B. was supported by NIH grant GM57756 and the Miescher Regents Professorship at the University of Texas. R.S. was supported by fellowship ASTF 37900-05 from the EMBO and grant GV06/031 from the Generalitat Valenciana. C.O.W. was supported by NIH grant AI 065960.
We thank Holly Wichman, Isabel Novella, and two anonymous reviewers for many helpful comments and suggestions.
Footnotes
Published ahead of print on 3 January 2007.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Anderson, J. P., R. Daifuku, and L. A. Loeb. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58:183-205. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, R. M., and R. M. May. 1991. Infectious diseases of humans: dynamics and control. Oxford University Press, Oxford, United Kingdom.
- 3.Bateman, A. J. 1959. The viability of near-normal irradiated chromosomes. Int. J. Radiat. Biol. 1:170-180. [Google Scholar]
- 4.Bell, G. 1982. The masterpiece of nature: the evolution and genetics of sexuality. University of California Press, Berkeley, CA.
- 5.Bordería, A. V., F. M. Codoñer, and R. Sanjuán. Selection drives organ compartmentalization in HIV-1: evidence from gag and pol genes. Evolution, in press. [DOI] [PubMed]
- 6.Bull, J. J., L. A. Meyers, and M. Lachmann. 2005. Quasispecies made simple. PLoS Comput. Biol. 1:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burch, C. L., and L. Chao. 2004. Epistasis and its relationship to canalization in the RNA virus φ6. Genetics 167:559-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger, R. 2000. The mathematical theory of selection, recombination, and mutation. Wiley, Chichester, United Kingdom.
- 9.Butcher, D. 1995. Muller's ratchet, epistasis and mutation effects. Genetics 141:431-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao, L. 1990. Fitness of RNA virus decreased by Muller's ratchet. Nature 348:454-455. [DOI] [PubMed] [Google Scholar]
- 11.Chao, L., C. U. Rang, and L. E. Wong. 2002. Distribution of spontaneous mutants and inferences about the replication mode of the RNA bacteriophage φ6. J. Virol. 76:3276-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 98:6895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domingo, E., C. K. Biebricher, M. Eigen, and J. J. Holland. 2001. Quasispecies and RNA virus evolution: principles and consequences. Landes Bioscience, Georgetown, TX.
- 14.Drake, J. W. 1993. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 90:4171-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duarte, E., D. Clarke, A. Moya, E. Domingo, and J. Holland. 1992. Rapid fitness losses in mammalian RNA virus clones due to Muller's ratchet. Proc. Natl. Acad. Sci. USA 89:6015-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duarte, E. A., I. S. Novella, S. Ledesma, D. K. Clarke, A. Moya, S. F. Elena, E. Domingo, and J. J. Holland. 1994. Subclonal components of consensus fitness in an RNA virus clone. J. Virol. 68:4295-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eigen, M. 1971. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 58:456-523. [DOI] [PubMed] [Google Scholar]
- 19.Eigen, M., J. McCaskill, and P. Schuster. 1988. Molecular quasi-species. J. Phys. Chem. 92:6881-6891. [Google Scholar]
- 20.Elena, S. F., and A. Moya. 1999. Rate of deleterious mutation and the distribution of its effects on fitness in vesicular stomatitis virus. J. Evol. Biol. 12:1078-1088. [Google Scholar]
- 21.Engen, S., R. Lande, B. E. Sæther, and H. Weimerskirch. 2005. Extinction in relation to demographic and environmental stochasticity in age-structured models. Math. Biosci. 195:210-227. [DOI] [PubMed] [Google Scholar]
- 22.Flanagan, E. B., L. A. Ball, and G. W. Wertz. 2000. Moving the glycoprotein gene of vesicular stomatitis virus to promoter-proximal positions accelerates and enhances the protective immune response. J. Virol. 74:7895-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fredericksen, B. L., and M. A. Whitt. 1998. Attenuation of recombinant vesicular stomatitis viruses encoding mutant glycoproteins demonstrate a critical role for maintaining a high pH threshold for membrane fusion in viral fitness. Virology 240:349-358. [DOI] [PubMed] [Google Scholar]
- 24.Furió, V., A. Moya, and R. Sanjuan. 2005. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. USA 102:10233-10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García-Dorado, A., and J. M. Marín. 1998. Minimum distance estimation of mutational parameters for quantitative traits. Biometrics 54:1097-1114. [PubMed] [Google Scholar]
- 26.Gessler, D. D. G. 1995. The constraints of finite size in asexual populations and the rate of the ratchet. Genet. Res. 66:241-253. [DOI] [PubMed] [Google Scholar]
- 27.Graci, J. D., and C. E. Cameron. 2004. Challenges for the development of ribonucleoside analogues as inducers of error catastrophe. Antivir. Chem. Chemother. 15:1-13. [DOI] [PubMed] [Google Scholar]
- 28.Graci, J. D., and C. E. Cameron. 2006. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 16:37-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grande-Pérez, A., A. Lázaro, P. Lowenstein, E. Domingo, and S. Manrubia. 2005. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 102:4448-4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grande-Pérez, A., S. Sierra, M. G. Castro, E. Domingo, and P. R. Lowenstein. 2002. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc. Natl. Acad. Sci. USA 99:12938-12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haigh, J. 1978. The accumulation of deleterious genes in a population—Muller's Ratchet. Theor. Popul. Biol. 14:251-267. [DOI] [PubMed] [Google Scholar]
- 32.Harris, T. E. 1963. The theory of branching processes. Springer, Berlin, Germany.
- 33.Holland, J. J., E. Domingo, J. C. de la Torre, and D. A. Steinhauer. 1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64:3960-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keightley, P. D. 1994. The distribution of mutation effects on viability in Drosophila melanogaster. Genetics 138:1315-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura, M., and T. Maruyama. 1966. The mutational load with epistatic gene interactions in fitness. Genetics 54:1337-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krakauer, D. C., and J. B. Plotkin. 2002. Redundancy, antiredundancy, and the robustness of genomes. Proc. Natl. Acad. Sci. USA 99:1405-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lande, R. 1988. Genetics and demography in biological conservation. Science 241:1455-1460. [DOI] [PubMed] [Google Scholar]
- 38.Lande, R. 1998. Risk of population extinction from fixation of deleterious and reverse mutations. Genetica 102-103:21-27. [PubMed] [Google Scholar]
- 39.Lee, C. H., D. L. Gilbertson, I. S. Novella, R. Huerta, E. Domingo, and J. J. Holland. 1997. Negative effects of chemical mutagenesis on the adaptive behavior of vesicular stomatitis virus. J. Virol. 71:3636-3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loeb, L. A., J. M. Essigmann, F. Kazazi, J. Zhang, K. D. Rose, and J. I. Mullins. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 96:1492-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luria, S. E., and M. Delbrück. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lynch, M., R. Bürger, D. Butcher, and W. Gabriel. 1993. The mutational meltdown in asexual populations. J. Hered. 84:339-344. [DOI] [PubMed] [Google Scholar]
- 43.Malpica, J. M., A. Fraile, I. Moreno, C. I. Obies, J. W. Drake, and F. García-Arenal. 2002. The rate and character of spontaneous mutation in an RNA virus. Genetics 162:1505-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansky, L. M. 1998. Retrovirus mutation rates and their role in genetic variation. J. Gen. Virol. 79:1337-1345. [DOI] [PubMed] [Google Scholar]
- 45.Maynard Smith, J. 1978. The evolution of sex. Cambridge University Press, Cambridge, United Kingdom.
- 46.Mukai, T. 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50:1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller, H. J. 1964. The relation of recombination to mutational advance. Mut. Res. 1:2-9. [DOI] [PubMed] [Google Scholar]
- 48.Pfeiffer, J. K., and K. Kirkegaard. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 100:7289-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poon, A., and S. P. Otto. 2000. Compensating for our load of mutations: freezing the meltdown of small populations. Evolution 54:1467-1479. [DOI] [PubMed] [Google Scholar]
- 50.Rouzine, I. M., J. Wakeley, and J. M. Coffin. 2003. The solitary wave of asexual evolution. Proc. Natl. Acad. Sci. USA 100:587-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schrag, S. J., P. A. Rota, and W. J. Bellini. 1999. Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J. Virol. 73:51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuster, P., and J. Swetina. 1988. Stationary mutant distributions and evolutionary optimization. Bull. Math. Biol. 50:635-660. [DOI] [PubMed] [Google Scholar]
- 54.Severson, W. E., C. S. Schmaljohn, A. Javadian, and C. B. Jonsson. 2003. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 77:481-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sierra, S., M. Dávila, P. R. Lowenstein, and E. Domingo. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 74:8316-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sniegowski, P. D., P. J. Gerrish, and R. E. Lenski. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387:703-705. [DOI] [PubMed] [Google Scholar]
- 57.Suárez, P., J. Varcárcel, and J. Ortín. 1992. Heterogeneity of the mutation rates of influenza A viruses: isolation of mutator mutants. J. Virol. 66:2491-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitlock, M. C. 2000. Fixation of new alleles and the extinction of small populations: drift load, beneficial alleles, and sexual selection. Evolution 54:1855-1861. [DOI] [PubMed] [Google Scholar]
- 59.Wiehe, T. 1997. Model dependency of error thresholds: the role of fitness functions and contrasts between the finite and infinite sites models. Genet. Res. 69:127-136. [Google Scholar]
- 60.Wilke, C. O. 2005. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilke, C. O., and C. Adami. 2003. Evolution of mutational robustness. Mut. Res. 522:3-11. [DOI] [PubMed] [Google Scholar]
- 62.Wilke, C. O., C. Ronnewinkel, and T. Martinetz. 2001. Dynamic fitness landscapes in molecular evolution. Phys. Rep. 349:395-446. [Google Scholar]
- 63.Wilke, C. O., J. L. Wang, C. Ofria, R. E. Lenski, and C. Adami. 2001. Evolution of digital organisms at high mutation rate leads to survival of the flattest. Nature 412:331-333. [DOI] [PubMed] [Google Scholar]
- 64.Yuste, E., S. Sánchez-Palomino, C. Casado, E. Domingo, and C. López-Galíndez. 1999. Drastic fitness loss in human immunodeficiency virus type 1 upon serial bottleneck events. J. Virol. 73:2745-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng, Q. 1999. Progress of a half century in the study of the Luria-Delbruck distribution. Math. Biosci. 162:1-32. [DOI] [PubMed] [Google Scholar]
- 66.Zhou, S., R. Liu, B. M. Baroudy, B. A. Malcolm, and G. R. Reyes. 2003. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310:333-342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.