

Abstract
Cells cured of persistent virus infection can be used to investigate cellular pathways of resistance to viral cytopathic effects. Persistent poliovirus (PV) infections were established in human intestinal Caco-2 cells, and spontaneously cured cell cultures were obtained. Two cell clones, cl6 and b13, cured of type 3 PV mutant infection and their parental Caco-2 cells were compared for susceptibility to PV infection, PV receptor CD155 expression, capacity to differentiate into polarized enterocytes, and PV-, staurosporine-, and actinomycin D-induced apoptosis. Our results strongly suggest that cells that are partially resistant to apoptosis can be selected during persistent virus infection.
Cells cured of persistent virus infection can be used to investigate cellular pathways of resistance to virus infection (5, 6, 11, 23, 24, 28). The spontaneous cure of persistent virus infection in vitro has been described only rarely (5, 9, 15, 22). Poliovirus (PV), the prototype member of the Enterovirus genus (25), is composed of a single-stranded positive-sense RNA enclosed in an icosahedral capsid. We have developed several models of persistent PV infection to elucidate the mechanisms of persistent infections and investigate cellular resistance to viral cytopathic effects (7). Mutated forms of the PV receptor, CD155, have been found in persistently PV-infected IMR-32 cells (4, 20). The allelic form with a threonine in position 67 (Thr67) of CD155 conferred partial resistance to PV-induced apoptosis on murine L cells, with reference to the other form (Ala67) expressed in the parental IMR-32 cell line (13). HEp-2 cells cured of PV infection have a variety of phenotypes often involving PV-CD155 interactions (5, 6). Persistent infections with PV mutants have also been established in human intestinal Caco-2 cells (15). We used PV type 3 mutant L2-2 (10) and Sabin 3 (S3) mutants H136-11, H442-11, and H637-11, which were isolated from a chronically infected hypogammaglobulinemic patient (17). Mutations differentiating these PV mutants and S3 have been described previously (15, 17). The mechanisms of persistent infection in human intestinal cells involve viral and cellular determinants (15). Caco-2 cultures (20 to 80%) stopped excreting infectious virus between 1 month and 1 year postinfection (p.i.) (15).
Clonal cell populations were grown from well-isolated colonies of cured cells, as described previously (6), and their resistance to de novo PV type 3 infection was tested. The absence of PV RNA in the cured clones was first verified by reverse transcription-nested PCR as described previously (24) (not shown). All cell clones were permissive to PV infection, but two out of five clones were partially resistant to virus-induced cell lysis. These clones, cl6 and b13, cured of L2-2 and H442-11 infection, respectively, were studied in comparison with parental Caco-2 cells. Cells (106) were seeded in duplicate in wells of 24-well plates in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and incubated overnight at 37°C. Cells were then mock infected or infected with a type 3 PV strain, S3, L2-2, or H442-11, at a multiplicity of infection (MOI) of 10−2 50% infectious doses (ID50) per cell. The percentage of cells that survived infection was determined at 1, 2, and 3 days p.i. by staining cells with trypan blue. The resistance of the clones to lysis was highly significant at 3 days p.i.: with all type 3 PV strains tested, more than 80% of parental Caco-2 cells were lysed, whereas 60 to 100% of cl6 and 40 to 80% of b13 cells survived infection (Fig. 1a to c). Similar results were obtained with infections with Sabin 1 and Sabin 2 PV strains (not shown), indicating that the resistance of cl6 and b13 cells to PV infection was not type specific. As S3 multiplication was slightly delayed in the clones (Fig. 1d), the resistance of the clones was tested at a higher MOI. At an MOI of 10 ID50 per cell, S3 multiplication in cl6 was delayed by 1 to 2 h during the exponential phase. However, at 24 h p.i., viral yields were similar for the three cell types, and cl6 cells were much more resistant to S3-induced lysis than Caco-2 cells (Fig. 1e and f). At this MOI, b13 was more susceptible to infection than cl6, in agreement with data at low MOIs.
FIG. 1.

Partial resistance of Caco-2 cell-derived clones cl6 and b13 to PV-induced cell lysis after de novo infection. PV type 3 strains Sabin 3 (S3), L2-2, and H442-11 were used at an MOI of 10−2 (a to d) or 10 (e and f) ID50/cell. Caco-2 (dark gray), cl6 (light gray), and b13 (white) cells were infected with the indicated virus strains, and the number of surviving cells was determined by trypan blue exclusion 1, 2, and 3 days p.i. at an MOI of 10−2 (a to c) and 1 day p.i. at an MOI of 10 (e). The results were obtained by calculating surviving cells in infected cultures as a percentage of those in mock-infected homologous cultures for the same day. S3 growth curves in Caco-2 (circles), cl6 (squares), and b13 (triangles) cells infected at an MOI of 10−2 (d) and 10 (f) ID50/cell are shown. The results are means ± standard errors of the means (SEM) from at least four cultures from two independent experiments.
Low levels of CD155 expression may contribute to resistance to virus-induced cell lysis (8). We quantified the expression of CD155 at the surface of Caco-2, cl6, and b13 cells over a period of 6 days between two cell passages. We used flow cytometry after indirect immunofluorescence labeling with anti-CD155 monoclonal antibody 404 (16) as described previously (13, 15). More than 90% of cells of each cell type were brightly stained, and the level of CD155 expression was slightly higher on the cured cells than on Caco-2 cells (Fig. 2a). This was confirmed by normalizing the results for ICAM-1 detected with control antibody 8.4A6 (Sigma) (Fig. 2b). Thus, the delay before the cytopathic effects in the clones was not due to a low level of CD155 expression.
FIG. 2.
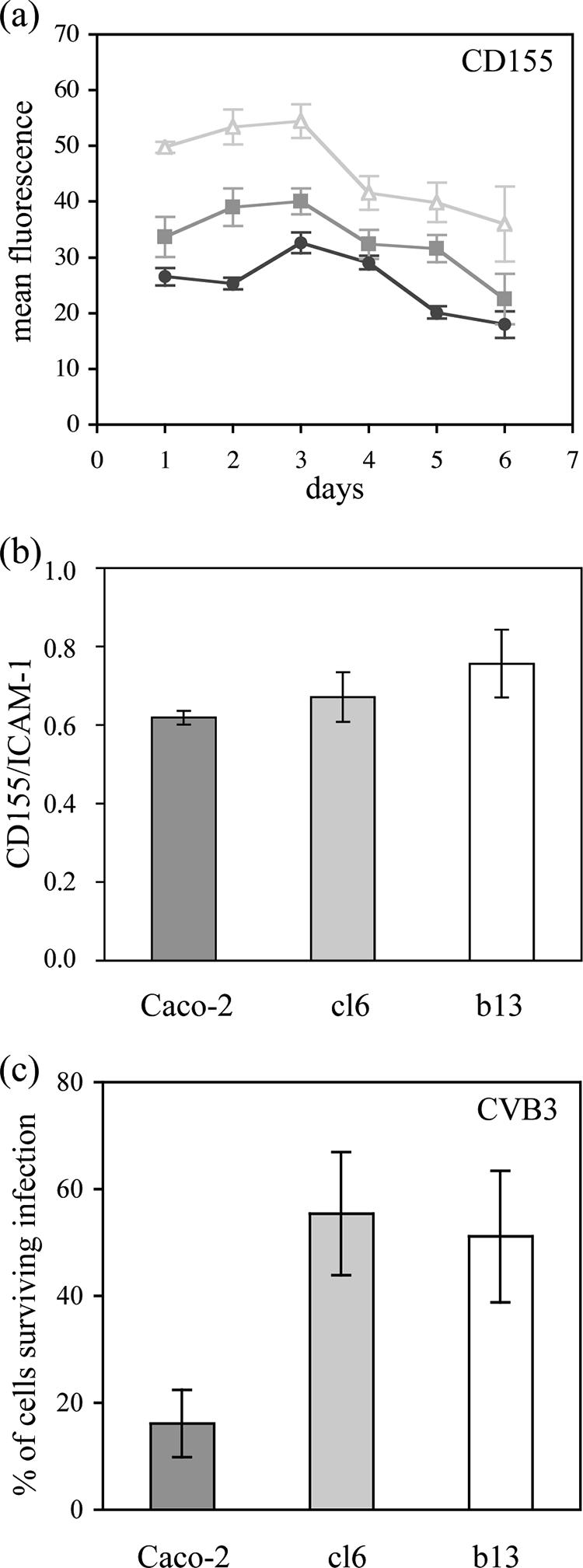
Resistance of clones to virus-induced lysis did not depend on CD155 expression. The level of CD155 expression at the surface of Caco-2 (circles), cl6 (squares), and b13 (triangles) cells was determined by indirect immunofluorescence, and the mean fluorescence per positive cell was quantified by flow cytometry in arbitrary units 1 to 6 days after cell passage by trypsinization (a). The ratio of CD155 to ICAM-1 expression, used as a reference, is shown for the three cell types (b). (c) Partial resistance of Caco-2 cell-derived clones cl6 and b13 to cell lysis after infection with coxsackievirus B3 (CVB3). Caco-2 (dark gray), cl6 (light gray), and b13 (white) cells were infected at an MOI of 10−2 ID50/cell, and the number of surviving cells was determined by trypan blue exclusion 2 days p.i. The results are means ± SEM from at least four cultures from two independent experiments.
We tested whether mutated forms of CD155 had been selected in cl6 and b13, because it was previously shown that PV multiplication could occur without cell lysis in cells expressing mutated CD155 (19). We determined the sequence of CD155 mRNA from these cells and from Caco-2 cells in the region corresponding to the domain of CD155 that includes the PV-binding site (14, 18), as described previously (20, 24). Nucleotide positions that were mutated in CD155 mRNA from persistently PV-infected IMR-32 cells were not mutated in Caco-2, cl6, or b13 cells: they were Gly39, Ala67, and Arg104. Also, no other mutation was detected, suggesting that the partial resistance of cl6 and b13 cells to PV-induced lysis was not due to the expression of CD155 forms mutated in the PV-binding domain. To exclude the role of other mutations in CD155, cells were infected with coxsackievirus B3, an enterovirus that uses the coxsackievirus and adenovirus receptor (3). Results were similar to those obtained with PV (Fig. 2c), confirming that the resistance of clones to virus-induced lysis did not depend on CD155.
We looked for a correlation between the partial resistance of clones to PV-induced lysis and their differentiation into polarized enterocytes, because enterocytes are more resistant to PV-induced lysis than undifferentiated Caco-2 cells (15, 27). No correlation between these phenotypes could be established (not shown).
PV infection induces apoptosis in several cell types, including Caco-2 cells (1, 2, 12, 21, 26). We therefore tested whether the resistance of clones to PV-induced lysis correlated with partial resistance to PV-induced apoptosis by comparing the percentages of the three cell types with DNA fragmentation 3 days after infection with S3 at an MOI of 10−2 ID50/cell: terminal deoxynucleotidyltransferase-mediated dUTP-rhodamine nick end labeling (TUNEL) was used as described previously (12). In mock-infected cultures, less than 1% of cells were TUNEL positive (not shown). In infected cultures, viral antigens were detected by indirect immunofluorescence with a rabbit anti-PV type 3 antiserum (5). The percentages of cells that were viral antigen positive did not differ significantly among the cell types (Fig. 3a). Interestingly, there were about threefold fewer viral antigen- and TUNEL-double-positive cells in cultures of cl6 and b13 cells than in Caco-2 cultures, suggesting that the clones were more resistant to PV-induced apoptosis than Caco-2 cells (Fig. 3a).
FIG. 3.

Cell clones cured of persistent PV infection were partially resistant to apoptosis. (a) Percentages of viral antigen- and TUNEL-positive cells in Caco-2, cl6, and b13 cultures 3 days p.i. by PV Sabin 3 (MOI of 10−2 ID50/cell). About 300 to 400 cells were analyzed under a microscope for each culture. (b and c) Specific enrichment in mono- and oligonucleosomes released into the cytoplasm of confluent Caco-2, cl6, and b13 cultures following treatment with AMD (8 μM for 22 h) (b) or staurosporine (ST) (2 μM for 18 h) (c). The enrichment factor was determined with a Cell Death Detection ELISAplus kit (Roche). The results are means ± SEM from at least two independent experiments.
To test whether cells cured of persistent infection were also partially resistant to apoptosis induced by nonviral inducers, we treated confluent cultures of each cell type with either actinomycin D (AMD) (8 μM for 22 h) or staurosporine (2 μM for 18 h). Like PV, both drugs activate the mitochondrial pathway of apoptosis, but staurosporine has a much more specific effect than AMD. DNA fragmentation was evaluated with a Cell Death Detection ELISAplus kit (Roche). Cells of cl6 and b13 appeared to be about two- and threefold more resistant to AMD- and staurosporine-induced apoptosis, respectively, than Caco-2 cells (Fig. 3b and c). These results indicate that the partial resistance of the cured clones to apoptosis is not exclusively virus specific.
In conclusion, our results strongly suggest that cells that are partially resistant to apoptosis can be selected during persistent virus infection. This may contribute to the mechanism of persistent infection.
Acknowledgments
We are grateful to I. Pelletier for her help and to J. Estaquier, A. Martin, C. Prehaud, S. Lay, S. Guillot, and N. Escriou for helpful suggestions. We thank B. Blondel for critical reading of the manuscript and Marc Lopez for the gift of anti-CD155 monoclonal antibody.
K.L. and A.S. were supported by fellowships from the Ministry (MENESR), and this work was supported by grants from the Institut Pasteur (PTR 120) and Danone Research Centre Daniel Carasso.
Footnotes
Published ahead of print on 27 December 2006.
REFERENCES
- 1.Agol, V. I., G. A. Belov, K. Bienz, D. Egger, M. S. Kolesnikova, L. I. Romanova, L. V. Sladkova, and E. A. Tolskaya. 2000. Competing death programs in poliovirus-infected cells: commitment switch in the middle of the infectious cycle. J. Virol. 74:5534-5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ammendolia, M. G., A. Tinari, A. Calcabrini, and F. Superti. 1999. Poliovirus infection induces apoptosis in Caco-2 cells. J. Med. Virol. 59:122-129. [PubMed] [Google Scholar]
- 3.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 4.Blondel, B., F. Colbère-Garapin, T. Couderc, A. Wirotius, and F. Guivel-Benhassine. 2005. Poliovirus, pathogenesis of poliomyelitis, and apoptosis. Curr. Top. Microbiol. Immunol. 289:25-56. [DOI] [PubMed] [Google Scholar]
- 5.Borzakian, S., T. Couderc, Y. Barbier, G. Attal, I. Pelletier, and F. Colbère-Garapin. 1992. Persistent poliovirus infection: establishment and maintenance involve distinct mechanisms. Virology 186:398-408. [DOI] [PubMed] [Google Scholar]
- 6.Calvez, V., I. Pelletier, T. Couderc, N. Pavio-Guedo, B. Blondel, and F. Colbère-Garapin. 1995. Cell clones cured of persistent poliovirus infection display selective permissivity to the wild-type poliovirus strain Mahoney and partial resistance to the attenuated Sabin 1 strain and Mahoney mutants. Virology 212:309-322. [DOI] [PubMed] [Google Scholar]
- 7.Colbère-Garapin, F., I. Pelletier, and L. Ouzilou. 2002. Persistent infections by picornaviruses, p. 437-450. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 8.Colston, E. M., and V. R. Racaniello. 1995. Poliovirus variants selected on mutant receptor-expressing cells identify capsid residues that expand receptor recognition. J. Virol. 69:4823-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de la Torre, J. C., M. Davila, F. Sobrino, J. Ortin, and E. Domingo. 1985. Establishment of cell lines persistently infected with foot-and-mouth disease virus. Virology 145:24-35. [DOI] [PubMed] [Google Scholar]
- 10.Duncan, G., I. Pelletier, and F. Colbère-Garapin. 1998. Two amino acid substitutions in the type 3 poliovirus capsid contribute to the establishment of persistent infection in HEp-2c cells by modifying virus-receptor interactions. Virology 241:14-29. [DOI] [PubMed] [Google Scholar]
- 11.Ebert, D. H., S. A. Kopecky-Bromberg, and T. S. Dermody. 2004. Cathepsin B is inhibited in mutant cells selected during persistent reovirus infection. J. Biol. Chem. 279:3837-3851. [DOI] [PubMed] [Google Scholar]
- 12.Girard, S., T. Couderc, J. Destombes, D. Thiesson, F. Delpeyroux, and B. Blondel. 1999. Poliovirus induces apoptosis in the mouse central nervous system. J. Virol. 73:6066-6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gosselin, A. S., Y. Simonin, F. Guivel-Benhassine, V. Rincheval, J. L. Vayssiere, B. Mignotte, F. Colbère-Garapin, T. Couderc, and B. Blondel. 2003. Poliovirus-induced apoptosis is reduced in cells expressing a mutant CD155 selected during persistent poliovirus infection in neuroblastoma cells. J. Virol. 77:790-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koike, S., H. Horie, I. Ise, A. Okitsu, M. Yoshida, N. Iizuka, K. Takeuchi, T. Takegami, and A. Nomoto. 1990. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 9:3217-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labadie, K., I. Pelletier, A. Saulnier, J. Martin, and F. Colbère-Garapin. 2004. Poliovirus mutants excreted by a chronically infected hypogammaglobulinemic patient establish persistent infections in human intestinal cells. Virology 318:66-78. [DOI] [PubMed] [Google Scholar]
- 16.Lopez, M., F. Jordier, F. Bardin, L. Coulombel, C. Chabannon, and P. Dubreuil. 1999. Identification of a new class of Ig superfamily antigens expressed in hemopoiesis, p. 1081. In T. Kishimoto, A. E. G. K. von dem Borne, S. M. Goyert, D. Y. Mason, M. Miyasaka, M. Moretta, K. Okumura, S. Shaw, T. A. Springer, K. Sugamura, and H. Zola (ed.), Leucocyte typing VI: white cell differentiation antigens. Garland Publishing, New York, NY.
- 17.Martin, J., G. Dunn, R. Hull, V. Patel, and P. D. Minor. 2000. Evolution of the Sabin strain of type 3 poliovirus in an immunodeficient patient during the entire 637-day period of virus excretion. J. Virol. 74:3001-3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendelsohn, C. L., E. Wimmer, and V. R. Racaniello. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence and expression of a new member of the immunoglobulin superfamily. Cell 56:855-865. [DOI] [PubMed] [Google Scholar]
- 19.Morrison, M. E., Y. J. He, M. W. Wien, J. M. Hogle, and V. R. Racaniello. 1994. Homolog-scanning mutagenesis reveals poliovirus receptor residues important for virus binding and replication. J. Virol. 68:2578-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavio, N., T. Couderc, S. Girard, J. Y. Sgro, B. Blondel, and F. Colbère-Garapin. 2000. Expression of mutated poliovirus receptors in human neuroblastoma cells persistently infected with poliovirus. Virology 274:331-342. [DOI] [PubMed] [Google Scholar]
- 21.Romanova, L. I., G. A. Belov, P. V. Lidsky, E. A. Tolskaya, M. S. Kolesnikova, A. G. Evstafieva, A. B. Vartapetian, D. Egger, K. Bienz, and V. I. Agol. 2005. Variability in apoptotic response to poliovirus infection. Virology 331:292-306. [DOI] [PubMed] [Google Scholar]
- 22.Ron, D., and J. Tal. 1986. Spontaneous curing of a minute virus of mice carrier state by selection of cells with an intracellular block of viral replication. J. Virol. 58:26-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez, A. B., M. Perez, T. Cornu, and J. C. de la Torre. 2005. RNA interference-mediated virus clearance from cells both acutely and chronically infected with the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 79:11071-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saulnier, A., I. Pelletier, K. Labadie, and F. Colbère-Garapin. 2006. Complete cure of persistent virus infections by antiviral siRNAs. Mol. Ther. 13:142-150. [DOI] [PubMed] [Google Scholar]
- 25.Stanway, G., T. Hovi, N. Knowles, and T. Hyypiä. 2002. Molecular and biological basis of picornavirus taxonomy, p. 17-26. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 26.Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A. Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69:1181-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker, S. P., C. L. Thornton, E. Wimmer, and R. W. Compans. 1993. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J. Virol. 67:4274-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallbracht, A., L. Hofmann, K. G. Wurster, and B. Flehmig. 1984. Persistent infection of human fibroblasts by hepatitis A virus. J. Gen. Virol. 65:609-615. [DOI] [PubMed] [Google Scholar]