

Abstract
As vertebrate genome sequences near completion and research refocuses to their analysis, the issue of effective genome annotation display becomes critical. A mature web tool for rapid and reliable display of any requested portion of the genome at any scale, together with several dozen aligned annotation tracks, is provided at http://genome.ucsc.edu. This browser displays assembly contigs and gaps, mRNA and expressed sequence tag alignments, multiple gene predictions, cross-species homologies, single nucleotide polymorphisms, sequence-tagged sites, radiation hybrid data, transposon repeats, and more as a stack of coregistered tracks. Text and sequence-based searches provide quick and precise access to any region of specific interest. Secondary links from individual features lead to sequence details and supplementary off-site databases. One-half of the annotation tracks are computed at the University of California, Santa Cruz from publicly available sequence data; collaborators worldwide provide the rest. Users can stably add their own custom tracks to the browser for educational or research purposes. The conceptual and technical framework of the browser, its underlying MYSQL database, and overall use are described. The web site currently serves over 50,000 pages per day to over 3000 different users.
We are fortunate to live in a time when the vast majority of the human genome has been sequenced, is freely available, and where work proceeds rapidly to fill in the remaining gaps. The public mapping and sequencing efforts have spanned a decade and involved thousands of people (Consortium 2001; McPherson et al. 2001). The end result of the sequencing efforts will be three billion A's, Cs, Gs, and Ts in a particular order that somehow contains instructions for building a human body. Over 2.7 billion bases are in the public databases today.
Finding which of the 2.7 billion bases are relevant to a particular aspect of biology or medicine can be a challenge. For the most part, researchers would prefer to view the genome at a higher level—at the level of an exon, a gene, a chromosome band, or a biochemical pathway. The base-by-base view is best reserved for preparing primers for experiments or looking for DNA motifs associated with particular functions. Interactive computer programs that can search and display a genome at various levels are very useful tools, and a number of these programs exist.
One of the earliest-such programs was a Caenorhabditis elegans database (ACEDB) (Eeckman and Durbin 1995; Kelley 2000). ACEDB began as a database to keep track of C. elegans strains and information from genetic crosses (J. Thierry-Mieg, pers. comm.). Soon ACEDB could display genetic maps. ACEDB was adopted by the C. elegans sequencing project at the Sanger Centre and Washington University (Consortium 1998). As cosmid and then sequence maps of C. elegans became available, these were added to ACEDB. ACEDB is a very flexible program and has been used in many other sequencing projects as well, including Arabidopsis and parts of the human genome project. Because of its use of the middle and right mouse buttons and other X-windows user interface features, ACEDB works best on a Unix or Linux system. The WormBase project (Stein et al. 2001) is actively adapting parts of ACEDB for use in their web-based display.
The Saccharomyces Genome Database (SGD) at http://genome-www.stanford.edu/Saccharomyces/ was designed with the web in mind. At SGD, it is possible to search for a gene either by name or by sequence, browse neighboring genes, retrieve the full sequence for a gene, look up functional summaries of most genes, and link into the literature all with a few clicks in a web browser. SGD was first described in 1998 (Cherry et al. 1998) and currently receives over 50,000 hits per week from biomedical researchers.
There are currently at least three sites that attempt to provide a similar service for the public working draft of the human genome. The open source Ensembl project at www.ensembl.org has been online since the very early days of the working draft (Birney et al. 2001). Ensembl was conceived before there were assemblies available of the draft human genome. Because the average size of the sequence contigs before assembly was considerably smaller than the average size of a human gene, initially Ensembl focused on identifying exons. Ensembl ran the Genscan program (Burge and Karlin 1997) to find genes in finished and draft clones. The contigs inside of draft clones were ordered when possible by mRNA information, but no attempt was made to merge overlapping clones. Genscan is a sensitive program but has a relatively high false rate of positive predictions. The putative exons Genscan identified were translated into protein, and when homologous proteins could be found in the EMBL database, the exons were marked as confirmed. When possible, exons were grouped together into genes. Ensembl produced a web-based display of their gene predictions and supporting evidence. When the University of California, Santa Cruz (UCSC) genome assemblies (Consortium 2001; Kent and Haussler 2001) became available, Ensembl quickly shifted to them and over time has added many additional annotations including Genewise gene predictions (Birney and Durbin 1997), homology with other species, positions of single nucleotide polymorphisms (SNPs) (Sachidanandam et al. 2001), and so forth. Ensembl recently has started to annotate the mouse genome as well.
The National Center for Biotechnology Information (NCBI) from the beginning has hosted the human genome as part of the BLAST-searchable GenBank (Benson et al. 1999). Inside GenBank, the genome is present as many separate records, mainly in records associated with bacterial artificial chromosome (BAC) clones. NCBI made their own assembly of the public human genome data available recently. Their assembly can be BLAST searched, and the relative positions of various features can be viewed on their map viewer. A page with links to NCBI's human genome-specific resources is at http://www.ncbi.nlm.nih.gov/genome/guide/human/. These resources include the RefSeq set of nonredundant mRNA sequences (Maglott et al. 2000; Pruitt and Maglott 2001). Functional descriptions of many of the RefSeq genes are available in the associated LocusLink and OMIM (Maglott et al. 2000; Pruitt and Maglott 2001) databases.
A third site that serves the human genome is the focus of this paper. The distinguishing features of the UCSC browser are the breadth of annotations, speed, stability, extensibility, and consistency of user interface. We actively seek data from third parties to display. Each set of annotations is shown graphically as a horizontal “track” over the genome sequence. Currently, one-half of the 31 annotation tracks in the browser are computed at UCSC while the other half are generated by collaborators worldwide. The browser is highly integrated with the BLAT sequence search tool (Kent 2002).
The UCSC browser had humble origins. The code originated with a small script in the C programming language, which displayed a splicing diagram for a gene prediction from the nematode C. elegans (Kent and Zahler 2000). This web-based splicing display later acquired tracks for mRNA alignments and for homology with the related nematode Caenorhabditis briggsae. This was published as the tracks display at http://www.cse.ucsc.edu/∼kent/intronerator (Kent and Zahler 2000a,b). It would have been difficult to move this browser to the human genome before the draft assembly because of the fragmented and redundant nature of the “Working Draft.” Because the human genome is 30 times larger than the C. elegans genome, even after the assembly, the software required substantial revision. In the end, we were able to maintain the same interactive response time we had on the worm on the vastly larger human data set via a series of algorithmic improvements, via use of the MySQL database, via a set of Linux pentium-class machines acting as web servers, and via systems tuning by our systems administrators. The result is a site that has become very popular with biologists. Currently, the UCSC Human Genome Browser at http://genome.ucsc.edu receives >50,000 hits per working day, from more than 3000 different users. In this paper, we describe the overall conceptual framework behind the browser and its use. We explain some of the algorithmic tricks behind the browser, demonstrate how to add your own tracks, and provide details on how some of the tracks were generated at UCSC.
RESULTS AND DISCUSSION
Using the Browser
To start a browser session, follow the “browser” link at http://genome.ucsc.edu. This will take you to a page where you can search for a gene by name, author, keyword, and so forth, or directly specify the region to view as either a chromosome band or a chromosome and range of bases. You can also enter the browser via a search for homologous regions to a DNA or protein sequence using the “BLAT” link. The BLAT search typically only takes a few seconds. The main browser display (Fig. 1) contains three main parts. On top is a series of controls for searching and for zooming and scrolling across a chromosome. In the middle is a dynamically generated picture that graphically displays genome annotations. On the bottom is another series of controls that fine-tune the graphic display.
Figure 1.

Part of the HOXA cluster as viewed in the University of California, Santa Cruz (UCSC) genome browser. The shortcut bar in blue provides quick access to BLAT searches, the DNA sequence, the annotations as text tables, earlier or later assemblies the genome, the corresponding NCBI and Ensembl views, and the user's guide. The controls directly beneath position the browser over a specific region in the genome. The large white picture in the middle displays various annotations. At the bottom are controls for fine-tuning the display and for the individual tracks. Only the first 15 of 31 available tracks are shown here.
This region contains three known genes that are all transcribed on the reverse strand as indicated by the arrowheads in the introns. Note the alternative splicing of HOXA1 in the Human RNA track. The Spliced EST track indicates that there is active transcription of a region between HOXA1 and HOXA2. Expressed sequence tag evidence for the presence of additional nonannotated genes in well studied regions like this often can be found using the UCSC browser. The Mouse Blat track indicated a high level of conservation between mouse and human in this region. Both the Mouse Blat and the Exofish ecores are based on translated alignments, but in highly conserved regions such as this it is not unusual for even translated alignments to paint conserved noncoding regions. The noncoding regions have diverged considerably more between human and pufferfish than between human and mouse.
The browser represents annotations as a series of horizontal tracks laid out over the genome. Each track displays a particular type of annotation, such as Genscan gene predictions, mRNA alignments, or interspersed repeats. Each track can be displayed in dense mode, fully expanded, or can be hidden. By default most tracks are displayed in dense mode where they take up a single line. Clicking on a dense track opens it up to a full mode, where there is a separate line for each item. Clicking on an item brings up detailed information on that item. Some particularly important tracks, such as the track for known genes, are fully open by default. The track display is useful at many scales, from a view of an entire chromosome down to the alternative splicing patterns of a single gene (Figs. 2–5). The notion of a track is important in the underlying database as well as in the browser itself. The tracks are relatively independent of each other both in the user interface and the underlying programming. As a consequence, it is very easy to add another track when new annotations become available. The tracks relate to each other simply by all being synchronized to the same underlying sequence. The user can see many lines of evidence in a single screen and on that basis quickly is able to make informed judgments about the biology of a particular region.
Figure 2.

All of chromosome 17. Generally, people work at smaller scales than this, but the browser is capable of displaying all of the annotations on a chromosome in a reasonable time. The centromere is depicted in red in the chromosome band track. The coverage track shows finished regions in black and draft regions in various shades of gray depending on the depth of coverage. There are two large gene deserts in chromosome bands q22 and q24.3. Tracks based on mRNAs, ESTs, and homology with Tetraodon all are quite sparse in these regions, though there is still quite a bit of mouse homology.
Figure 5.

A known gene and an unknown gene or two. ITGB3, the integrin β chain, β 3 precursor is on the left. To the right is a relatively small gene, C17001176, predicted by the Fgenesh++ program, which is supported by mouse and fish homology. Between ITGB3 and C17001176 is a region quite likely to contain another gene judging by the EST and mouse homology evidence.
The graphic display of the browser is invaluable for getting a quick overview of a particular region in the genome and for visually correlating various types of features. However, there is a limit to what can be displayed in a single window. As mentioned above, clicking on an individual item in a fully opened track brings up further information on the track as a whole and on the specific item. In many cases, this includes links to other databases such as those at NCBI and Ensembl. Figure 6 shows the details page for the known gene VLDLR (the very-low-density lipoprotein receptor). It's possible to retrieve the mRNA and protein sequence for this gene from this page, as well as the genomic sequence with exons in upper case.
Figure 6.

Details page on the known gene VLDLR.
At times, the user might want a list of features in a particular section of the genome in a text rather than a graphical format. The Table Browser, which is accessible from the “tables” link, extracts information in a tab-delimited format suitable for import into text editors, spreadsheets, or your own databases. The database behind both the graphical and table browsers is described further in a later section.
Correlations Between Tracks
A common use of the browser is to look for evidence of previously unidentified genes. The EST, cross-species homology, and ab initio gene prediction tracks in particular are very useful for this purpose. Table 1A provides a summary of how well these various tracks correlate with the RefSeq-based Known Gene tracks across the entire genome and Table 1B provides a summary of how well the various tracks correlate with the Sanger Centre gene annotations on chromosome 22 (Dunham et al. 1999). The Exofish track, based on homology with the pufferfish Tetraodon nigroviridis (Roest Crollius et al. 2000) is exceedingly specific, but covers less than half of bases in known coding regions. This coverage will increase somewhat as more pufferfish sequence is added. The Genscan track, on the other hand, covers well over three-quarters of bases in known coding regions, but has only moderate specificity. The Ensembl, Fgenesh++ (Salamov and Solovyev 2000) and Genie (Kulp et al. 1996, 1997) gene prediction tracks available in some versions of the browser integrate ab initio gene-finding techniques with homology evidence. Currently, there is no gene prediction tool that integrates all of the evidence displayed in the browser into a definitive track. The genome assembly and annotations found on the April 2001 version of the browser were used for these tables.
Table 1.
Correlations between various tracks and experimentally verified gene annotations
| Track | Covers | Yield Tx | Yield Co | Enrich Tx | Enrich Co |
|---|---|---|---|---|---|
| Human EST | 5.83% | 83.7% | 82.8% | 14.4 | 14.2 |
| Spliced EST | 1.11% | 59.2% | 72.3% | 53.3 | 65.1 |
| Mouse Blat | 3.62% | 60.4% | 82.3% | 16.7 | 22.7 |
| Other mRNA | 0.77% | 49.3% | 67.4% | 64.0 | 87.5 |
| Other EST | 0.95% | 53.0% | 69.6% | 55.8 | 73.3 |
| Exofish | 0.40% | 23.7% | 36.6% | 59.3 | 91.5 |
| Genscan | 1.65% | 57.0% | 86.0% | 34.5 | 52.1 |
| RefSeq Tx | 0.79% | 100% | 100% | 126.6 | 126.6 |
| RefSeq Co | 0.50% | 63.3% | 100% | 126.6 | 200.0 |
| A—Whole genome using RefSeq Annotations | |||||
| Track | Covers | Yield Tx | Yield Co | Enrich Tx | Enrich Co |
|---|---|---|---|---|---|
| Human EST | 8.40% | 78.1% | 74.7% | 9.3 | 8.9 |
| Spliced EST | 1.80% | 43.9% | 55.9% | 24.4 | 31.0 |
| Mouse Blat | 2.89% | 44.3% | 65.3% | 15.3 | 22.6 |
| Other mRNA | 1.05% | 27.8% | 41.3% | 26.5 | 39.3 |
| Other EST | 1.37% | 37.6% | 53.1% | 27.4 | 38.8 |
| Exofish | 0.61% | 16.3% | 27.1% | 26.7 | 44.4 |
| Genscan | 3.00% | 47.7% | 76.4% | 15.9 | 25.5 |
| Sanger Tx | 2.80% | 100% | 100% | 35.7 | 35.7 |
| Sanger Co | 1.60% | 57.1% | 100% | 35.7 | 62.5 |
| B—Chromosome 22 using Sanger Centre Annotations | |||||
The Covers column shows the percentage of the genome (A) or chromosome 22 (B) covered by a particular track. The Yield Tx column describes the percentage of bases in the annotated gene transcripts (from known genes in RefSeq in A and the Sanger Centre annotated genes in B) covered by the track, while the Yield Co column describes the percentage of the annotated protein coding regions covered. The Enrich Tx and Enrich Co columns show how many times enriched the track is for transcribed and coding regions compared to the genome as a whole. The yield columns correspond directly to sensitivity of the feature for detecting genes. Because the annotations, particularly the whole-genome annotations, are incomplete, it is not possible to do traditional specificity calculations. However, the enrichment columns allow one to compare the relative specificity of the tracks. The rows for the tracks RefSeq Tx (transcribed regions in RefSeq), RefSeq Co (coding regions in RefSeq), Sanger Tx (transcribed regions for Sanger annotated genes), and Sanger Co (coding regions from Sanger annotated genes) are included to show the maximum possible yields and enrichments for transcript and coding tracks.
Tracks Based on Human mRNA
There are several tracks based on alignments of human mRNA sequences with the genome. All human mRNAs from the primate database in GenBank are used to make the Human mRNA track. All human ESTs from dbEST in GenBank are used to make the Human EST and Spliced EST tracks. In all cases, the alignment is done with the BLAT program (Kent 2002) using the default nucleotide alignment parameters.
In many cases, a single mRNA will align in multiple places in the draft human genome. This can be a result of psuedogenes, genes that share a common domain, recent duplication events in the human genome, and assembly errors in the draft. We filter the alignments to help focus on the genes rather than the psuedogenes and paralogs. The first filter is based on percentage identity. For ESTs, the threshold is 93%. For mRNAs, the threshold is 96%. These thresholds were chosen to be ∼2% below the mean error rate observed in the first large-scale cDNA sequencing projects in the 1990s. Because the error rate of modern cDNA projects is considerably less, we are considering increasing these thresholds in the future. Note that because exons frequently are missing from the draft genome, the percentage identity is only calculated within the blocks that do align. The second filter is a “near best in genome” filter. A score based largely on percent identity is assigned to each alignment. The best-scoring alignment for each base of the mRNA sequence is recorded. Alignments that do not score within 1% of the best score for at least 20 bases in a row are filtered out. The combination of filters reduces the number of alignments by fivefold to tenfold, however most of the alignments eliminated are quite short involving repeat elements and short conserved motifs. Occasionally, a nearly full-length alignment to a paralogous gene also will be eliminated by these filters. The EST alignments then are analyzed for signs of splicing, specifically for gaps of at least 32 bases that have ends matching the GT/AG intron consensus. These EST alignments then are selected to make the spliced EST track.
Known Genes
The known gene track is created from human RefSeq mRNAs. These are aligned with BLAT as above, but with more stringent filtering. Because RefSeq mRNA sequences tend to be quite clean, they are required to match at 98% identity, and the near best in genome filter is set to pass only those within 0.2% of the best alignment. The alignment then is turned into a gene prediction by mapping the protein coding (CDS) portion of the mRNA to the genome, and merging blocks in the alignment separated by gaps of five bases or less into exons. The HUGO gene name, if any, is mapped to the gene prediction by way of tables downloaded from NCBI. These same tables provide us with the raw materials to make hyperlinks into the OMIM, RefSeq, and LocusLink (http://www.ncbi.nih.gov).
Tracks Based on Homology with Other Species
The browser has a number of tracks that show homology with other species. Some of these are generated by third parties, as detailed in the Acknowledgments section. The Mouse Blat, Nonhuman mRNA, and Nonhuman EST are all generated at UCSC using the BLAT program in translated mode using the default score settings. The human genome was run through RepeatMasker (Smit 1999; Jurka 2000) and Tandem Repeat Finder (Benson 1999) before the alignments. The current version of the Mouse Blat track is based on random whole genome shotgun reads deposited in the NCBI/EBI trace archive by the Mouse Sequencing Consortium. There are ∼13 million of these reads covering the mouse genome to an ∼2.5× depth. The Nonhuman mRNA and ESTs are taken from GenBank.
Gene Expression Tracks
In addition to the extensive nucleotide annotation available in the browser, two new tracks present information about the experimental behavior of mRNA transcripts as determined by Serial Analysis of Gene Expression (SAGE) and DNA microarrays.
The track incorporating SAGE data is the SAGE/UniGene track, which presents data indicating the transcriptional level of different UniGene clusters (http://www.ncbi.nlm.nih.gov/UniGene/) from the SAGEMap project at the NCBI (Lal et al. 1999; Lash et al. 2000). In the browser window, the UniGene clusters are represented by the alignment of the longest sequence in the cluster to the draft sequence using BLAT. The clusters are colored by the average expression level of that cluster over the different SAGE experiments. Clicking on a UniGene cluster presents a summary table for individual SAGE experimental results for each of the clusters in the current browser window. From the details page, it is also possible to view the SAGE results as a graph or to go directly to the SAGEMap's virtual northern page for that cluster.
The first tracks incorporating DNA microarray data are the Rosetta tracks, which contain DNA probes for every predicted and confirmed exon on chromosome 22 as previously described (Shoemaker et al. 2001). The predicted and confirmed exons are represented by separate tracks in the browser. The same sequences that were used to select probes are aligned to the draft genome using BLAT. In full mode, these tracks present both the location of the exons in the genome and a red and green banding pattern that corresponds to the log ratio of expression in the 69 experiments used. Clicking on an individual exon presents a more detailed view of all of the exons present in the current browser window over all of the experiments. For each exon in each experiment, the average log ratio of all of the probes in a particular experiment is presented as a red and green false color display. If the actual intensities are of interest for a particular experiment, these can be displayed graphically for each probe in each exon in the browser window by filling out the form presented.
Tracks Based on Genome-Wide Maps
High-level maps of the human genome existed for many years prior to the existence of sequence-based maps (Caspersson et al. 1968; Hudson et al. 1995; Dib et al. 1996; Broman et al. 1998; Deloukas et al. 1998; http://shgc-www.stanford.edu/Mapping/TNGMAPS/). We have a Chromosome Band track and a Fluorescent In Situ Hybridization (FISH) Clones track that display information related to the cytogenetic map (Trask 1999). There is also a Sequence-Tagged Site (STS) Markers track with data from genetic, radiation hybridization (RH), and yeast artificial chromosome (YAC) maps.
The BAC Resource Consortium has identified the positions of several thousand BAC clones on the cytogenetic map using FISH experiments (Cheung et al. 2001). We have determined the locations of these clones on the sequence assembly in one of several ways. If the clone is fully sequenced and is used in the construction of the assembled draft genome, its location simply can be looked up. If both the BAC end sequences are known, they are aligned using BLAT and again the position of the full extent of the clone can be determined. For the remaining clones, if an STS is known to be contained within the sequence or at least one of the end sequences is available, the locations of these determined by BLAT are used to approximate the location of the clone without giving the exact boundaries. These clones and more information about them can be seen on the FISH Clones track.
The locations of the FISH-mapped clones on the cytogenetic map and the sequence assembly are used to approximate the boundaries of the chromosome bands at the 800-band resolution. A dynamic programming algorithm developed at UCSC determines these boundary locations by maximizing the concordance between the chromosome band or bands assigned by FISH experiments and that assigned to the region of the sequence assembly where the clone has been placed. Clones placed at NCI are weighted slightly more because of the higher resolution FISH experiments being performed (Kirsch et al. 2000). Constraints have been implemented to ensure that the length of the predicted bands do not deviate too substantially from the standard percentage lengths as set forth by the International System for Human Cytogenetic Nomenclature (ISCN) (Mitelman 1995).
The STS Markers track displays the positions of markers used in constructing the Genethon genetic map (Dib et al. 1996), Marshfield genetic map (Broman et al. 1998), Whitehead Institute YAC map (Hudson et al. 1995), GeneMap99, GB4, and G3 RH maps (Deloukas et al. 1998), Stanford TNG RH map (http://shgc-www.stanford.edu/Mapping/TNGMAPS/), and the Whitehead Institute RH map (Hudson et al. 1995). Additional markers contained in the Homo sapiens portion of the UniSTS database (http://www.ncbi.nlm.nih.gov/genome/sts/index.html) at NCBI also are contained in this track. For many of these STS markers, the full sequence is known, and we use BLAT to determine a location in the sequence assembly. For others, only the 3′ and 5′ primer sequences are known. In previous versions, we employed Greg Schuler's e-PCR program (Schuler 1998) to determine locations. We are now using BLAT for these placements as well. Many markers are mapped to multiple locations equally well, and only those with three or less placements are shown in the browser. The details page for an individual marker on this track gives additional information such as aliases, primer sequences, and locations on the maps mentioned above, as well as links to UniSTS, GenBank, and GDB.
BAC End Sequence Pairs
BAC end sequences available from GenBank's dbGSS division are aligned to the genome sequence assembly using BLAT. The alignments are searched for pairs that constitute the 5′ and 3′ end sequences for a single BAC clone. Those pairs for which the end sequences are oriented correctly and that are at least 50 Kb but no more than 600 Kb apart are considered valid pairs. These are displayed as the BAC End Pairs track. In the full view, the orientation of the corresponding clone is shown by arrows between the sequence pairs. The details page provides the accessions of the end sequences with links to GenBank and information on the alignment of the end sequences to the assembly sequence.
Adding and Publishing Your Own Tracks
Since August 2001, it has become possible for users to upload their own annotations for display in the browser. These annotations can be in the standard GFF format (http://www.sanger.ac.uk/Software/formats/GFF), or in some formats designed specifically for the human genome project including GTF, PSL, and BED. The formats are described in detail in the web page http://genome.cse.ucsc.edu/goldenPath/help/customTrack.html. Note that the GFF and GTF files must be tab delimited rather than space delimited. Uploaded annotations can be seen only on the machine from which they were uploaded and are only kept for 8 h after the last time they were accessed.
It is possible to make custom tracks in a more permanent and public fashion as well. To do this, the track provider puts a file in one of the supported formats onto a web site. The URL for this file can be pasted into the browser's custom track control. It also is possible to construct links from your own web pages into the browser in such a way that the custom track is automatically included. The following is an example of such a link:
The position specifies where the browser should open. The db variable specifies the database version. It is always of the form hgN, where N is incremented for each version. For the August 2001 version, the db variable is “hg8”. The customText contains the URL for the custom track file. Tracks produced in this method are not as fast as tracks loaded into the database at UCSC, but if the size of the track file is less than 1 or 2 Mb, the performance is usually very good.
The Challenge of Change—Keeping Up with the Working Draft
One of the challenges of annotating the human genome is that there are so many versions of it. At UCSC, we try to assemble a new version approximately every three months to incorporate new sequence. The chromosomal coordinates of genes and other features change with each version. Occasionally, a chunk of sequence will even get moved from one chromosome to another as the map is refined. We have recently put in a feature to help jump between the most recent three versions. This feature is available from the “convert” button at the top of the browser. It works by performing a BLAT search on the first 1000, last 1000, and middle 1000 bases in the current window. If all three searches land uniquely in the same order on the other version, the program announces a successful conversion. If the search results are not so straightforward, the user is given various options to find the corresponding sequence. Frequently, if the feature the user is looking for is tied to an mRNA, it is simplest just to BLAT the mRNA.
The Database
The genome.ucsc.edu database is built on top of MySQL (www.mysql.com). We initially chose this database to be compatible with the Ensembl project. MySQL has turned out to be very well suited to our purposes. It is extremely efficient at retrieving data from indexed files. We use MySQL as a “read-mostly” database. We load the database in large batches and the rest of the time treat it as read-only. Each of our seven web servers has a copy of the database on local disk.
To create the graphical display, the browser queries MySQL track by track, asking for data that overlaps the display window. The SQL query to fetch these data for the cpgIsland track on a window covering from base 10,000 to base 20,000 on chromosome 3 is as follows:
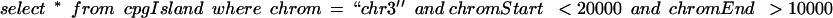 |
We created indices on chrom,chromStart and chrom,chromEnd for this table. The query went reasonably fast for small tables, such as the 29,005-item cpgIsland table. Even for relatively small tables, sorting the data by chrom,chromStart before loading the database turned out to be critical for performance. If the indices are small enough to fit into RAM, this presorting reduces the number of disk seeks needed to load the data from one track to a very small number, often to a single seek.
For larger tables, such as the 4.2-million-item EST alignment table, more complicated schemes were needed for good interactive performance. As a first step, we split such tables between chromosomes so that the basic query becomes something like this:
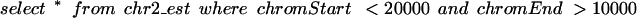 |
This reduced the size of the indices by eliminating the need to index the chromosome field, making it more likely for the indices to fit into RAM. In general, the database had to scan the index for half of the chromosome when the query was formulated in this fashion. As a consequence, the browser was slower on large chromosomes than on smaller ones. The performance was still tolerable we felt (response time was typically <5 sec even on the largest chromosome), but as we added more data, the performance degraded. When the large mouse homology tables were added, it was clear that we needed a more intelligent scheme.
We settled on a binning scheme suggested by Lincoln Stein and Richard Durbin. A simple version of this scheme is shown in Figure 7. In the browser itself, we use five different sizes of bins: 128 kb, 1 Mb, 8 Mb, 64 Mb, and 512 Mb.
Figure 7.

Binning scheme for optimizing database accesses for genomic annotations that cover a particular region of the genome. This diagram shows bins of three different sizes. Features are put in the smallest bin in which they fit. A feature covering the range indicated by line A would go in bin 1. Similarly, line B goes in bin 4 and line C in bin 20. When the browser needs to access features in a region, it must look in bins of all different sizes. To access all the features that overlapped or were enclosed by line A, the browser looks in bins 1, 2, 3, 7, 8, 9, 10, and 11. For B the browser looks in bins 1, 4, 14, 15, 16, 17. For C, the browser looks in bins 1, 5, and 20.
The query in the previous paragraph using this binning scheme becomes:
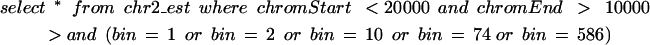 |
Though the query itself is more complex than before, it executes much faster. Typically, almost all features are in the smaller bins, and in the most common usage scenarios only the contents of a few of these smaller bins need to be examined. This binning scheme is relatively simple to implement and seems to have sufficient performance to meet our needs indefinitely. A modest improvement we have yet to implement would be to stagger the bin boundaries so that small features that happen to span the point at 64 Mb do not necessarily end up in the largest bin, and similarly for other bin boundaries that occur at multiple levels.
In addition to the tables that contain positional information and that may be split between chromosomes and/or binned as described above, there are nonpositional tables. These contain auxiliary information that is not needed for the graphical display, but which may be useful when examining a particular feature in the details page. Some examples of nonpositional tables include the DNA sequence, author, cell type, and library name of ESTs. At the time we designed the database, file sizes on Linux machines were limited to −2 gigabases. Largely for this reason, most of the actual DNA data are stored in external files. The external files are still indexed through the database.
A detailed table-by-table and field-by-field description of the database is at http://genome.ucsc.edu/goldenPath/gbdDescriptions.html. The entire database is dumped weekly into tab-delimited files that can be downloaded either a table at a time or as a single large zip file at genome.ucsc.edu. With the Table Browser at http://genome.ucsc.edu/goldenPath/hgText.html, it is possible to extract subsets of the database, in many cases eliminating the need to set up your own copy of the MySQL database.
Enhanced functions are provided for downloading DNA sequence data itself. At any point while browsing, the user can use the “DNA” link at the top of the browser to download the genome sequence for the region currently being viewed. Basic options include reverse complementation, upper/lower case, and masking of repeats by RepeatMasker (Smit 1999; Jurka 2000), possibly using lower case. Here, the output is a simple text file. Advanced options produce an HTML file containing the sequence. These options allow users to use a variety of combinations of case, underlining, bold, italic, and color to represent one or more kinds of annotation on the genome sequence. Any track of annotation that is available on the browser can be represented in the sequence using any combination of these representation modes. Multiple tracks of annotation can be represented simultaneously in the sequence by choosing a different mode or combination of modes for each track.
The Programming Interface Between the Database and the Browser
There is a natural tension between how an object is represented in the database and in computer programs such as the scripts that make up the browser. A program in the C language typically will represent an object as a “struct” of some sort and have a family of functions that operate on this structure. An object in a relational database may be represented as a row in a table, as an entire table, or even as an abstract entity spanning multiple tables that are joined together by an appropriate SQL query at run time. Some programmers have even resorted to converting their objects to some sort of complex text format such as XML, and storing the object as a “blob” in the database. A disadvantage of this last approach is that it becomes difficult to index the fields of the object separately.
In the browser database, we found a pragmatic compromise that works very well for us. We have a program, autoSql, which takes a data definition as an input. From this definition, autoSql creates a C structure, a C function to load the structure from an array of strings (which is how a MySQL query returns a row in a table), a C function to save the structure as a line in a tab-separated file (which can be used to load the database), a C function to free up the dynamic memory used by the structure, and a SQL create statement. There is thus a one-to-one correspondence between a structure in memory and a row in a table on disk, and likewise a one-to-one correspondence between the fields in structure in memory and the fields in a row. The autoSql definitions can include arrays and substructures. The arrays are represented in the database as comma-separated lists stored as blobs. While autoSql is capable of generating code to handle substructures, these also end up stored in blobs. Because blobs are difficult to index, we have not actually used this feature in the genome.ucsc.edu database, though arrays are fairly common. See http://www.soe.ucsc.edu/∼kent/exe/doc/autoSql.doc for more information on autoSql.
Most of the browser database also can be accessed via the Distributed Annotation Service (DAS) protocol (Dowell et al. 2001). DAS is a rapidly evolving open source standard for distributing genomic annotations over the web. It is similar in function to the publishing-your-own tracks system we describe here, but the data is transmitted in an XML rather than a tab-separated format. Further information on DAS can be found at http://www.biodas.org. The web address of our DAS server is http://genome.ucsc.edu/cgi-bin/das. Because of the large size of the annotations, particularly when represented in DAS-GFF XML format, for best results, enable compression on the DAS clients when accessing our DAS server.
Other Features
The UCSC browser is linked with the Ensembl human genome browser at http://www.ensembl.org in such a way that users viewing any region of the genome at UCSC can switch easily to viewing the same region in the Ensembl browser and vice versa. Like the facility for user-published tracks described above, this is another way the power of the web can be exploited to enrich the variety of information about a gene or region of interest that is rapidly accessible to a user. Mirrors of the UCSC browser in Europe and Asia provide faster access to the information contained in the browser and its database to researchers in these parts of the world and serve as redundant sites for all users on occasions where a power outage or some other exceptional condition puts genome.ucsc.edu temporarily off line. Because the browser runs on Linux with a MySQL database, we are able to help academic and nonprofit institutions set up mirror sites at no cost to the mirroring institution. Finally, help and frequently-asked-questions pages are available to assist users with features of the browser and database that are not evident from self-exploration. This information is supplemented by a moderated and archived e-mail discussion group.
Conclusion
The web site at http://genome.ucsc.edu is a valuable tool for exploring the human genome. It provides fast sequence and text-based search facilities. The graphical display is relatively simple to use yet quite powerful and is able to handle huge annotation sets such as those describing human repeats or human/mouse homologies smoothly. The underlying database has a relatively simple yet robust design and can be accessed by many methods. It is possible for visitors to incorporate custom annotations in the context of the annotations built into the browser either in a public or a private fashion. In the coming years, we plan to continue adding to this site and to adapt it to other genomes. We have already adapted it to the mouse genome.
WEB SITE REFERENCES
http://genome.ucsc.edu; The UCSC Human Genome Browser. A web tool for display of any requested portion of the genome at any scale, together with several dozen aligned annotation tracks.
http://shgc-www.stanford.edu/Mapping/TNGMAPS/; Radiation hybrid maps at Stanford University.
http://genome-www.stanford.edu/Saccharomyces/; The Saccharomyces Genome Database (SGD) at Stanford University.
http://www.biodas.org; Distributed Annotation System web site.
http://www.cse.ucsc.edu/∼kent/intronerator; C. elegans genome browser with an emphasis on alternative splicing.
http://www.ensembl.org; Ensembl human genome browser.
http://www.ncbi.nlm.nih.gov/genome/guide/human/; A page with links to NCBI's human genome-specific resources.
http://www.sanger.ac.uk/Software/formats/GFF; Description of the Gene Finder Format (GFF).
www.mysql.com; The main web site for the MySQL database.
Figure 3.

Chromosome 17 band q21.32. This region spans several million bases and is covered by a mix of finished and draft clones. The large blocks in the gap track indicate gaps between clones, while the small ticks indicate gaps within draft clones. Where there is evidence for the relative order and orientation of the contigs on either side of a gap, a white line is drawn though the gap. Most of the contigs in this region are ordered. At this scale, it is possible to resolve most individual genes but not necessarily individual exons.
Figure 4.

One million bases in the middle of 17q21.32. This is a scale frequently used when trying to positionally clone a gene. Many of the genes in this region are already known, but the EST, mouse, and fish homology evidence suggest the presence of additional genes as well, particularly between ITGB3 and NPEPPS.
Acknowledgments
We acknowledge the following individuals and institutions who contributed programs and/or data for tracks: Barbara Trask, Vivian Cheung, Norma Nowak, and colleagues for the FISH data that was used to create the chromosome bands and FISH Clones tracks; Greg Schuler, Arek Kasprzyk, Wonhee Jang, and Sanja Rogic for helping process the map information to generate the STS track, and Genethon, the Marshfield Clinic, the David Cox lab, Whitehead Institute, and the International RH Mapping Consortium for generating the data; Bob Waterston, John McPhearson, Asif Chinwalla, LaDeana Hillier, Shiaw-Pyng Jang, John Wallis, and colleagues at Washington University for the map that drove the assembly and that formed the basis for the FPC Contig track and also for their work on the CpG Island track; Deanna Church for the Mouse Synteny track; Jeff Bailey and Evan Eichler for the Genomic Duplications track; Kim Pruitt, Donna Maglott, and colleagues for the RefSeq and LocusLink project, which forms the basis of our Known Genes track; David Kulp, Ray Wheeler, Alan Williams, and Affymetrix Corp. for the Genie gene prediction tracks; Ewan Birney, Michelle Clamp, Tim Hubbard, Elia Stupka, Imre Vastrik and the Ensembl project for the Ensembl gene prediction track and help with the TPF maps; Victor Solovyev and A. Salamov for the Fgenesh++ gene prediction and the TSSW Promoter tracks; Danielle-et-Jean Thierry-Mieg and Vahan Simonyan for the Acembly gene prediction tracks; Ian Dunham and colleagues at the Sanger Centre for the chromosome 22 annotations, and Victoria Haghighi and Bill Noble for remapping these annotations; Greg Schuler, Lukas Wagner, and colleagues at NCBI for the Unigene database and the EST 3′ end track; John Quackenbush, Foo Cheung, and colleagues at TIGR for the TIGR Gene Index track; Hugues Roest Crollius, Olivier Jaillon, Jean Weissenbach, and colleagues at Genoscope for the Exofish track; Guy Slader and the Mouse Sequencing Consortium for the Exonerate Mouse track; Ming Li and colleagues at Bioinformatics Solutions for the Pattern Hunter Mouse track; Lincoln Stein, Steve Sherry, the SNP Consortium, and the NIH for the SNP tracks; Arian Smit, Victor Pollara, and J. Jurka for the RepeatMasker track; Sean Eddy, Todd Lowe, and colleagues for the RNA Genes track; G. Benson for the trf program, which is the basis of the Simple Repeats track; and Kim Worley, James Durbin, John Bouck, and Richard Gibbs for introducing us to trf and executing the early runs of that program and the CpG island finder. We also thank all the members of the International Human Genome Project and everyone who has ever contributed data to Genbank for the sequence that forms the basis of this work. W.J.K, T.F., K.R., A.Z., and D.H. acknowledge support from NHGRI Award 1 P41 HG02371–01. T.F also acknowledges support from DOE Grant DE-FG03–99ER62849. C.S. acknowledges support from Howard Hughes Medical Institute Award SC-00–63.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kent@biology.ucsc.edu
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.229102. Article published online before print in May 2002.
REFERENCES
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 1999;27:12–17. doi: 10.1093/nar/27.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Bateman A, Clamp ME, Hubbard TJ. Mining the draft human genome. Nature. 2001;409:827–828. doi: 10.1038/35057004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Durbin R. Dynamite: A flexible code generating language for dynamic programming methods used in sequence comparison. Ismb. 1997;5:56–64. [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic maps: Individual and sex-specific variation in recombination. Am J Hum Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Caspersson T, Farber S, Foley GE, Kudynowski J, Modest EJ, Simonsson E, Wagh U, Zech L. Chemical differentiation along metaphase chromosomes. Exp Cell Res. 1968;49:219–222. doi: 10.1016/0014-4827(68)90538-7. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, et al. SGD: Saccharomyces genome database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, Furey TS, Kim UJ, Kuo WL, Olivier M, et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409:953–958. doi: 10.1038/35057192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TCES. Genome sequence of the nematode C. elegans: A platform for investigating biology. The C. elegans Sequencing Consortium. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Consortium TIHGS. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Deloukas P, Schuler GD, Gyapay G, Beasley EM, Soderlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, et al. A physical map of 30,000 human genes. Science. 1998;282:744–746. doi: 10.1126/science.282.5389.744. [DOI] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Jokerst RM, Day A, Eddy SR, Stein L. The distributed annotation system. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Eeckman FH, Durbin R. ACeDB and macace. Methods Cell Biol. 1995;48:583–605. [PubMed] [Google Scholar]
- Hudson TJ, Stein LD, Gerety SS, Ma J, Castle AB, Silva J, Slonim DK, Baptista R, Kruglyak L, Xu SH, et al. An STS-based map of the human genome. Science. 1995;270:1945–1954. doi: 10.1126/science.270.5244.1945. [DOI] [PubMed] [Google Scholar]
- Jurka J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Kelley S. Getting started with Acedb. Brief Bioinform. 2000;1:131–137. doi: 10.1093/bib/1.2.131. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT the BLAT-like alignment tool. Gen Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Haussler D. Assembly of the working draft of the human genome with GigAssembler. Genome Res. 2001;11:1541–1548. doi: 10.1101/gr.183201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. Conservation, regulation, synteny, and introns in a large-scale C. briggsae-C. elegans genomic alignment. Genome Res. 2000a;10:1115–1125. doi: 10.1101/gr.10.8.1115. [DOI] [PubMed] [Google Scholar]
- ————— The intronerator: Exploring introns and alternative splicing in C. elegans. Nucleic Acids Res. 2000b;28:91–93. doi: 10.1093/nar/28.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch IR, Green ED, Yonescu R, Strausberg R, Carter N, Bentley D, Leversha MA, Dunham I, Braden VV, Hilgenfeld E, et al. A systematic, high-resolution linkage of the cytogenetic and physical maps of the human genome. Nat Genet. 2000;24:339–340. doi: 10.1038/74149. [DOI] [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. Ismb. 1996;4:134–142. [PubMed] [Google Scholar]
- ———. 1997. Integrating database homology in a probabilistic gene structure model. Pac. Symp. Biocomput. 232–244. [PubMed]
- Lal A, Lash AE, Altschul SF, Velculescu V, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Polyak K, et al. A public database for gene expression in human cancers. Cancer Res. 1999;59:5403–5407. [PubMed] [Google Scholar]
- Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, Altschul SF. SAGEmap: A public gene expression resource. Genome Res. 2000;10:1051–1060. doi: 10.1101/gr.10.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, et al. A physical map of the human genome. Nature. 2001;409:934–941. doi: 10.1038/35057157. [DOI] [PubMed] [Google Scholar]
- Mitelman F. An international system for human cytogenetic nomenclature. Basel: S. Karger; 1995. [Google Scholar]
- Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roest Crollius H, Jaillon O, Bernot A, Dasilva C, Bouneau L, Fischer C, Fizames C, Wincker P, Brottier P, Quetier F, et al. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat Genet. 2000;25:235–238. doi: 10.1038/76118. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey WL, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler GD. Electronic PCR: Bridging the gap between genome mapping and genome sequencing. Trends Biotechnol. 1998;16:456–459. doi: 10.1016/s0167-7799(98)01232-3. [DOI] [PubMed] [Google Scholar]
- Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G, et al. Experimental annotation of the human genome using microarray technology. Nature. 2001;409:922–927. doi: 10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Stein L, Sternberg P, Durbin R, Thierry-Mieg J, Spieth J. WormBase: Network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 2001;29:82–86. doi: 10.1093/nar/29.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask B. Genome analysis: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Press; 1999. [Google Scholar]