

Abstract
The nonstructural protein 1 (nsp1) of the severe acute respiratory syndrome coronavirus has 179 residues and is the N-terminal cleavage product of the viral replicase polyprotein that mediates RNA replication and processing. The specific function of nsp1 is not known. Here we report the nuclear magnetic resonance structure of the nsp1 segment from residue 13 to 128, which represents a novel α/β-fold formed by a mixed parallel/antiparallel six-stranded β-barrel, an α-helix covering one opening of the barrel, and a 310-helix alongside the barrel. We further characterized the full-length 179-residue protein and show that the polypeptide segments of residues 1 to 12 and 129 to 179 are flexibly disordered. The structure is analyzed in a search for possible correlations with the recently reported activity of nsp1 in the degradation of mRNA.
After the major outbreak of severe acute respiratory syndrome (SARS) in the beginning of 2003, the SARS coronavirus (SARS-CoV) became a major topic of coronavirus research. The coronavirus genome is composed of a single plus-strand RNA of about 30 kb, which is the largest nonsegmented genome among known RNA viruses. About two-thirds of the coronavirus genome is devoted to encoding the replicase that mediates viral RNA synthesis (64). The replicase gene comprises two large open reading frames (ORFs) located at the 5′ end of the genome. The first one, ORF1a, encodes a polyprotein of 450 to 500 kDa (polyprotein 1a), and the second one, ORF1b, is translated together with ORF1a after a −1 ribosomal frameshift, leading to the expression of the “polyprotein lab,” which has a size of 750 to 800 kDa. The replicase polyprotein is processed by ORF1a-encoded viral proteinases, which leads to about 16 nonstructural proteins (nsp), which are numbered consecutively from the N terminus to the C terminus of the polyprotein (14, 53).
The large number of mature proteins produced from the polyprotein indicates a high level of complexity of the viral replication process. Some of the enzymatic activities that were detected or predicted in SARS-CoV to date include the main protease (nsp5), a papain-like proteinase (nsp3d; PLpro), an RNA-dependent RNA polymerase (nsp12), an RNA helicase (nsp13), an endoribonuclease (nsp15), an ADP-ribose-1"-phosphatase (nsp3b), a deubiquitinase (nsp3d), a 3′→5′ exoribonuclease (nsp14), and a ribose-2′-O-methyltransferase (nsp16) (3, 5, 18, 21, 22, 44, 59, 70). For the two proteases, the endoribonuclease, and the ADP-ribose-1"-phosphatase, three-dimensional structures have been solved, which together with biochemical data have revealed some aspects of the enzyme mechanisms (27, 54, 56, 58, 63, 67; reviewed in references 4, 35, and 65). The physiological functions of several of the other nonstructural proteins remain to be determined, but high-resolution structure determinations have allowed the identification of possible functional sites and provided the basis for further biochemical studies (15, 30, 52, 61, 62, 68). Other replicase proteins still remain to be characterized.
Nsp1 is the N-terminal cleavage product of the replicase polyprotein and is produced by the action of PLpro. It is among the least well-understood nsps, and other than in coronaviruses, no viral or cellular homologs are known. Levels of sequence conservation among the different coronaviruses are highest at the 3′ end of the genome, and the sequences are very divergent at the 5′ end, especially in nsp1 to nsp3, which are products of PLpro cleavage. nsp1 has been proposed to be useful as a group-specific marker (59). In the group 1 coronaviruses, nsp1 (also known as p9) is a protein of about 110 residues, with 20 to 50% sequence identity among all group 1 CoVs. The viruses of subgroup 2a, such as murine hepatitis virus (MHV) and human coronavirus OC43, encode an nsp1 protein of about 245 residues, also known as p28, while the group 3 viruses (avian) do not encode an nsp1. The nsp1 of SARS-CoV, which has been classified as the only member to date of the subgroup 2b (19, 20, 59), comprises 180 residues, with a molecular mass of 20 kDa. nsp1 sequences are divergent between groups 2a and 2b, and no sequence similarity between SARS-CoV nsp1 and group 2a nsp1 proteins could be identified using standard searching tools such as BLAST.
Biochemical experiments demonstrated interactions between MHV nsp1 and two other replication proteins (nsp7 and nsp10) and colocalization with nonstructural proteins and the nucleocapsid protein at viral replication complexes in the cytoplasm during the early stages of infection (6). In contrast, during the later stages of infection, MHV nsp1 was found to colocalize with structural proteins at virion assembly sites (6). Mutations at the nsp1/nsp2 cleavage site of MHV that prevented the cleavage of nsp1 from the polyprotein caused slower growth and reduced RNA synthesis relative to wild-type viruses (13). Deletion of the nsp1-coding region in infectious clones of MHV yielded viruses that were unable to productively infect cultured cells (7). Furthermore, exogenous expression of MHV nsp1 in mammalian cells arrested the cell cycle in the G0/G1 phase and inhibited cell proliferation (8). A point mutation in the proteolytic cleavage site between nsp1 and nsp2 in the full-length genome, and in minigenomes of the group 1 CoV porcine transmissible gastroenteritis virus, blocked the release of nsp1 from the nascent polyprotein and caused a dramatic reduction in virus viability (17). SARS-CoV nsp1 was shown to specifically accelerate the degradation of mRNA and thus lead to a reduction in cellular protein synthesis, which may provide a survival advantage for the virus (31). Overall, these observations indicate that nsp1 might participate in multiple stages of the coronavirus life cycle, and they implicate this protein as a potentially important virulence factor.
This paper describes the nuclear magnetic resonance (NMR) structure of SARS-CoV nsp1. Before this study, no information about the three-dimensional structure of nsp1 was available, and SARS-CoV nsp1 does not have significant amino acid sequence similarity with any protein with known three-dimensional structure. SARS-CoV nsp1 was therefore selected for NMR structure determination by the Consortium for Functional and Structural Proteomics of SARS-CoV-Related Proteins (http://sars.scripps.edu). The availability of a high-resolution solution structure will help to guide further investigations of the biochemical and physiological functions of nsp1.
MATERIALS AND METHODS
Target optimization strategy.
Six constructs truncated at different positions were created based on secondary structure prediction, using the amino acid sequence as input for the software Jpred (12). The truncated nsp1 variants were cloned in an Escherichia coli expression plasmid derived from pET-28 under the control of the T7 promoter and in frame with 5′ coding sequences for a His6 tag and followed by a spacer sequence which ends with ENLYFQG. This strategy allows the preparation of proteins that have only one extra glycine at the N terminus after proteolysis with tobacco etch virus protease. This expression strategy was selected after tests of different E. coli strains and different temperatures during the induction in 10-ml cultures. The samples were expressed in a microexpression device (M. S. Almeida, M. Geralt, R. Horst, and K. Wüthrich, unpublished), purified using Ni2+ affinity chromatography, concentrated with ultrafiltration centrifugal devices, and subjected to one-dimensional (1D) 1H NMR screening using a Bruker DRX700 spectrometer with a 1-mm TXI HCN z-gradient microprobe. Based on the high-quality 1D 1H NMR spectrum, the construct consisting of nsp1 residues 13 to 128 [nsp1(13-128)] was selected for a NMR structure determination. In an attempt to further improve this sample, variant constructs of nsp1(13-128) with Cys 52 replaced by Ala, Ser, Arg, or Asp were prepared by site-directed mutagenesis using the QuikChange kit (Stratagene) according to the manufacturer's instructions. The variants were evaluated by 1D 1H NMR screening for a globular fold and by circular dichroism spectroscopy to determine their stability.
Protein preparation.
Large-scale expression of uniformly 15N-labeled or 13C- and 15N-labeled nsp1(13-128) in E. coli BL21(DE3) cells was carried out at 18°C in 500 ml of M9 minimal medium containing either 0.5 g 15NH4Cl or 0.5 g 15NH4Cl and 2 g [13C6]-d-glucose as the sole nitrogen and carbon sources, respectively. For the protein purification, the cells were disrupted by sonication in the presence of 25 mM HEPES at pH 8.0, 250 mM NaCl, 2 mM dithiothreitol, 0.03% NaN3, and EDTA-free Complete protease inhibitor tablets (Roche). The cell lysate was loaded onto a 10-ml HisTrap FF column equilibrated with 50 mM imidazole in the same buffer system as mentioned above. The retained proteins were eluted with a 50 to 500 mM imidazole gradient and incubated with recombinant tobacco etch virus protease at 22°C for 2 days. The resulting solution was loaded onto a 300-ml Superdex 75 column equilibrated with 25 mM sodium phosphate at pH 7.0, 250 mM NaCl, and 0.03% NaN3. The protein eluted with a retention volume equivalent to about 13 kDa. The solution was concentrated with ultrafiltration centrifugal devices and supplemented with 10% D2O to a final sample volume of about 300 μl.
NMR spectroscopy and structure calculation.
The NMR samples contained 2 mM of nsp1(13-128). NMR spectra were collected at 298 K with Bruker Avance 600-MHz and Avance 800-MHz spectrometers equipped with TXI HCN z-gradient probes. The sequence-specific resonance assignment (66) has been described elsewhere (1). The input for the structure calculation consisted of the chemical shift list obtained from the resonance assignment, a 3D 15N-resolved 1H,1H nuclear Overhauser effect spectroscopy (NOESY) spectrum, and two 3D 13C-resolved 1H,1H NOESY spectra optimized for the aliphatic and aromatic 13C regions. The nuclear Overhauser effect (NOE) data were measured at 800 MHz with a mixing time of 60 ms. For the peak picking of the NOESY spectra, NOE assignment, and structure calculation, the stand-alone ATNOS/CANDID program (24, 25) was used in conjunction with the CYANA torsion angle dynamics algorithm (23). The standard protocol with seven cycles of peak picking, NOE assignment, and 3D structure calculation with simulated annealing in torsion angle space (24, 25) was applied. Backbone ϕ and ψ dihedral angle constraints derived from the Cα chemical shifts (40, 60) were used as supplementary data in the structure calculation. The 20 conformers with the lowest residual CYANA target function values obtained from cycle 7 of the ATNOS/CANDID/CYANA calculation were energy minimized in a water shell with the program OPALp (34, 39), using the AMBER force field (9). The program MOLMOL (33) was used to analyze the protein structure and to prepare the figures showing the NMR structures. Analysis of the stereochemical quality of the models was accomplished using the Joint Center for Structural Genomics validation central suite (http://www.jcsg.org) and the Protein Data Bank validation server (http://deposit.pdb.org/validate).
Steady-state 15N{1H} NOEs were measured with transverse relaxation-optimized spectroscopy (TROSY)-based experiments (55, 69) on a Bruker Avance 600-MHz spectrometer, using a saturation period of 3 s and an interscan delay of 5 s.
Accession numbers.
The chemical shifts have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu) under accession number 7014. The atomic coordinates of the bundle of 20 conformers used to represent the nsp1 structure have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb) with the code 2GDT, and those of the conformer closest to the mean coordinates have the code 2HSX.
RESULTS AND DISCUSSION
The 179-residue nsp1 of SARS-CoV was included in a high-throughput proteomics characterization strategy by the consortium “Functional and Structural Proteomics of the SARS-CoV” (unpublished). The full-length protein and the fragment consisting of residues 1 to 159 were cloned, expressed and purified by the Protein Production Core, and given to us for NMR screening (50). The 1D 1H NMR spectra of both constructs showed characteristics of a globular fold as well as of disordered regions (data not shown). Based on these results, the protein was transferred to us for an NMR structure determination.
Since nsp1 has no identifiable sequence similarity with proteins with known three-dimensional structures, it was not possible to predict the domain structure of this protein based on sequence comparisons. However, the presence of flexibly disordered regions in the protein identified by 1H NMR spectroscopy (see “Characterization of the full-length SARS-CoV nsp1” below) was consistent with the results of secondary structure prediction, which indicated that a few residues at the N terminus, as well as a greater number of residues in the C-terminal one-third of the protein, would not adopt regular secondary structure. To investigate the boundaries of the globular domain and to optimize conditions for protein expression, sample preparation, and NMR structure determination, we designed a set of truncated variants of nsp1, bearing in mind the results of secondary structure predictions for this protein. The variant constructs have molecular masses of 12.7 kDa to 19.5 kDa, not including the N-terminal tag of 3.5 kDa (Table 1). These constructs were used to transform E. coli strains Rosetta(DE3), BL21(DE3) RIL, and BL21(DE3), and the recombinant proteins were expressed in a microshaker at 37°C, 27°C, and 18°C. The best growth rates and expression levels were obtained with the strain BL21(DE3). Table 1 provides a survey of the expression results with six different nsp1 constructs. Most of the protein in the samples expressed at 37°C was insoluble for all six variants. For two constructs, higher yields of soluble protein were obtained at 27°C, but the best results were achieved with expression at 18°C, where most of the expressed protein was in the soluble fraction.
TABLE 1.
Summary of the recombinant production of nsp1 variants in BL21(DE3) E. coli cells
| Name | Molecular massa (kDa) | Soluble expressionb at:
|
Protein recovery (mg)c | Expression level (μM)c | ||
|---|---|---|---|---|---|---|
| 37°C | 27°C | 18°C | ||||
| nsp1 | 19.5 | −/+ | −/+ | + | 3 | 15 |
| nsp1(13-179) | 18.3 | −/+ | −/+ | ++ | 2 | 11 |
| nsp1(1-149) | 16.1 | −/+ | −/+ | ++ | 3 | 19 |
| nsp1(13-149) | 15.0 | −/+ | −/+ | ++ | 2 | 13 |
| nsp1(1-128) | 13.8 | −/+ | + | ++ | 4 | 29 |
| nsp1(13-128) | 12.7 | −/+ | + | ++ | 3 | 24 |
Without the expression tag of 3,500 Da (see text).
The soluble expression levels were evaluated by denaturing polyacrylamide gel electrophoresis of the total cell lysate and of the soluble fraction thereof. −/+, less than 25% soluble protein; +, about 50% soluble protein; ++, more than 85% soluble protein.
The value is for purified protein in a 10-ml volume of buffer. The proteins were purified from cells grown at 18°C in 10 ml of culture.
Based on the results in Table 1, BL21(DE3) E. coli cells in cultures at 18°C were used for the protein production. The proteins were purified by Ni2+ affinity chromatography and gel filtration chromatography. The final protein recovery in 10 ml of culture was in the range of 2 to 4 mg, which represents expression levels in the range of 11 to 29 μM (Table 1). Samples were concentrated for 1D 1H NMR screening with a microcoil probe (Almeida et al., unpublished). The two shortest constructs, nsp1(1-128) and nsp1(13-128), exhibited the highest expression levels. The nsp1(13-128) construct was selected for the structure determination, based on its high-quality 1H NMR spectrum and on its greater stability in comparison to the other five constructs of Table 1.
Since cysteine residues are susceptible to oxidation and formation of intermolecular disulfide bonds, which can lead to unstable and heterogeneous protein samples, we also investigated variant constructs of nsp1(13-128) with Cys 52 replaced by Ala, Ser, Arg, or Asp as part of our initial target optimization strategy, using 1D 1H NMR and circular dichroism spectroscopy to evaluate their foldedness and stability. The variants with Ser 52, Asp 52, or Arg 52 were thus found to be unstable. The variant with Cys 52 replaced by Ala led to a stable, folded protein. However, after observing excellent sample stability of the wild-type protein despite the single Cys residue, we chose the wild-type protein for the structure determination.
The parameters in Table 2 show that a well-defined NMR structure of nsp1(13-128) was obtained. Above-average local disorder is limited to the C-terminal heptapeptide segment of residues 122 to 128 and to a disordered loop of residues 77 to 86 (Fig. 1a). The structure of intact nsp1 includes a globular domain of residues 13 to 121 and the disordered regions of residues 1 to 12 and 122 to 179 (Fig. 1b).
TABLE 2.
Input for the structure calculation and characterization of the bundle of 20 energy-minimized CYANA conformers representing the NMR structure of nsp1(13-128)
| Parameter | Valuea |
|---|---|
| NOE upper distance limits (intraresidual, short-range, medium-range, long-range) | 2,659 (566, 692, 393, 1008) |
| Dihedral angle constraints | 100 |
| Residual target function (Å2) | 1.96 ± 0.35 |
| Residual NOE violations | |
| No. ≥0.1Å | 28 ± 5 |
| Maximum (Å) | 0.19 ± 0.16 |
| Residual dihedral angle violations | |
| Number ≥2.5° | 1 ± 1 |
| Maximum (°) | 4.66 ± 1.27 |
| Amber energies (kcal/mol) | |
| Total | −3,966.42 ± 91.80 |
| van der Waals | −331.44 ± 14.88 |
| Electrostatic | −4,633.80 ± 89.58 |
| RMSDb from ideal geometry | |
| Bond lengths (Å) | 0.0075 ± 0.0002 |
| Bond angles (°) | 2.039 ± 0.047 |
| RMSD to the mean coordinates (Å)c | |
| bb (14-74, 85-125) | 0.45 ± 0.06 |
| ha (14-74, 85-125) | 0.91 ± 0.07 |
| Ramachandran plot statistics (%)d | |
| Most favored regions | 73 |
| Additional allowed regions | 22 |
| Generously allowed regions | 3 |
| Disallowed regions | 2 |
Except for the NOE upper distance limits, dihedral angle constraints, and Ramachandran plot statistics, the average values for the 20 energy-minimized conformers with the lowest residual CYANA target function values and the standard deviations among them are listed.
RMSD, root mean square deviation.
bb, backbone atoms N, Cα, and C′; ha, all heavy atoms. The numbers in parentheses indicate the residues for which the RMSD was calculated.
As determined by PROCHECK (45).
FIG. 1.

(a) Bundle of 20 energy-minimized CYANA conformers of nsp1(13-128). In this stereo view, the polypeptide backbone is shown as a gray spline function through the Cα positions. Selected sequence positions are identified by numerals. (b) Ribbon representation of the closest conformer to the mean coordinates of the bundle of 20 conformers used to represent the NMR structure. The β-strands are cyan, the helices are red, and polypeptide segments with nonregular secondary structure are gray. The regular secondary structures are further identified by lettering. The polypeptide segments shown in green represent the additional, structurally disordered polypeptide segments of the full-length nsp1.
The structure of nsp1(13-128) represents a new fold.
The sequential arrangement of the regular secondary structures in the globular domain of nsp1 is β1-α1-β2-310-β3-β4-β5-β6. There is a mixed parallel/antiparallel six-stranded β-barrel, where the spatial arrangement of the β-strands is β1-β2-β5-β3-β4-β6, and β1 makes contact with β6 (Fig. 2 and 3). The β-strands consist of residues 15 to 21, 52 to 56, 69 to 73, 87 to 92, 104 to 110, and 117 to 124. The helix α1 with residues 36 to 49 is located across one barrel opening, and the 310-helix of residues 62 to 64 is positioned alongside the barrel. A search of the Protein Data Bank using the structure of nsp1 as input for the DALI server (26) did not indicate statistically significant structural similarity to any other protein described to date.
FIG. 2.

Two stereo views of the globular domain of nsp1. (a) Ribbon presentation of the closest conformer of nsp1 to the mean coordinates of the bundle in Fig. 1a, shown in the same orientation as in Fig. 1a. The organization of the β-strands in the barrel is indicated by the labels. (b) Same as panel a after rotation about a horizontal axis, so that one looks at one side of the β-barrel; the axes of the β-barrel and the helix α1 are nearly perpendicular to each other, and those of the barrel and the 310-helix are nearly parallel to each other.
FIG. 3.

Two topology diagrams of the nsp1 mixed parallel/antiparallel six-stranded β-barrel (see text). The numbering indicates the first and last residues of each β-strand.
For the continued discussion it is helpful to adopt a systematic analysis of β-barrels, using the number of strands (n); the shear number (S), which measures the stagger of the strands; and the tilt angle (α) of each strand, which is the angle between the barrel axis and the line adjusted for best fit to the N, Cα, and C′ atoms of each strand (43, 46, 47). S must be an even integer because of the hydrogen bonding pattern between the β-strands (46). Using standard values for the mean Cα-Cα distance along the strands (a = 3.3 Å) and between the strands (b = 4.4 Å), the following geometric relations characteristic of β-barrel structures in proteins have been proposed (43):
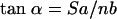 |
(1) |
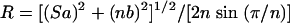 |
(2) |
R is the barrel radius, which is defined as the average of the distances between the Cα atoms of the three residues in opposite strands that are closest to the central part of the barrel (47).
The β-barrel in nsp1 contains six strands and has a shear number (S) of 10. The measured tilt of the strands to the barrel axis (α) ranges from 38o (β2) to 78o (β4), with an average value of 60o. The wide variation among the tilt angles of the individual strands reflects that the β-barrel of nsp1 is pronouncedly irregular (Fig. 2a and b).
The residues used to calculate the radius of the nsp1 barrel are 18 to 20, 53 to 55, 71 to 73, 86 to 88, 106 to 108, and 122 to 124, which give a mean barrel radius (R) of 7 ± 1 Å. Overall, we thus have for nsp1 that the theoretical tilt angle value of 51o calculated from equation 1 shows a discrepancy with the observed average of 60o, whereas the theoretical value of the mean barrel radius of 7 Å, as calculated from equation 2, is in close agreement with the observed value of 7 ± 1 Å.
The interior of the nsp1 barrel and the interfaces between the two helices and the barrel surface consist primarily of hydrophobic residues. The arrangement of the side chains inside the barrel is highly compact, as expected for a barrel of six strands, but the inspection of space-filling models suggests that there is a tight cavity along the center of the barrel, with a radius of about 1.2 Å (not shown). The inside of the barrel consists of 17 side chains, which are contributed by all six strands and which are arranged in three layers. One layer contains L105 and the four hydrophilic residues E56, R74, K85, and R120. The four peripheral hydrophilic groups mediate the contacts with the solvent at the barrel opening opposite to helix α1 (Fig. 4) (in the orientation of Fig. 2b, these residues would be at the bottom of the structure). The charged groups of the side chains of these residues are fully solvent exposed, and E56 makes a salt bridge with R120. The side chain of V21 in the second layer and the βCH2-γCH2 fragment of R120 are located between this first layer and the other side chains of the second layer, which is in the narrowest portion of the barrel and includes the all-hydrophobic side chains of residues L54, I72, V87, and V122. A third layer consists of the side chains of residues L17, L19, V70, L89, A91, L108, and L124, which make hydrophobic contacts with the side chains of residues V36, A39, L40, A43, and L47 from the amphipathic helix α1, and the side chains of residues C52, F32, P110, and P68. The second and third layers of β-strand side chains thus combine with the inner side of the helix α1 to form a large hydrophobic core (Fig. 4). It is worth noting that the variant proteins with Cys 52 replaced by Ser, Asp, or Arg were unstable, which is consistent with a disruption of the β-barrel core, as one would predict from the NMR structure.
FIG. 4.

Stereo view of nsp1(13-128) in the same orientation as in Fig. 2b. The side chains in the interior of the barrel are differently colored to visualize their arrangement in three layers, as discussed in the text. The polypeptide backbone is shown as a gray spline function through the Cα positions. Amino acid side chains are shown as stick drawings. Color code: red, residues of layer 1 at the barrel opening opposite to helix α1, where the four hydrophilic residues are in solvent contact; green, residues in the central layer 2; blue, residues of the third layer, which make hydrophobic contacts to the residues shown in magenta at the top, where V36, A39, L40, A43, and L47 originate from the amphipathic helix α1.
In the SCOP database (48), there are 13 folds with β-barrels of n = 6 and S = 10. Nine of these folds contain antiparallel β-barrels, i.e., the QueA-like fold (41), the hypothetical protein HI1480 (37), the ferredoxin reductase-like fold (10), the phage tail protein (42), the flavin mononucleotide-binding split barrel (36), the reductase/isomerase/elongation factor common domain (10), the elongation factor/aminomethyltransferase common domain (32), the core binding factor β (28), and the surface presentation of antigens fold (16). Four folds have mixed parallel/antiparallel β-barrels similar to the nsp1 fold, but none has the topology observed in nsp1 (Fig. 3). These include the ribosomal protein L25-like (57), the β and β′ subunits of DNA-dependent RNA polymerase (11), the double-ψ β-barrel (38), and the acid protease fold (51). In addition to the apparently unique β-strand topology and the irregular β-barrel geometry, another interesting feature of the nsp1 fold is that the polypeptide chains connecting the β-strands run along the side of the barrel, except for the loop between β3 and β4 (Fig. 2 and 3). This is a rare feature for barrels with n = 6 and S = 10, and besides nsp1, it has been observed only between two β-strands in the ribosomal protein L25-like fold.
It is intriguing that none of the aforementioned folds are quite as irregular as that of nsp1. The distortion of the nsp1 structure seems to be related to the polypeptide segments connecting the β-strands across the side of the barrel. Interestingly, although the adjoining ends of strands β5 and β6 are the furthest apart in space of all strand combinations in nsp1 (approximately 15 Å between P110 and I117), they are connected by the shortest polypeptide segment across the side of the barrel (Fig. 2 and 3). This imposes a lower limit on the shear between these strands. The shear number of 10 seems to be the result of a balance between tight hydrophobic packing inside the nsp1 barrel, which is favored by lower shear numbers, and unstrained arrangement of the linker polypeptide segments on the outside the barrel, which is favored by larger shear numbers. We discuss the β-barrel topology in much detail in order to advance the hypothesis that the outstanding irregularity of the nsp1 β-barrel might be related to a so-far-unknown, possibly entirely novel physiological function of nsp1.
The arrangement of the linker polypeptide segments on the outside of the barrel is puzzling also with regard to the folding pathway of nsp1. For example, if the strand β1 formed hydrogen bonds with β2 early during translation, this would also fix the first linker across the barrel, which might limit the ease with which β6 could make hydrogen bonds with β4 and β1. Schemes representing the topology of the β-barrel (Fig. 3) would intuitively suggest that folding starts midway during translation with the formation of a β-hairpin of the strands β3 and β4. In the folded protein, this pair of β-strands forms the least distorted part of the β-barrel, with highly regular hydrogen bonds, and the loop between β3 and β4 is the only one that does not run along the barrel surface. In subsequent folding steps the two-stranded sheets of β2 and β5 and of β1 and β6, respectively, might be formed, which also have quite regular hydrogen bonding in the nsp1 structure. The linkers between β2 and β3 and between β4 and β5 have almost the same lengths, which should support to position β5 close to β2 if β4 is arranged close to β3. The three regular two-stranded β-sheets (Fig. 3a) are connected in the barrel by the formation of irregular hydrogen bonding patterns.
Characterization of the full-length SARS-CoV nsp1.
The full-length nsp1 was characterized by comparison of the numbers of backbone 15N-1H correlation peaks and the HN and 15N chemical shifts with those of nsp1(13-128) and by heteronuclear NOE measurements of the truncated and full-length nsp1. The truncated construct nsp1(13-128) has an NMR spectrum with large 1H and 15N chemical shift dispersion (Fig. 5a), which is typical for a well-folded globular domain, where the atoms of different individual amino acid residues experience different local microsusceptibilities due to the nonperiodic nature of the interiors of globular proteins. The spectrum of the full-length protein, nsp1(1-179) (Fig. 5b), contains a set of peaks that overlays very closely with those of nsp1(13-128), showing that the globular domain is contained in both constructs. All the additional peaks have HN chemical shifts of 7.9 to 8.5 ppm, which is the region characteristic of “random-coil” polypeptide chains (66).
FIG. 5.

(a) 2D 15N,1H heteronuclear single-quantum coherence (HSQC) spectrum of nsp1(13-128). (b) 2D 15N,1H HSQC spectrum of full-length nsp1(1-179). (c) 2D TROSY-based 15N{1H} NOE experiment with full-length nsp1(1-179), with negative peaks shown in red. The spectra were recorded at a 1H frequency of 600 MHz at 298 K.
A direct measure of intramolecular mobility is provided by the heteronuclear 2D 15N{1H} NOE experiment, which is routinely used to access protein dynamics on the picosecond to nanosecond timescale (55, 69). Positive signals with intensities of ∼0.8 identify residues in the folded cores of small and medium-size globular proteins, with mobility of the individual 15N-1H moieties restricted to the overall rotational tumbling of the molecule. This is illustrated with the 15N{1H} NOE data for nsp1(13-128) (Fig. 6), which also serve as a reference for assessing the state of the additional chain segments in nsp1(1-179). 15N{1H} NOE values of about 0.8 are seen for most of the residues in the regular secondary structure elements (Fig. 6). Increased flexibility of the polypeptide chain that causes reduced NOE intensities is found in the disordered loop between residues 75 and 87 and in the region of residues 94 to 103, which forms nonregular secondary structure with one γ-turn of residues 97 to 99 and a type II β-turn of residues 98 to 101 (Fig. 1). Most of the resonances in full-length nsp1 that are not present in nsp1(13-128) have either small positive or negative 15N{1H} NOEs (Fig. 5c), showing that the polypeptide segments of residues 1 to 12 and 129 to 179 are best described as a short N-terminal and a long C-terminal flexibly disordered tail, respectively (Fig. 1b). Interestingly, it has been determined that the carboxy-terminal half of the related protein MHV nsp1 is not needed for viral replication in culture but is important for efficient proteolytic cleavage between nsp1 and nsp2 and for optimal viral replication (7).
FIG. 6.

Plot of the 15N{1H} NOE intensities versus the sequence of nsp1(13-128). The data were collected at a 1H frequency of 600 MHz at 298 K. The positions of the regular secondary structure elements are indicated. Each point represents the mean of three measurements, and the error bars represent the standard deviations of the three measurements.
Structure-based search for nsp1 functions.
In an initial attempt to identify leads to possible nsp1 functions, Fig. 7a identifies the solvent-exposed residues of nsp1, which would be sterically accessible for intermolecular contacts with reaction partners. These residues give rise to an uneven electrostatic surface charge distribution (Fig. 7b), with a large negative surface on one face and hydrophobic, polar, and positively charged residues forming the opposite surface. The large contiguous patches of positive and negative surface charge could mediate specific as well as nonspecific intermolecular interactions by electrostatic forces. For example, considering that it has been shown that nsp1 promotes mRNA degradation (31), the area of positive charge on the molecular surface formed by K48, R125, and K126 (Fig. 7b) is of interest as a potential site for a direct interaction with mRNA. Alternatively, the positively and negatively charged areas of the protein surface might be involved in protein-protein interactions, and nsp1 might then exert its biological effect not by direct interactions with the mRNA but by interacting with other proteins involved in the regulation of cellular mRNA stability (49). In this context it seems worth mentioning that the NMR structure determination of nsp1(13-128) was performed in 250 mM NaCl because the protein precipitated at lower ionic strengths, which might be due to self-aggregation of nsp1 caused by the uneven charge distribution.
FIG. 7.

(a) Amino acid sequence of nsp1, with solvent-exposed residues highlighted in green. A residue is considered to be exposed if at least one atom of its side chain has more than 50% surface accessibility to the solvent. For glycines, the CO and HN exposure is considered. (b) Surface views of nsp1 in a space-filling representation. In the surface view shown on the left, the structure has the same orientation as in Fig. 2b. Some of the surface-exposed side chains discussed in the text are identified with the one-letter amino acid code and the residue number. Color code: gray, hydrophobic and polar residues; red, negatively charged; blue, positively charged. (c) Sequence alignment between SARS-CoV nsp1 and MHV nsp1 identified with the FFAS server. Identical residues are shown in red. Arrows indicate single-amino-acid replacements in MHV p28 that were generated and studied by Brockway et al. (7). Mutations that are detrimental to the viral replication are identified by boldface, while those that are not detrimental are in italic. Residues removed in the truncated variant protein MHV1 nsp1ΔC are shown in lowercase (see text). Residues in β-strands and in helical secondary structures are underlined with solid and dashed lines, respectively.
For comparisons with the nsp1 proteins of other coronaviruses, data are available for the p9 proteins of group 1 CoVs and for the p28 proteins of group 2a CoVs. Using database searches such as BLAST or PSI-BLAST (2), no significant sequence similarity between nsp1 of SARS-CoV and proteins of group 2a coronaviruses could be identified. However, pairwise alignment with the FFAS server (29), employing a profile/profile-based method that is able to detect distant relationships, identified 20% sequence identity between SARS-CoV nsp1 and MHV nsp1 (p28) over 174 aligned residues (Fig. 7c). The FFAS score of −9.6 indicates that these two proteins might share the novel nsp1 three-dimensional fold, but the remaining sequence divergence leaves open the possibility that these proteins might perform different functions even if they had a common fold. To our knowledge, it has not yet been determined whether the p28 proteins of group 2a coronaviruses also promote mRNA degradation, but MHV p28 expression has been shown to cause cell cycle arrest in cultured cells.
The most striking result of the alignment of SARS-CoV nsp1 with the polypeptide fragment consisting of residues 46 to 247 of MHV p28 is the observation of a consensus sequence, LRKxGxKG, positioned at the end of strand β6 of the globular domain of SARS-CoV nsp1, which is conserved not only in MHV p28 (Fig. 7c) but also in human CoV OC43 p28. It includes the two residues R125 and K126, which contribute to the positively charged patch on the nsp1 molecular surface (Fig. 7b). If future studies of the p28 proteins of group 2a CoVs should show that these proteins share mRNA degradation activity with SARS-CoV nsp1, this conserved region could be a candidate for mRNA interaction.
Analysis of the nsp1 structure also provides indications for functional differences between the p28 proteins and SARS-CoV nsp1. For example, the motif K109-R110-L111 in MHV p28 was identified by Chen et al. as a potential cyclin-binding motif (8), and SARS-CoV nsp1 lacks residues corresponding to R110 and L111. In addition, Chen et al. identified residues 30 to 33 (S/NPER) of p28 as a potential site for phosphorylation by cyclin-dependent kinases (8). These residues occur in an N-terminal 45-residue segment of p28 that appears not to be homologous to SARS-CoV nsp1. The propensity to induce cell cycle arrest may therefore be unique to MHV p28, or possibly to the group 2a p28 proteins in general, and it might not be shared by SARS-CoV nsp1 even if it turned out that these proteins all share a similar fold.
In other comparisons, no significant sequence identity between SARS-CoV nsp1 and the nsp1 (p9) proteins of the group 1 CoVs could be detected. These results are consistent with the analysis by Snijder et al. (59), who described nsp1 as a specific marker of group 2 CoVs. The p9 proteins of group 1 CoVs most likely differ from those of group 2 CoVs in both structure and function.
The MHV1 p28 protein was subjected to a mutagenesis study by Brockway et al. (7), who generated single-amino-acid replacements and truncated versions of this protein and studied their impact on viral replication in cultured cells. Among the mutations found to affect viral replication, only some occur in residues conserved between MHV1 p28 and SARS-CoV nsp1 (Fig. 7c). Deletion of the entire p28 protein or of the polypeptide segment from residue 87 to 164 of MHV p28 prevented the virus from productively infecting cultured cells (7). If MHV p28 and SARS-CoV nsp1 did indeed share a similar fold, the latter construct would lack most of the globular domain. In contrast, the carboxy-terminal half of MHV p28 (residues 124 to 241) has been shown to be dispensable for replication in culture, but it is important for efficient proteolytic cleavage of the protein and for optimal viral replication. If MHV p28 were to contain regular secondary structures similar to those of SARS-CoV nsp1, removal of the polypeptide segment from residue 124 to 241 would correspond to the loss of the strands β3, β4, β5, and β6, as well as of the flexibly disordered C-terminal tail, which would appear to entail a considerable disruption of the protein fold. The following considerations might help to resolve the apparent ensuing discrepancies. First, the increased flexibility and lack of a globular fold in the C-terminal region of the protein may ensure accessibility of the protease recognition site between nsp1 and nsp2 but may not be directly involved with the activity exerted by the protein. Second, it appears that the strands β1 and β2 and the helix α1 might provide for a sufficiently stable fold to maintain the so-far-unidentified biological activity, in particular if one assumes that the additional N-terminal 45-residue segment of MHV p28, which is not homologous to SARS-CoV nsp1, could participate in a globular fold and help to stabilize the shortened protein.
In conclusion, this paper shows that the SARS-CoV protein nsp1, which is encoded at the 5′ terminus of the genome, forms a previously unknown complex β-barrel fold with several unique structural features. We hypothesize that the uniqueness of the irregular β-barrel fold may be related to a so-far-unknown, unique biological function of nsp1. The definition of the globular region of nsp1 and the identification of residues on the molecular surface likely to contribute to mRNA degradation activity may provide a platform for continued research on the role of this protein in SARS-CoV and in other coronaviruses.
Acknowledgments
We thank Kumar Saikatendu, Jeremiah Joseph, Vanitha Subramanian, Benjamin W. Neuman, Michael J. Buchmeier, Raymond C. Stevens, and Peter Kuhn of the Consortium for Functional and Structural Proteomics of the SARS-CoV for providing us with samples of nsp1(1-179) and nsp1(1-159) for the initial NMR screening. The use of the high-performance computing facility at The Scripps Research Institute is gratefully acknowledged.
This study was supported by NIAID/NIH contract no. HHSN266200400058C “Functional and Structural Proteomics of the SARS-CoV” to P. Kuhn and M. J. Buchmeier and by the Joint Center for Structural Genomics through NIH/NIGMS grant no. U54-GM074898. Additional support was obtained for M.S.A. through the Pew Latin American Fellows Program in the Biological Sciences and the Skaggs Institute for Chemical Biology and for M.A.J. through a fellowship from the Canadian Institutes of Health Research and the Skaggs Institute for Chemical Biology. Kurt Wüthrich is the Cecil H. and Ida M. Green Professor of Structural Biology at TSRI and a member of the Skaggs Institute for Chemical Biology.
Footnotes
Published ahead of print on 3 January 2007.
REFERENCES
- 1.Almeida, M. S., M. A. Johnson, and K. Wüthrich. 2006. NMR assignment of the SARS-CoV protein nsp1. J. Biomol. NMR 36(Suppl.):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barretto, N., D. Jukneliene, K. Ratia, Z. Chen, A. D. Mesecar, and S. C. Baker. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189-15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlam, M., H. Yang, and Z. Rao. 2005. Structural insights into SARS coronavirus proteins. Curr. Opin. Struct. Biol. 15:664-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhardwaj, K., J. Sun, A. Holzenberg, L. A. Guarino, and C. C. Kao. 2006. RNA recognition and cleavage by the SARS coronavirus endoribonuclease. J. Mol. Biol. 361:243-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockway, S. M., X. T. Lu, T. R. Peters, T. S. Dermody, and M. R. Denison. 2004. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J. Virol. 78:11551-11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockway, S. M., and M. R. Denison. 2005. Mutagenesis of the murine hepatitis virus nsp1-coding region identifies residues important for protein processing, viral RNA synthesis, and viral replication. Virology 340:209-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, C. J., K. Sugiyama, H. Kubo, C. Huang, and S. Makino. 2004. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 78:10410-10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, J. K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179-5197. [Google Scholar]
- 10.Correll, C. C., C. J. Batie, D. P. Ballou, and M. L. Ludwig. 1992. Phthalate dioxygenase reductase: a modular structure for electron transfer from pyridine nucleotides to [2Fe-2S]. Science 258:1604-1610. [DOI] [PubMed] [Google Scholar]
- 11.Cramer, P., D. A. Bushnell, and R. D. Kornberg. 2001. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 292:1863-1876.11313498 [Google Scholar]
- 12.Cuff, J. A., M. E. Clamp, A. S. Siddiqui, M. Finlay, and G. J. Barton. 1998. JPred: a consensus secondary structure prediction server. Bioinformatics 14:892-893. [DOI] [PubMed] [Google Scholar]
- 13.Denison, M. R., B. Yount, S. M. Brockway, R. L. Graham, A. C. Sims, X. Lu, and R. S. Baric. 2004. Cleavage between replicase proteins p28 and p65 of mouse hepatitis virus is not required for virus replication. J. Virol. 78:5957-5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denison, M. R., P. W. Zoltick, S. A. Hughes, B. Giangreco, A. L. Olson, S. Perlman, J. L. Leibowitz, and S. R. Weiss. 1992. Intracellular processing of the N-terminal ORF 1a proteins of the coronavirus MHV-A59 requires multiple proteolytic events. Virology 189:274-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egloff, M.-P., F. Ferron, V. Campanacci, S. Longhi, C. Rancurel, H. Dutartre, E. J. Snijder, A. E. Gorbalenya, C. Cambillau, and B. Canard. 2004. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. USA 101:3792-3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fadouloglou, V. E., A. P. Tampakaki, N. M. Glykos, M. N. Bastaki, J. M. Hadden, S. E. V. Phillips, N. J. Panopoulos, and M. Kokkinidis. 2004. Structure of HrcQB-C, a conserved component of the bacterial type III secretion systems. Proc. Natl. Acad. Sci. USA 101:70-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galán, C., L. Enjuanes, and F. Almazán. 2005. A point mutation within the replicase gene differentially affects coronavirus genome versus minigenome replication. J. Virol. 79:15016-15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorbalenya, A. E., and E. V. Koonin. 1989. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. 17:8413-8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorbalenya, A. E., E. J. Snijder, and W. J. Spaan. 2004. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J. Virol. 78:7863-7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorbalenya, A. E., L. Enjuanes, J. Ziebuhr, and E. J. Snijder. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117:17-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorbalenya, A. E. 2001. Big nidovirus genome. When count and order of domains matter. Adv. Exp. Med. Biol. 494:1-17. [PubMed] [Google Scholar]
- 22.Guarino, L. A., K. Bhardwaj, W. Dong, J. Sun, A. Holzenburg, and C. Kao. 2005. Mutational analysis of the SARS virus nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 353:1106-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Güntert, P., C. Mumenthaler, and K. Wüthrich. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273:283-298. [DOI] [PubMed] [Google Scholar]
- 24.Herrmann, T., P. Güntert, and K. Wüthrich. 2002. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24:171-189. [DOI] [PubMed] [Google Scholar]
- 25.Herrmann, T., P. Güntert, and K. Wüthrich. 2002. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319:209-227. [DOI] [PubMed] [Google Scholar]
- 26.Holm, L., and C. Sander. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233:123-138. [DOI] [PubMed] [Google Scholar]
- 27.Hsu, M.-F., C.-J. Kuo, K.-T. Chang, H.-C. Chang, C.-C. Chou, T.-P. Ko, H.-L. Shr, G.-G. Chang, A. H.-J. Wang, and P.-H. Liang. 2005. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 280:31257-31266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang, X., J. W. Peng, N. A. Speck, and J. H. Bushweller. 1999. Solution structure of core binding factor beta and map of the CBF alpha binding site. Nat. Struct. Biol. 6:624-627. [DOI] [PubMed] [Google Scholar]
- 29.Jaroszewski, L., L. Rychlewski, Z. Li, W. Li, and A. Godzik. 2005. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 33:W284-W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joseph, J. S., K. S. Saikatendu, V. Subramanian, B. W. Neuman, A. Brooun, M. Griffith, K. Moy, M. K. Yadav, J. Velasquez, M. J. Buchmeier, R. C. Stevens, and P. Kuhn. 2006. Crystal structure of nonstructural protein 10 from the severe acute respiratory syndrome coronavirus reveals a novel fold with two zinc-binding motifs. J. Virol. 80:7894-7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamitani, W., K. Narayanan, C. Huang, K. Lokugamage, T. Ikegami, N. Ito, H. Kubo, and S. Makino. 2006. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 103:12885-12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kjeldgaard, M., P. Nissen, S. Thirup, and J. Nyborg. 1993. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure 1:35-50. [DOI] [PubMed] [Google Scholar]
- 33.Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics 14:51-55. [DOI] [PubMed] [Google Scholar]
- 34.Koradi, R., M. Billeter, and P. Güntert. 2000. Point-centered domain decomposition for parallel molecular dynamics simulation. Comp. Phys. Commun. 124:139-147. [Google Scholar]
- 35.Liang, P.-H. 2006. Characterization and inhibition of SARS-coronavirus main protease. Curr. Top. Med. Chem. 6:361-376. [DOI] [PubMed] [Google Scholar]
- 36.Liepinsh, E., M. Kitamura, T. Murakami, T. Nakaya, and G. Otting. 1997. Pathway of chymotrypsin evolution suggested by the structure of the FMN-binding protein from Desulfovibrio vulgaris (Miyazaki F). Nat. Struct. Biol. 4:975-979. [DOI] [PubMed] [Google Scholar]
- 37.Lim, K., E. Sarikaya, A. Galkin, W. Krajewski, S. Pullalarevu, J. H. Shin, Z. Kelman, A. Howard, and O. Herzberg. 2004. Novel structure and nucleotide binding properties of HI1480 from Haemophilus influenzae: a protein with no known sequence homologues. Proteins 56:564-571. [DOI] [PubMed] [Google Scholar]
- 38.Ludvigsen, S., and F. M. Poulsen. 1992. Three-dimensional structure in solution of barwin, a protein from barley seed. Biochemistry 31:8783-8789. [DOI] [PubMed] [Google Scholar]
- 39.Luginbühl, P., P. Güntert, M. Billeter, and K. Wüthrich. 1996. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J. Biomol. NMR 8:136-146. [DOI] [PubMed] [Google Scholar]
- 40.Luginbühl, P., T. Szyperski, and K. Wüthrich. 1995. Statistical basis for the use of 13Cα chemical shifts in protein structure determination. J. Magn. Reson. 109:229-233. [Google Scholar]
- 41.Mathews, I., R. Schwarzenbacher, D. McMullan, P. Abdubek, E. Ambing, H. Axelrod, T. Biorac, J. M. Canaves, H. J. Chiu, A. M. Deacon, M. DiDonato, M. A. Elsliger, A. Godzik, C. Grittini, S. K. Grzechnik, J. Hale, E. Hampton, G. W. Han, J. Haugen, M. Hornsby, L. Jaroszewski, H. E. Klock, E. Koesema, A. Kreusch, P. Kuhn, S. A. Lesley, I. Levin, M. D. Miller, K. Moy, E. Nigoghossian, J. Ouyang, J. Paulsen, K. Quijano, R. Reyes, G. Spraggon, R. C. Stevens, H. van den Bedem, J. Velasquez, J. Vincent, A. White, G. Wolf, Q. Xu, K. O. Hodgson, J. Wooley, and I. A. Wilson. 2005. Crystal structure of S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) from Thermotoga maritima at 2.0 Å resolution reveals a new fold. Proteins 59:869-874. [DOI] [PubMed] [Google Scholar]
- 42.Maxwell, K. L., A. A. Yee, C. H. Arrowsmith, M. Gold, and A. R. Davidson. 2002. The solution structure of the bacteriophage lambda head-tail joining protein, gpFII. J. Mol. Biol. 318:1395-1404. [DOI] [PubMed] [Google Scholar]
- 43.McLachlan, A. D. 1979. Gene duplications in the structural evolution of chymotrypsin. J. Mol. Biol. 128:49-79. [DOI] [PubMed] [Google Scholar]
- 44.Minskaia, E., T. Hertzig, A. E. Gorbalenya, V. Campanacci, C. Cambillau, B. Canard, and J. Ziebuhr. 2006. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 103:5108-5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris, A. L., M. W. MacArthur, E. G. Hutchinson, and J. M. Thornton. 1992. Stereochemical quality of protein structure coordinates. Proteins 12:345-364. [DOI] [PubMed] [Google Scholar]
- 46.Murzin, A. G., A. M. Lesk, and C. Chothia. 1994. Principles determining the structure of β-sheet barrels in proteins. I. A theoretical analysis. J. Mol. Biol. 236:1369-1381. [DOI] [PubMed] [Google Scholar]
- 47.Murzin, A. G., A. M. Lesk, and C. Chothia. 1994. Principles determining the structure of β-sheet barrels in proteins. II. The observed structures. J. Mol. Biol. 236:1382-1400. [DOI] [PubMed] [Google Scholar]
- 48.Murzin, A. G., S. E. Brenner, T. Hubbard, and C. Chothia. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247:536-540. [DOI] [PubMed] [Google Scholar]
- 49.Newbury, S. F. 2006. Control of mRNA stability in eukaryotes. Biochem. Soc. Trans. 34:30-34. [DOI] [PubMed] [Google Scholar]
- 50.Page, R., W. Peti, I. A. Wilson, R. C. Stevens, and K. Wüthrich. 2005. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc. Natl. Acad. Sci. USA 102:1901-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pearl, L., and T. Blundell. 1984. The active site of aspartic proteinases. FEBS Lett. 174:96-101. [DOI] [PubMed] [Google Scholar]
- 52.Peti, W., M. A. Johnson, T. Herrmann, B. W. Neuman, M. J. Buchmeier, M. Nelson, J. Joseph, R. Page, R. C. Stevens, P. Kuhn, and K. Wüthrich. 2005. Structural genomics of the severe acute respiratory syndrome coronavirus: nuclear magnetic resonance structure of the protein nsP7. J. Virol. 79:12905-12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prentice, E., J. McAuliffe, X. Lu, K. Subbarao, and M. R. Denison. 2004. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 78:9977-9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ratia, K., K. S. Saikatendu, B. D. Santarsiero, N. Barretto, S. C. Baker, R. C. Stevens, and A. D. Mesecar. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 103:5717-5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renner, C., M. Schleicher, L. Moroder, and T. A. Holak. 2002. Practical aspects of the 2D 15N-{1H}-NOE experiment. J. Biomol. NMR 23:23-33. [DOI] [PubMed] [Google Scholar]
- 56.Ricagno, S., M.-P. Egloff, R. Ulferts, B. Coutard, D. Nurizzo, V. Campanacci, C. Cambillau, J. Ziebuhr, and B. Canard. 2006. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 103:11892-11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rould, M. A., J. J. Perona, D. Soll, and T. A. Steitz. 1989. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNA(Gln) and ATP at 2.8 Å resolution. Science 246:1135-1142. [DOI] [PubMed] [Google Scholar]
- 58.Saikatendu, K. S., J. S. Joseph, V. Subramanian, T. Clayton, M. Griffith, K. Moy, J. Velasquez, B. W. Neuman, M. J. Buchmeier, R. C. Stevens, and P. Kuhn. 2005. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1"-phosphate dephosphorylation by a conserved domain of nsP3. Structure 13:1665-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snijder, E. J., P. J. Bredenbeek, J. C. Dobbe, V. Thiel, J. Ziebuhr, L. L. Poon, Y. Guan, M. Rozanov, W. J. Spaan, and A. E. Gorbalenya. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spera, S., and A. Bax. 1991. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113:5490-5492. [Google Scholar]
- 61.Su, D., Z. Lou, F. Sun, Y. Zhai, H. Yang, R. Zhang, A. Joachimiak, X. C. Zhang, M. Bartlam, and Z. Rao. 2006. Dodecamer structure of severe acute respiratory syndrome coronavirus nonstructural protein nsp10. J. Virol. 80:7902-7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sutton, G., E. Fry, L. Carter, S. Sainsbury, T. Walter, J. Nettleship, N. Berrow, R. Owens, R. Gilbert, A. Davidson, S. Siddell, L. L. M. Poon, J. Diprose, D. Alderton, M. Walsh, J. M. Grimes, and D. I. Stuart. 2004. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure 12:341-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan, J., K. H. G. Verschueren, K. Anand, J. Shen, M. Yang, Y. Xu, Z. Rao, J. Bigalke, B. Heisen, J. R. Mesters, K. Chen, X. Shen, H. Jiang, and R. Hilgenfeld. 2005. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 354:25-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thiel, V., J. Herold, B. Schelle, and S. G. Siddell. 2001. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 75:6676-6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu, Y.-S., W.-H. Lin, J. T.-A. Hsu, and H.-P. Hsieh. 2006. Antiviral drug discovery against SARS-CoV. Curr. Med. Chem. 13:2003-2020. [DOI] [PubMed] [Google Scholar]
- 66.Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley, New York, NY.
- 67.Yang, H., M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld, and Z. Rao. 2003. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 100:13190-13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhai, Y., F. Sun, X. Li, H. Pang, X. Xu, M. Bartlam, and Z. Rao. 2005. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat. Struct. Mol. Biol. 12:980-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu, G., Y. Xia, L. K. Nicholson, and K. H. Sze. 2000. Protein dynamics measurements by TROSY-based NMR experiments. J. Magn. Reson. 143:423-426. [DOI] [PubMed] [Google Scholar]
- 70.Ziebuhr, J. 2005. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 287:57-94. [DOI] [PMC free article] [PubMed] [Google Scholar]