

Abstract
Varicella-zoster virus (VZV) open reading frame 10 (ORF10) is a determinant of virulence in SCIDhu skin xenografts but not in human T cells in vivo. In this analysis of the regulation of ORF10 transcription, we have identified four ORF10-related transcripts, including a major 1.3-kb RNA spanning ORF10 only and three other read-through transcripts. Rapid-amplification-of-cDNA-ends experiments indicated that the 1.3-kb transcript of ORF10 has single initiation and termination sites. In transient expression assays, the ORF10 promoter was strongly stimulated by the major VZV transactivator, IE62. Deletion analyses revealed approximate boundaries for the full ORF10 promoter activity between −75 and −45 and between +5 and −8, relative to the ORF10 transcription start site. The recombinant virus POKA10-Δpro, with the ORF10 promoter deletion, blocked transcription of ORF10 and also of ORF9A and ORF9 mRNAs, whereas expression of read-through ORF9A/9/10 and ORF9/10 transcripts was increased, compensating for the loss of the monocistronic mRNAs. The cellular factor USF bound specifically to its consensus site within the ORF10 promoter and was required for IE62 transactivation, whereas disrupting the predicted TATA boxes or Oct-1 binding elements had no effect. The USF binding site was disrupted in the recombinant virus, POKA10-proΔUSF, and no ORF10 protein was produced. Both ORF10 promoter mutants reduced VZV replication in SCIDhu skin xenografts. These observations provided further evidence of the contribution of the ORF10 protein to VZV pathogenesis in skin and demonstrated that VZV depends upon the cellular transcriptional factor USF to support its virulence in human skin in vivo.
Varicella-zoster virus (VZV) is an alphaherpesvirus that causes varicella, establishes latency in sensory ganglia, and may reactivate, resulting in herpes zoster (1, 42). As is characteristic of herpesviruses, VZV genes are predicted to be transcribed in an ordered and temporally controlled cascade of putative immediate-early, early, and late gene expression during lytic infection. VZV immediate-early proteins are essential for activating viral genes, early genes are required for VZV genome synthesis, and many late genes are involved in virion morphogenesis and maturation (40). Much of the control of the expression of these different classes of viral proteins is at the level of transcription. Both the kinetics and the levels of expression of transcripts that encode viral proteins are dictated by features of the promoter controlling the particular transcript (15). VZV gene expression also appears to occur in latently infected cells in sensory ganglia, but it is restricted to a subset of viral genes, which may include open reading frames (ORFs) 4, 21, 29, 62, 63, and 66 (7, 9). The differential expression of VZV genes during productive and persistent infection is likely to be related to specific cis-acting regulatory mechanisms, alternative promoter usage, and neuron-specific cellular factors. Therefore, a better understanding of the characteristics of VZV gene promoters may lead to insights about VZV infection of differentiated human cells and new strategies for controlling primary and recurrent VZV infections.
The IE62 protein is the major viral transactivator and induces expression of all classes of VZV genes (11, 16, 33). In addition, several cellular transcription factors, including specificity factor 1 (Sp1), upstream stimulatory factor (USF), activator protein 1, octamer-binding protein 1 (Oct-1), TATA element, as well as other viral regulatory proteins, including IE4, IE63, ORF10, and ORF61, participate in regulating the activities of VZV gene promoters in reporter plasmid assays (7, 18, 20, 25, 30, 38, 43). One such cellular factor, USF, is particularly important for VZV replication, because USF has been demonstrated to have a direct physical interaction with IE62 in the activation of VZV promoters in transient-transfection assays (25, 39, 51).
USF is a family of evolutionarily conserved basic-helix-loop-helix-leucine zipper transcription factors that interact with DNA through the consensus sequence, 5′-GGTCACGTGACC-3′ (3, 13, 19, 23, 44). Purified human USF is composed of two polypeptides, with apparent molecular masses of 43 and 44 kDa, which exist as both homo- and heterodimers, with the heterodimer being the major species (44, 45, 46). Nearly one-fourth of the putative VZV regulatory elements that control expression of the 71 predicted or known VZV open reading frames contain USF consensus binding sites, indicating that USF is likely to play an important role in VZV replication (43). USF transactivates the promoters of VZV IE4 and ORF10 in conjunction with the IE62 protein in reporter plasmid assays (27). Previous work by Meier and Straus (26) showed that USF1 is the dominant isoform bound to the VZV ORF28/29 regulatory element, and further study of this dual promoter demonstrated that the activation domain of USF1 is necessary and sufficient for synergistic activation with IE62 (52). Transcription of both USF1 and USF2 is detected and remains relatively stable throughout the course of VZV replication in melanoma (MeWo) cells, and USF1 and USF2 protein levels are maintained without significant variation during VZV replication (39). USF and IE62 also function synergistically in activation of the VZV gI promoter. However, analysis of VZV gI promoter mutants in the SCIDhu model showed that disrupting binding sites for both USF and Sp1 abolished VZV replication in skin xenografts, while disrupting the USF binding site alone had only a minor effect on VZV virulence in skin xenografts (17).
The VZV late gene product, the ORF10 protein, is a regulatory/tegument protein that has homology with herpes simplex virus type 1 (HSV-1) VP16 (8). However, in contrast to VP16, ORF10 protein is dispensable for VZV replication in vitro and in SCIDhu T-cell xenografts in vivo, but it is critical for VZV growth in human skin in vivo. An ORF10-null mutant, POKAΔ10, exhibited deficiencies in virion formation and cell-cell spread in infected skin (6), but deleting ORF10 had no consequences for VZV replication in T-cell xenografts in SCIDhu mice compared to results with wild-type POKA. In addition to equal infectious virus yields, POKAΔ10-infected T-cell xenografts showed the same pattern of T-cell depletion observed with fully virulent VZV at late time points after infection of T-cell xenografts (6). Given this evidence of the specific importance of ORF10 for the pathogenesis of VZV infection of skin and the lack of information about the functional architecture of its promoter region and the regulation of ORF10 expression in the context of the VZV genome during replication, we undertook a detailed analysis of the ORF10 promoter and its responsiveness to VZV and cellular transactivators both in vitro and in SCIDhu skin xenografts in vivo.
In the present study, we have identified the major ORF10 transcription initiation and termination sites and have mapped transcripts regulated by the ORF10 promoter. The DNA sequences that act in cis to regulate ORF10 expression were defined by mutagenesis. We also confirmed that IE62 transactivates the ORF10 regulatory element and markedly increases ORF10 expression. In addition, we demonstrated that production of bicistronic RNA, including ORF9 and ORF10, can compensate functionally for the loss of monocistronic ORF9 RNA and allow production of the ORF9 protein. Finally, we found that USF is essential for optimal IE62-mediated effects on the ORF10 promoter in cultured cells, and this USF function is a cellular determinant of virulence in SCIDhu skin xenografts in vivo.
MATERIALS AND METHODS
Generation of POKA recombinant viruses with ORF10 promoter mutations.
VZV recombinants were made as described previously (6). Briefly, two- or three-step PCRs with the pT7Blue-3 SacI-EcoRI plasmid (6) as a template were used to delete a fragment or mutate nucleotides within the ORF10 intergenic region. To delete nucleotides (nt) −87 to −97, two fragments were amplified by PCR with primers P1 (5′-GGATTCCCGAAGCGAGCTC-3′), corresponding to POKA nt 11436 to 11454 and including a SacI restriction site, and P2 (5′-ATAAACACAAACCACGACTG-3′), located at −63 to −87 of the ORF10 regulatory element; P3 (5′-GTATTATATGTCACATATTA-3′), located at −97 to −111 of the ORF10 regulatory element, and P4 (5′-GGAAGTGCGTAGACGGAATTC-3′), containing an EcoRI site and corresponding to nt 14647 to 14667. The resulting PCR products were inserted by triple ligation into the pT7Blue-3 SacI-EcoRI vector, which had been digested with the enzymes SacI and EcoRI, to obtain the pT7Blue-3 SacI-EcoRI plasmid with the ORF10 regulatory element −87 to −97 deleted. This fragment was cloned back into the pvFsp73 cosmid as described previously (6). Similar strategies were used to delete +39 to +63, −179 to +63, and a six-nucleotide substitution in the USF binding site within the ORF10 regulatory element. Two pairs of primers, P1 and P5 (5′-CGACTGTCTTTTATACGTTTATTTA-3′) and P4 and P6 (5′-ATGGAGTGTAATTTAGGAACCG-3′), were used to delete nt −179 to +63. One set of primers, P1 and P7 (5′-CGATTCCCTTTATCAAAACCCC-3′), plus P4 and P6, was used to delete nt +39 to +63. To disrupt the USF binding site, three pairs of primers, P1 and P8 (5′-CTGTTTAAATAGTACTCTACCGTATTATGAACAG-3′), P4 and P9 (5′-CTGTTCATAATACGGTAGAGTACTATTTAAACAG-3′), and P1 and P4, were used (underlined sequences show the positions of the USF consensus binding site). The PCR primers were manufactured by Operon Technologies, Inc. (Birmingham, AL).
Recombinant viruses, designated POKA10-proΔ−87/−97, POKA10-Δpro, POKA10-proΔ+39/+63, and POKA10-proΔUSF, were isolated by transfection of human MeWo cells with the mutated pvFsp73 cosmid and three intact cosmids, pvSpe14, pvPme2, and pvSpe23 (6). MeWo cells were maintained in minimum essential medium (Mediatech, Washington, DC) supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA), nonessential amino acids, and antibiotics. To confirm nucleotide deletions or substitutions, DNA was isolated from MeWo cells or xenograft tissues with DNAzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. PCR was performed using Pfu DNA polymerase (Stratagene, La Jolla, CA) with primers P1 and P4. The resulting PCR products were separated on 1% agarose gels, isolated with GeneClean (QIAGEN, Inc.), and confirmed by sequencing. Viruses were propagated in human embryonic lung fibroblasts (HELF) for infection of SCIDhu mouse xenografts. The replication kinetics and peak titers of recombinant viruses were assessed by infectious focus assay. MeWo cells were seeded in six-well plates and infected with an inoculum of ∼1 × 103 PFU. Cells were trypsinized on days 1 to 6, and titers were determined as described previously (28).
Northern blots.
Total single-stranded RNA was extracted from cells infected with POKA or ORF10 promoter mutants by using Tri-Reagent (Invitrogen, Carlsbad, CA). RNAs were separated by electrophoresis in formamide-formaldehyde denaturing 1.1% agarose gels in morpholinepropanesulfonic acid buffer and transferred to positively charged nylon membranes (Roche, Inc.). RNA detection on membranes was done with nonradioactive digoxigenin-labeled riboprobes; the membranes exposed to nonradioactive digoxigenin-labeled riboprobes were further reacted with an antidigoxigenin-alkaline phosphatase Fab fragment (antibody) and developed with the CSPD chemiluminescent substrate (Roche, Inc.). Riboprobes were prepared to detect ORF9A, ORF9, ORF10, and ORF11 transcripts: (i) the ORF9A probe was generated by primers P10 (5′-CATATCACCTGTATTTACTGCCG-3′) and P11 (5′-GCTAACGAAATAAGGGCTACAC-3′), covering ORF9A from nt 10610 to 10849; (ii) the ORF9 probe was made with primers P12 (5′-CGTAATGGCATCTTCCGACG-3′) and P13 (5′-CGCATCAGTTCTTGATGCCG-3′), including a region of ORF9 from nt 11004 to 11907; (iii) the ORF10 probe was prepared with primers P14 (5′-GGAGTGTAATTTAGGAACCGAAC-3′) and P15 (5′-GTATCTCGAGCGGGTGTGCAGATTGAC-3′), covering nt 12161 to 12821; (iv) the ORF11 probe was synthesized with primers P16 (5′-CGTCACACACACGGAACACG-3′) and P17 (5′-CGGGTACATTCGCGCATAGC-3′) and included ORF11 from nt 13661 to 14394. The fragments were synthesized by PCR using pvFsp73 or pvSpe23 cosmid DNA as a template; ORF-specific products inserted into the pCRII-TOPO clone vector (Invitrogen, Carlsbad, CA) were used to synthesize positive-stranded-RNA-specific probes by using T7 or Sp6 RNA polymerase (Ambion, Inc.). Northern blots were assessed using ImageJ, an image processing and analysis program.
Immunofluorescence.
Infected HELF cells were fixed in 2% paraformaldehyde-0.05% Triton X-100 48 h after infection. Cells were washed five times in phosphate-buffered saline (PBS) for 5 min, blocked with 5% normal goat serum-1× PBS and 0.02% bovine serum albumin for 1 h, and incubated overnight at 4°C with murine anti-IE62 monoclonal antibody and rabbit anti-ORF9 polyclonal antibody. Cells were washed five times with 1× PBS; fluorescein isothiocyanate-labeled antirabbit antibody and Texas Red-labeled antimouse antibody (Jackson ImmunoResearch, Inc.) were added for 1 h. After five PBS washes, coverslips were mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA) and stored at 4°C in the dark. Imaging was performed with a MultiProbe 2010 laser confocal microscope.
EMSA.
The histidine-tagged USF1 protein was made as described previously (52). For probe labeling, oligonucleotides for targeting the VZV ORF10 promoter containing the wild-type (wtUSF-fw [5′-CTG TTT AAA TAG TAC CAC GTG GTA TTA TGA ACA-3′]; and wtUSF-rev [5′-TGT TCA TAA TAC CAC GTG GTA CTA TTT A-3′]) or the mutant (mUSF-fw [5′-CTG TTT AAA TAG TAC TCT ACC GTA TTA TGA ACA-3′]; mUSF-rev [5′-TGT TCA TAA TAC GGT AGA GTA CTA TTT A-3′]) sequence (the underlined sequences show the positions of the wild-type consensus and mutated nucleotide) were annealed, and 100 μg of the double-stranded oligonucleotide probes were labeled with 30 μCi of [α32P]dCTP (Perkin-Elmer) using the Klenow enzyme (Promega). Electrophoretic mobility shift assay (EMSA) was performed with a final volume of 10 μl with 5 ng of the recombinant protein and 10 fmol of [α32P]dCTP-labeled double-stranded oligonucleotide probe incubated in 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 50 μg/μl poly(dI-dC) as nonspecific competitor. Samples were incubated at 25°C for 20 min. For competition, a 10-fold or 100-fold molar excess of unlabeled wild-type or mutated double-stranded oligonucleotide was preincubated with the protein for 5 min at 25°C and then incubated with the wild-type probe for an additional 15 min. Samples were resolved on a 6% polyacrylamide gel in 0.5× Tris-borate EDTA and analyzed by autoradiography.
Immunoblot analysis of protein expression.
Whole-cell lysates were prepared by lysing infected or uninfected MeWo cells with 1 ml radioimmunoprecipitation assay buffer (50 mM Tris [pH 8], 150 mM NaCl, 1% Igepal CA-360 [Sigma, St. Louis, MO], 0.1% sodium dodecyl sulfate [SDS] [Bio-Rad, Hercules, CA], 0.5% deoxycholic acid [Sigma], and a cocktail of protease inhibitor [Roche]) per T75 flask, followed by sonication and centrifugation. Lysates were boiled in sample buffer and separated by SDS-polyacrylamide gel electrophoresis in 10% gels. Proteins were transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, Bedford, MA). The amount of total protein in 10 μl of each sample was equivalent, as verified by Bradford assay (Bio-Rad) and amido black staining. Immunoblotting was performed using rabbit polyclonal antibody to the ORF9 or ORF10 protein (kindly provided by Jeffery I. Cohen, National Institutes of Health, Bethesda, Maryland). Immunoblots of the same protein extracts was also analyzed by using anti-ORF4 polyclonal antibody as a loading control for viral proteins.
5′ and 3′ RACE.
Total RNAs extracted from POKA-infected cells with Tri-Reagent (Invitrogen, Carlsbad, CA) were subjected to 5′ and 3′ rapid amplification of cDNA ends (RACE). For 5′ RACE, a primer (5′-GCCACCTCACGATAATATCTAAG-3′) specific for the gene was used to synthesize the first-strand cDNA. A homopolymeric tail was then added to the 3′ end of the cDNA using terminal deoxynucleotidyl transferase and dCTP, and then an ORF10 gene-specific primer (5′-GAATCCTGGACCTCCTGAACG-3′) and a deoxyinosine-containing adapter primer (Invitrogen, Carlsbad, CA) were used for the next PCR. For 3′ RACE, an oligo(dT)-containing adapter primer (5′-ATGACCAATCAGATGGCAC (T)14-3′) was used to compose the first-strand cDNA; PCR was then followed with an ORF10 gene-specific primer (5′-CTGACCAGAAAGCTTACACGC-3′) and an adapter primer (5′-ATGACCAATCAGATGGCAC-3′). The resulting PCR products from 5′ and 3′ RACE were cloned and sequenced.
Luciferase reporter plasmids.
A set of luciferase reporter plasmids was constructed by using the pGL3-Basic vector (Promega). The full-length 242 bp and deletions of the ORF10-ORF11 intergenic region were amplified by PCR using primers containing NcoI sites at their 5′ end and XhoI sites at 3′ end, respectively. The pvFsp73 cosmid was used as a template. The PCR product was digested and inserted into the pGL3-Basic plasmid upstream of the firefly luciferase reporter gene. To disrupt the USF binding site or TATA box within the ORF10 promoter, two constructs with site-directed mutations were generated in a three-step PCR protocol. The first PCR was done with a 5′ primer containing an NcoI restriction site and the antisense primer containing mutated nucleotides; the second PCR used a 3′ primer containing an XhoI restriction site and a sense primer complementary to its antisense primer; the pvFsp 73 cosmid was used as a template. The resulting PCR products, which are complementary at the junction sites, were used as the template for the third PCR with 5′ and 3′ primers described above. Each mutated fragment was digested by NcoI and XhoI and inserted in the pGL3-Basic vector. ORF10 regulatory element luciferase reporter constructs were verified by sequencing. Plasmids pCMV4, pCMV10, pCMV61, pCMV62, and pCMV63, in which the VZV transactivator ORF4, ORF10, ORF61, ORF62, or ORF63 is expressed under control of the cytomegalovirus promoter, were kindly provided by P. R. Kinchington (University of Pittsburgh, Pittsburgh, PA).
Transient transfections and reporter gene assays.
MeWo cells were transfected with the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) following the manufacture's instructions. Transfections were performed with 24-well plates; 2 × 105 MeWo cells per well were seeded in 1 ml of complete growth medium 1 day before the experiment. In experiments determining the effects of VZV transactivators on ORF10 promoter activity, 0.95 μg of reporter plasmid containing the full-length ORF10 regulatory element (pFL) was transfected either alone or together with 0.02 or 0.05 μg of the pCMV4, pCMV10, pCMV61, pCMV62, and pCMV63 plasmids, and the pBluescript plasmid (Stratagene, La Jolla, CA) was added to reach one microgram of total DNA for each transfection. For the IE62 dose-dependent experiment, 0.9 μg of pFL plasmid was cotransfected with various amounts of the pCMV62 plasmid and complementary amounts of the pBluescript plasmid to equalize the total amounts of DNA to 1 μg in each transfection. In experiments mapping the ORF10 promoter boundaries and to determine essential cellular transcriptional factors, a fixed (0.05 μg) amount of pCMV62 was transfected together with 0.95 μg of each ORF10 regulatory element reporter construct. In all transfection experiments, luciferase activities were standardized to Renilla luciferase activities by using a constant amount (0.07 ng) of the plasmid pRL-TK(−) (Promega pRL-TK plasmid in which TK promoter was removed) in the transfections. Three microliters of lipofectamine reagent was used per microgram of transfected DNA in each transfection. Cells were collected 24 h after transfection and were lysed in 100 μl of passive lysis buffer provided in the dual-luciferase reporter assay system (Promega), followed by dual-luciferase assays. Dual-luciferase assays were performed using 20 μl of cell extract, 100 μl of LARII reagent, and 100 μl of Stop & Glo reagent in each assay. All transfections were performed in triplicate, and all experiments were confirmed by independent experiments. Luciferase activities are presented as n-fold induction of the measured activity over that with the promoterless reporter vector.
RESULTS
ORF10 encodes a dominant 1.3-kb transcript.
To determine the transcriptional pattern of ORF10, total RNA was extracted from POKA-infected MeWo cells for Northern hybridization. Preliminary mapping of the ORF10 transcripts was done by Northern blotting with an ORF10 RNA-specific probe (Fig. 1, top). The riboprobe spanning ORF10 nt 3 to 661 hybridized to one abundant RNA of 1.3 kb, two larger RNAs of approximately 2.6 kb and 4.4 kb, and an additional, fainter signal at approximately 3 kb. To further investigate the composition of the transcripts containing ORF10 sequence, Northern hybridizations were done with probes specific for the adjacent genes: ORF9A, ORF9, and ORF11 (Fig. 1, top). Two transcripts of about 1.7 and 3 kb were identified by the ORF9A probe. Previous transcriptional analysis had indicated that RNAs of 1.2 and 1.7 kb mapped to the ORF9 region, with the 1.7-kb RNA containing ORF9A/9 and the 1.2-kb RNA including ORF9 alone (41). In addition to the 1.2- and 1.7-kb RNAs, the probe specific for ORF9 identified two larger transcripts of about 2.6 and 3 kb in length. The probe specific to ORF11 identified a minor 4.4 kb-RNA and a major 2.8-kb RNA. No ORF10 transcripts were detected in mock-infected MeWo cells (data not shown).
FIG. 1.

Transcription of ORF10 and adjacent genes in VZV-infected cells. (Top) Northern blot hybridizations of total RNA extracted from POKA-infected MeWo cells, using riboprobes specific for ORF9A, ORF9, ORF10, or ORF11, are shown. The positions of transcripts related to ORF9A, ORF9, ORF10, ORF11, and their corresponding sizes are indicated. (Bottom) Schematic diagram of VZV genomic organization from ORF8 to ORF12. The directions of transcription from these ORFs are indicated by arrows. Lines below the diagram of the ORF8-to-ORF12 segment of the VZV genome represent transcripts detected for ORF9A, ORF9, ORF10, and ORF11 and their approximate sizes. Transcripts generated from the ORF9A, ORF9, ORF10, and ORF11 promoters are indicated as groups a, b, c, and d. The bold line represents the major ORF10 transcript.
These hybridization results showed that the 1.3-kb transcript, which is consistent with the expected size of the ORF10 gene transcript, is detected by the ORF10 probe but not by the ORF9A, ORF9, or ORF11 probe and is the dominant ORF10 product. It originates between the ORF9 stop and ORF10 start codons and terminates within the ORF10-ORF11 intergenic region (Fig. 1). The 4.4-kb transcript hybridized with the ORF10 and ORF11 probe but not the ORF9A and ORF9 probes and represents a bicistronic transcript that includes ORF10 and ORF11; this transcript could be 5′-coterminal with the 1.3-kb ORF10 transcript and 3′-coterminal with the ORF11 transcript (Fig. 1). The 3-kb read-through RNA detected by the OR9A, ORF9, and ORF10 probes spans ORF9A, ORF9, and ORF10 and appears to start from the ORF9A mRNA initiation site and stop at the ORF10 mRNA termination point (Fig. 1). In addition, the 2.6-kb RNA is recognized by probes specific for ORF9 and ORF10 but not by the ORF9A and ORF11 probes, begins at the ORF9 mRNA start site, and terminates at same point as the 1.3-kb and 3-kb RNAs (Fig. 1). The ORF11 probe recognized a 2.8-kb transcript initiated within the ORF11 intergenic region, in accordance with the predicted size of a 2.4-kb coding sequence for ORF11 (Fig. 1).
Determination of ORF10 RNA transcription termini.
To identify the ORF10 promoter, 5′ RACE was carried out to map the 5′ end of the 1.3-kb ORF10 mRNA. cDNA synthesized with a primer specific for a sequence located 950 bp downstream of the ORF10 translational start site was used to add homopolymeric dCTP tails at their 3′ ends and then amplified by a nested PCR with a ORF10 gene-specific primer and a deoxyinosine-containing adapter primer. A unique PCR product with an estimated size of 650 bp was isolated, cloned, and sequenced (Fig. 2a). No RACE products were observed using RNA from uninfected cells. Sequencing indicated a single transcription initiation site for ORF10 at 63 bp upstream of the ORF10 start codon.
FIG. 2.

Mapping the major ORF10 transcriptional terminus and the intergenic region of ORF10. RACE products were visualized by electrophoresis using 1% agarose gels stained with ethidium bromide. Lanes 1 in panels a and b show a 100-bp DNA ladder, and lanes 2 represent the 5′ and 3′ RACE products, respectively. (c) The major ORF10 transcripts with the 5′and 3′termini genomic locations indicated (section 1), the genomic organization of VZV ORF9 to ORF11, directions of ORFs, and positional locations of start and stop codons for ORF9 to ORF11 (section 2), and the sequence of the 242-bp ORF10 intergenic region located between the stop codon of ORF9 and the start codon of ORF10 (section 3) are shown. Potential transcriptional factor binding sites within the putative ORF10 regulatory element predicted by the computer programs MatInspector V2.2 and Motif are indicated in italics. The arrow indicates the initial transcription site; the corresponding nucleotide is referred as +1 and shown in italics.
The Northern blot data suggested that multiple transcripts are homologous to the 3′ end sequences of ORF10. To determine the 3′ end of the dominant 1.3-kb ORF10 transcript, total RNA extracted from POKA-infected MeWo cells was used to synthesize cDNA using an oligo(dT) adapter primer. The 3′ portion of the RNA was then amplified by PCR with a specific primer located from nt 607 to 627 of ORF10 and a primer composed of the same sequence as the adapter, followed by cDNA cloning and sequencing (Fig. 2b). Sequencing of 15 separate cDNA clones revealed a single termination site. This termination site is located at genomic position 13433, 42 nt distal to the ORF10 stop codon (TAA) and 13 nt downstream from a consensus polyadenylation signal (TAAATAAA). This result was consistent with the Northern blot analysis (Fig. 1) and the 5′ terminus mapping assay (Fig. 2), which identified one dominant ORF10 mRNA, 1.3 kb in size, that extends from 63 nt upstream of ORF10 ATG through 1,233 nt of ORF10. In addition, the existence of a single termination site demonstrates that the 1.3-kb ORF10 transcript is 3′ coterminal with the other two 2.6- and 3-kb RNA species, which were observed in Northern blot hybridization via the ORF9 and ORF10 probes or by the ORF9A, ORF9, and ORF10 probes, respectively.
Effects of VZV trans-activating proteins on ORF10 regulatory element reporter constructs.
Having mapped the 5′ terminus of the major ORF10 1.3-kb transcript, we were able to locate sequences that might constitute the ORF10 promoter. The sequences within the ORF10 intergenic region including the ORF10 mRNA initiation site and potential binding sites for the cellular transcription factors TATA-binding protein (TBP), Oct-1, USF, and TATA elements were identified using MatInspector V2.2 (50) and Motif (http://motif.genome.jp/) (Fig. 2). To determine whether the ORF10 regulatory element is trans activated by VZV trans-activating proteins, the full-length 242-bp fragment of the ORF10 intergenic region was inserted into the pGL3-Basic plasmid upstream of the firefly luciferase reporter gene, and the resultant construct was designated pFL. The plasmid pFL was transfected into MeWo cells, either alone or with various amounts (0.02 and 0.05 μg) of pCMV plasmids, from which expression of VZV genes 4, 10, 61, 62, and 63 was driven by the cytomegalovirus immediate-early promoter. The plasmid pRL-TK(−) constitutively expressing Renilla luciferase was added to each transfection reaction to serve as an internal control, and expression was analyzed by the dual-luciferase assay. The transient-transfection assay results showed that the basal expression level of luciferase produced by the ORF10 regulatory element was very low (<3-fold); this low level of expression did not increase when plasmids expressing IE4, ORF10, ORF61, or IE63 were cotransfected with the ORF10 promoter construct (pFL). However, when 0.02 μg and 0.05 μg of the plasmid expressing IE62 was cotransfected with this reporter construct, luciferase expression increased significantly, to 53-fold and 85-fold above the basal activity, respectively (data not shown). These results suggested that IE62 alone efficiently transactivates the ORF10 promoter. Promoter expression was not altered by the VZV transactivators IE4, IE63, ORF10 and ORF61 when expressed in the absence of IE62, but these experiments did not examine their potential effects in enhancing the IE62-dependent activation of ORF10.
Mapping boundaries of the ORF10 promoter.
To define the ORF10 promoter region and to establish its limits, a series of 5′ and 3′ regulatory element deletion constructs were generated by cloning amplified regions into a pGL3-Basic luciferase reporter plasmid (Promega) (Fig. 3a). The resulting constructs, as well as pFL and promoterless control plasmids, were transfected into MeWo cells in the presence of 0.05 μg of pCMV62. A 0.07-ng amount of the pRL-TK(−) plasmid was added to each transfection to normalize the basal and IE62-mediated luciferase activities expressed from each reporter construct. The results from the experiments using the progressively smaller fragments are presented as n-fold induction of the luciferase activity compared to that of the promoterless control in the absence of IE62, which was normalized to 1 (Fig. 3b). Deletion to −75 from the 5′ end (Fig. 3, constructs 1 to 6) or +5 from the 3′ end (Fig. 3, constructs 10 to 15) of the putative ORF10 regulatory element had little or no effect on promoter strength. However, deletion to −45 from the 5′ end (Fig. 3, constructs 7 and 8) or deletion to −8 from the 3′ end (Fig. 3, construct 16) failed to induce expression of the reporter gene. These analyses delineated the tentative boundaries of the fully active promoter to a 79-nt sequence between −75 and +5, relative to the ORF10 transcription start site.
FIG. 3.

Mapping the boundaries and cis-acting elements of the ORF10 promoter. (a) Diagram of the strategy to map the boundaries and to determine the essential cis-acting elements of the ORF10 promoter. A series of 5′and 3′ regulatory element deletions and mutations of the USF binding site or TATA3 box element were inserted into a pGL3-Basic luciferase reporter plasmid. The designations of constructs are shown at the right. The arrow indicates the transcription start site; the corresponding nucleotide is referred as +1. (b) Transient-transfection assays of ORF10 promoter reporter constructs in the presence of 0.05 μg of pCMV62. The results from luciferase assays of ORF10 promoter deletions or mutations are presented as n-fold induction of the luciferase activity relative to that of the promoterless control (control), which was normalized to 1. The results represent triplicate experiments for each construct.
Identification of cis-acting elements required for ORF10 promoter activity.
The experiments showing that the deletion to −75 from the 5′ end of the ORF10 regulatory element had no effect on promoter activity indicate that the two Oct-1 binding sites (Oct-1a and Oct-1b) and two TATA boxes (TATA1 and TATA2) located within this region (Fig. 2c) are not important for promoter activity in tissue culture. In contrast, deletion to −45, which removed the putative USF binding site, dramatically reduced reporter expression, suggesting that the USF element may be critical for the response to IE62 trans-activation. To investigate whether the USF binding site and the TATA box (designated TATA3 [Fig. 2c]) located in the minimal ORF10 promoter have a functional role, we generated constructs 9 and 10 (Fig. 3a). These constructs retained the full promoter region but had a mutated USF binding sequence (changed from CACGTG to TCTACA) or had a disrupted TATA3 binding motif (changed from TATAA to TCTCC) which eliminated the predicted TBP binding site within the minimal ORF10 promoter region. The six-base substitution in the predicted USF sequence greatly reduced reporter IE62-mediated transactivation (Fig. 3b, lane 9). In contrast, normal reporter expression was induced in the presence of the mutant TATA3 sequence, suggesting that this TATA box is not important for ORF10 promoter function (Fig. 3b, lane 10).
To examine whether the predicted USF binding sequence was capable of binding USF protein, we performed EMSA with purified recombinant USF protein and 32P-labeled double-stranded oligonucleotide probes corresponding to VZV genome sequences, nt 12030 to 12062, containing the wild-type or mutated USF site. USF bound efficiently to oligonucleotides containing the wild-type site (Fig. 4, lane II) but did not bind to the mutated USF motif (Fig. 4, lane VII). Binding of USF to the wild-type sequence was inhibited by the addition of increasing amounts of unlabeled wild-type oligonucleotide (Fig. 4, lanes III and IV) but not by the unlabeled oligonucleotides that contained the mutated USF site (Fig. 4, lane V). This analysis, together with the site-directed mutagenesis experiments, demonstrated that USF specifically binds to the consensus USF site present in the ORF10 promoter and that this USF binding element is essential for IE62-mediated promoter activity.
FIG. 4.
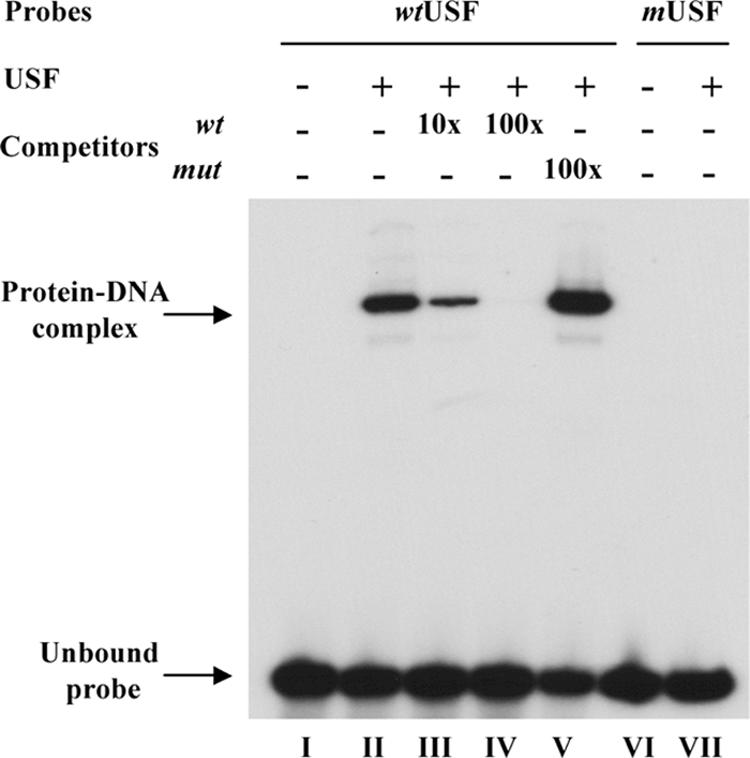
USF protein binding to the ORF10 regulatory element. Electrophoretic mobility shift assays were done to assess the effect of the six nucleotides substitutions in USF consensus sequence on USF protein binding. Wild-type oligonucleotides (wtUSF) containing the ORF10 USF consensus site or oligonucleotides with a mutated USF (mUSF) binding site were labeled and incubated with (+) or without (−) 5 ng USF protein, followed by polyacrylamide gel electrophoresis. The probes used are shown above each lane. The unlabeled wild-type oligonucleotides (wt) or mutated oligonucleotides (mut) used as competitors and the molar excesses used in the reaction are also indicated.
Generation of POKA recombinant viruses with ORF10 promoter mutations.
Based on the information obtained from mutational and deletional analyses using reporter constructs, several regions and residues that regulated promoter activity or mediated effects of cellular and viral regulatory proteins were selected for evaluation in the context of the viral genome. Two pvFsp73 cosmids, from which the regions from −87 to −97 or +39 to +63 of the ORF10 intergenic region had been deleted, were transfected along with the remaining cosmids, pvSpe14, pvPme2, and pvSpe23, into MeWo cells; these transfections yielded infectious viruses, which were designated POKA10-proΔ−87/−97 and POKA10-proΔ+39/+63 (Fig. 5A). Based on transient-transfection assays, deleting these two small regions within the ORF10 promoter should have no effect on the expression of ORF10. Transfection with the pvFsp73 cosmid from which 179 bp (−117 to +63) of the ORF10 intergenic region had been deleted, along with pvSpe14, pvPme2, and pvSpe23, also yielded infectious virus, which was designated POKA10-Δpro (Fig. 5A). A recombinant virus, POKA10-proΔUSF, was derived by transfecting pvFsp73, in which the putative USF binding motif was mutated as described for the reporter assays, together with the other intact cosmids (Fig. 5A). The deletions and the presence of the targeted mutations in the ORF10 regulatory element in these VZV recombinants were confirmed by sequencing. The replication of these four ORF10 promoter mutants was equivalent to that of POKA in a 6-day growth assay with MeWo cells (data not shown).
FIG. 5.

Construction of POKA recombinants with ORF10 promoter deletion or mutations. (A) Diagrammatic representation of POKA and recombinant viruses with ORF10 promoter mutations. Filled boxes represent the segment of the VZV genome up- or downstream of the ORF10 regulatory element, which is shown as open boxes. The gaps connected with dashes represent deletions, and the designations for the recombinant viruses are given at the left. The nucleotide numbers within the ORF10 regulatory element are as shown in Fig. 3. The location of the USF binding motif is indicated. (B) Northern blot hybridizations of total RNA extracted from POKA-infected (lane 1), POKA10-proΔ−87/−97-infected (lane 2), POKA10-Δpro-infected (lane 3), POKA10-proΔ+39/+63-infected (lane 4), or POKA10-proΔUSF-infected (lane 5) MeWo cells, using riboprobes specific to ORF9A, ORF9, ORF10, or ORF11, respectively. The positions of transcripts are indicated at the margins. The read-through transcripts that show increased expression in POKA10-Δpro-infected cells are indicated by asterisks. Representative Northern blots from three independent experiments are shown. The density of the bands of the read-through transcripts from POKA or POKA10-Δpro was measured with ImageJ, an image processing and analysis program. Detection of ORF4 transcripts with an ORF4 probe served as a loading control. An additional, indistinct band (lane 3) was detected with three different probes, indicating that it is nonspecific. (C) Detection of ORF10 protein in MeWo cells infected with POKA or an ORF10 promoter mutant. Lysates from cells infected with POKA, POKA10-proΔ−87/−97, or POKA10-proΔ+39/+63 express a protein of 50 kDa that reacts with antibody to ORF10; lysates from uninfected cells or cells infected with POKA10-Δpro or POKA10-proΔUSF do not contain the 50-kDa protein. IE4 protein detected by ORF4 antibody was used as a loading control for VZV proteins.
Effects of ORF10 promoter mutants on the expression of ORF10 and adjacent genes.
To determine whether the deletion or mutations of the ORF10 regulatory element affected the expression of ORF10 or its adjacent genes, Northern blotting was performed with probes specific to ORF9A, ORF9, ORF10, or ORF11. POKA10-proΔ−87/−97 had the same pattern of expression of ORF9A, ORF9, ORF10, and ORF11 as POKA (Fig. 5B, lanes 1 and lanes 2). As expected, transcripts generated from the ORF10 promoter were not detected in cells infected with POKA10-Δpro using the ORF10 or the ORF11 probe (Fig. 5B, lane 3). In addition, neither ORF9A nor ORF9 mRNAs were detected, whereas the 2.6-kb and 3-kb RNAs, representing ORF9/10 or ORF9A/9/10 read-through transcripts, were overexpressed (Fig. 5B, lane 3). These transcripts were only weakly present in POKA and the other ORF10 promoter mutants. Further, ratios of the values for the overexpressed ORF9A/9/10 and ORF9/10 RNAs to those of POKA on the same blot were calculated. Analysis of Northern blots with the ImageJ program revealed a ∼7-fold increase in read-through transcripts of ORF9A/9/10 or ORF9/10 in cells infected with POKA10-Δpro compared with those for POKA. Cells infected with POKA10-proΔ+39/+63 showed less expression of transcripts generated from the ORF10 promoter using the ORF10 probe, but the levels of other RNA species generated from the ORF9A, ORF9, and ORF11 promoters did not differ from those for POKA (Fig. 5B, lane 4). No transcripts were produced from the ORF10 promoter in cells infected with POKA10-proΔUSF, but this mutant had no effect on the expression of neighboring genes, ORF9A, ORF9, and ORF11 (Fig. 5B, lane 5).
Immunoblots using rabbit antiserum against ORF10 protein showed expression of a protein of ∼50 kDa in lysates of cells infected with POKA, POKA10-proΔ−87/−97, and POKA10-proΔ+39/+63 (Fig. 5C, lanes 1, 2, and 4) but not in cells infected with POKA10-Δpro or POKA10-proΔUSF (Fig. 5C, lanes 3 and 5). All of the cell lysates had equivalent levels of VZV IE4 protein (Fig. 5C). Taken together, these results demonstrated that removing a 179-bp region of the ORF10 regulatory element or disrupting the USF motif within the ORF10 promoter region is lethal for ORF10 promoter function.
Functional assay of gene-combining read-through transcripts.
Immunoblots with ORF9 antibody were used to determine whether POKA10-Δpro, which lacks the major ORF9 RNA but expresses increased amounts of read-through ORF9/10 transcripts, was capable of producing ORF9. Rabbit antiserum against the ORF9 protein recognized a polypeptide of approximately 40 kDa in POKA- and POKA10-Δpro-infected cells (Fig. 6A), although less ORF9 protein was expressed in cells infected with POKA10-Δpro than in those infected with POKA, suggesting diminished translation of ORF9 from the read-through transcripts. No protein was detected by immunoblotting with the ORF9 antibody in mock-infected cells (data not shown).
FIG. 6.

Expression of ORF9 and the distribution of ORF9 and IE62 in POKA- and POKA10-Δpro-infected cells. (A) Immunoblot with rabbit anti-ORF9. An approximately 40-kDa ORF9 protein was detected in both POKA- and POKA10-Δpro-infected cells. ORF4 protein detected by ORF4 rabbit polyclonal antibody was used to assess equal loading for viral proteins. (B) Distribution of IE62 and ORF9 in HELF cells infected with POKA or POKA10-Δpro. At 48 h after infection, cells were examined for ORF9 (A and D) and IE62 (B and E) by confocal microscopy. ORF9 was detected with ORF9 rabbit polyclonal antiserum and secondary fluorescein isothiocyanate-conjugated antibody (green). IE62 was detected with mouse monoclonal anti-IE62 antibody and Texas Red-labeled anti-mouse immunoglobulin G (red). Merged images are shown in panels C and F. Cell nuclei were stained with 4′, 6′-diamidino-2-phenylindole (blue).
Confocal microscopy was used to examine the effects of the reduction in ORF9 gene expression and the absence of the ORF10 protein that were observed in POKA10-Δpro-infected cells on the intracellular distribution of ORF9 and IE62. POKA10-Δpro and POKA plaque morphologies were indistinguishable, and the localization of IE62 and ORF9 was similar at 48 h in POKA- and POKA10-Δpro-infected HELF cells (Fig. 6B). In contrast to IE62, the ORF9 protein accumulated primarily in the cytoplasm, with enhanced expression surrounding nuclei.
Effects of ORF10 promoter mutants on VZV replication in skin xenografts in vivo.
When its replication was evaluated in skin xenografts in SCID mice, POKA10-Δpro +39/+63 exhibited growth characteristics typical of POKA in skin implants, with titers of 2.1 × 103 and 4.2 × 103 per implant at 12 and 19 days after infection (Fig. 7 ). In contrast, the production of infectious virus by POKA10-Δpro and POKA10-proΔUSF was decreased and delayed significantly in skin compared to that of POKA (P < 0.05). On day 12, the mean titers for POKA10-Δpro and POKA10-proΔUSF were approximately 1.0 × 102 PFU per implant. On day 19, only half of the xenografts yielded infectious virus; the mean titer in virus-positive specimens remained approximately 1.0 × 102 PFU per implant (Fig. 7). The POKA10-Δpro and POKA10-proΔUSF mutants exhibited a 10-fold decrease in replication in skin, which was equivalent to the effect that was observed with our previously characterized mutant POKAΔ10. This reduction has the biological consequence of decreasing the size of VZV skin lesions in vivo (6). This pattern was as predicted, because removing the putative ORF10 promoter or mutating the USF consensus site eliminated ORF10 protein synthesis. The expected sequences were confirmed in viruses recovered from xenografts infected with the three ORF10 promoter mutants (data not shown).
FIG. 7.
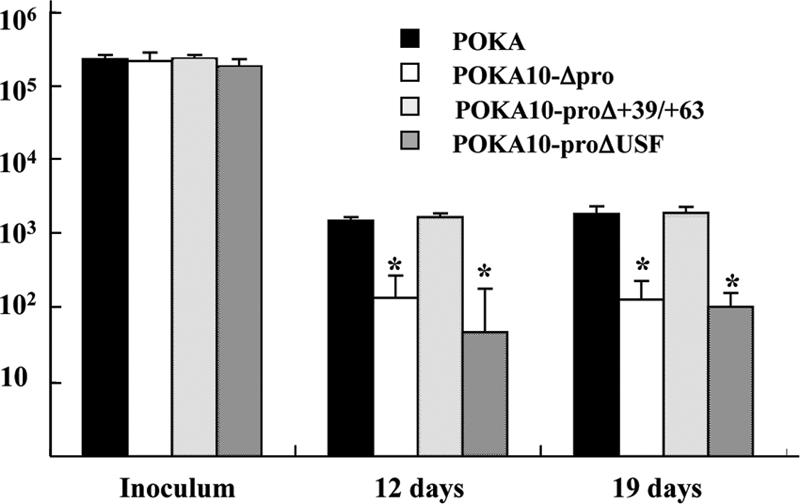
Replication of POKA or POKA ORF10 promoter mutants in skin xenografts in SCIDhu mice. Skin tissue was infected with POKA, POKA10-proΔ+39/+63, POKA10-Δpro, or POKA10-proΔUSF grown in HELF. Each bar represents the mean titer of infectious virus recovered from five to seven implants harvested at 12 and 19 days after inoculation. Inoculum titers are indicated at left. The asterisk above the error bar indicates that titers were significantly lower than those of POKA (P < 0.05) at the same time point. Error bars represent standard deviations.
DISCUSSION
In this study of VZV ORF10 and its flanking genes, we have shown that the transcription of ORF9, ORF10, and ORF11 is mono-, bi-, or polycistronic, with each of the respective monocistronic transcripts being predominant. In contrast, ORF9A, a small VZV gene which encodes an 87-amino-acid protein, shared a polyadenylation signal with its neighboring gene, ORF9, and the bicistronic transcript was dominant among ORF9A RNA species. The transcriptional patterns of ORF9A, ORF9, and ORF10, which encode at least two transcripts, indicate that theses genes are expressed within complex VZV transcriptional units, whereas we found that ORF11 encodes a single transcript.
Expression of polycistronic RNA is typical of the many gene clusters of HSV-1, pseudorabies virus, and Marek's disease virus, in which one polyadenylation site is shared by several genes (2, 24, 47, 48). Although more than one ORF is contained in the polycistronic transcripts, the monocistronic nature of eukaryotic RNAs implies that such transcripts are unlikely to be translated into protein. Our analysis of the ORF9, ORF10, and ORF11 gene cluster made it possible to investigate whether the polycistronic RNAs can play any role during VZV replication. To address this question, we generated recombinant POKAORF10-Δpro, which contains a disruption of the ORF9 transcriptional termination signal because of removal of the GT-rich region of ORF9 poly(A) motifs and a deletion of the ORF10 promoter. This VZV mutant had the capacity to produce the ORF9 protein. This result demonstrated that a bicistronic RNA is translated, albeit at a reduced level, to compensate for the loss of a monocistronic RNA during herpesvirus replication.
The VZV ORF9 protein, like its homologue in Marek's disease virus (12; K. Osterreider, personal communication), appears to be essential, whereas the related proteins in HSV-1 and pseudorabies virus, designated VP22, are not (10, 35). Therefore, one likely hypothesis is that the polycistronic transcript is translationally quiescent in lytic infection, but this transcript can be selectively translated under certain conditions, particularly when the function of the protein encoded by the ORF is essential and cannot be fully compensated by other viral proteins. Such a mechanism would ensure that enough of the ORF9 protein could be produced to allow viral replication even if the monocistronic transcript was not made.
The transcriptional initiation and termination sites for ORF10 appear to be single sites, unlike VZV ORF14 and ORF63 RNAs, which contain multiple transcriptional start or stop sites and appear to have a complex pattern of gene expression (21, 22). A single initiation site for monocistronic ORF10 RNA was identified 63 bases upstream of the ORF10 AUG, and a single unique 3′ end was located at 42 bases distal to the ORF10 stop codon or 13 bases downstream from a typical eukaryotic termination signal, TAAATAAA. The detailed mapping of the ORF10 transcripts enabled us to examine the regulatory sequences governing ORF10 transcription. Transient-transfection experiments documented that the ORF10 promoter was strongly responsive to the major VZV immediate-early transactivator, IE62, as has been observed with many other VZV genes (43). Recently studies of several VZV promoters showed that their activities induced by VZV infection didn't differ from that mediated by IE62 alone, suggesting that the synergistic effects on the promoter activations induced by VZV infection could be reflected by IE62-mediating activation (49). There was little or no response to other known VZV transactivators, including IE4, the ORF61 protein, and IE63. Thus, the ORF10 protein seems to be expressed only as a result of IE62 transactivation and does not require the enhancing activity of the other VZV transactivators, although synergistic effects of these other VZV transactivators with IE62 were not excluded.
We found that the sequence required for ORF10 promoter activity extends from up to −75 to +5, relative to its transcription initiation site. The TATA box and Oct-1 binding sites located beyond this core promoter had no role in ORF10 promoter activity. Further, altering the TATA site within the core promoter did not affect expression. However, when the USF binding site was disrupted, expression from the mutated ORF10 promoter construct was reduced significantly, indicating that USF binding is required for full IE62-mediated transactivation of the ORF10 promoter and suggesting that this USF site alone is sufficient for IE62 activation.
The finding that IE62 could activate the ORF10 promoter upon mutation of the TATA3 element was an unexpected result. Prior to this study, work by Perera et al. (34) and Yang et al. (52) showed that IE62 required a TATA element for activation of model promoters, including those containing sites for USF. Further, all of the native VZV promoters analyzed thus far have been shown to be dependent on the presence of canonical or noncanonical TATA elements for activation by IE62 (32, 43, 51). In addition, the placement of the TATA3 element relative to the USF site and start site of transcription in the ORF10 promoter is similar to that observed in the ORF28 promoter portion of the ORF28/29 regulatory element (25, 51). Thus, an as yet unidentified cis-acting sequence within the ORF10 minimal promoter appears to be required for interaction with TBP. Alternatively, IE62 and USF, both of which increase the presence of TBP at promoters and have activation domains that capture TBP in glutathione S-transferase pull-down assays (32, 52), may recruit and correctly position TBP/TFIID independently of any specific DNA element in the case of this promoter.
Since transient-transfection assays may not necessarily reflect the functional extent of viral promoters within the context of the viral genome during replication, VZV mutants were generated that had deletions or substitutions altering the ORF10 promoter. Notably, the effects of transferring the ORF10 promoter mutations into VZV recombinants were as predicted from data obtained with luciferase reporter constructs. ORF10 transcription was blocked in cells infected with POKA10-Δpro, from which the ORF10 promoter was deleted, or with POKA10-proΔUSF, in which the USF binding motif was disrupted. In contrast, a mutant with a deletion less than 30 bases beyond the ORF10 core promoter but within the intergenic region had levels of ORF10 transcription that were similar to those for POKA. None of the POKA recombinants with ORF10 promoter mutations had impaired replication in cultured cells compared to POKA, which was expected, since the ORF10 protein is dispensable for POKA replication in cultured cells (6). The amount of the ORF9 protein made by the POKA10-Δpro mutant was reduced, but VZV replication was not affected in vitro.
While ORF10 is dispensable for viral growth in vitro, it is required for efficient virion formation, viral replication, and cell-cell spread in human skin xenografts in SCIDhu mice in vivo (6). As with the full deletion of ORF10, the POKA10-Δpro promoter mutant exhibited reduced growth in skin xenografts in SCIDhu mice in vivo. The 10-fold decrease in peak titers between POKA and POKA10-Δpro was comparable to the difference observed when the ORF10 gene was deleted from POKA (6), which may indicate that the diminished synthesis of the ORF9 protein has little additional effect on the impairment of VZV replication in skin relative to that of blocking ORF10 expression alone.
The most important observation from our evaluation of viruses with ORF10 promoter mutations was that mutating a single USF binding site totally abolished IE62-dependent ORF10 promoter activity and had the same consequences for virus replication as the complete deletion of ORF10 in differentiated skin cells in vivo. Previous studies indicate that a USF site within the VZV ORF28/29 dual promoter is required for IE62-mediated activation (25, 51, 52). However, other cis elements, in addition to the USF site, must be present for full activation of the 28/29 dual promoter by IE62 in vitro. The USF site in the ORF10 promoter also appears to be more important for VZV replication than the USF site in the gI promoter (17). The gI promoter has both USF and Sp1 binding sites. A minor effect on VZV virulence in skin xenografts was observed if the USF binding site alone was disrupted, indicating that IE62 together with Sp1 was sufficient to induce enough gI expression for VZV replication in differentiated epidermal cells in vivo (17). USF is not the only cellular factor that can bind to the CACGTG (E-box) motif, which is also recognized by other transcription factors, including c-Myc (4, 14, 36) and Max/Myn (5, 37). Although we demonstrated that USF1 binds to the consensus sequence in the ORF10 promoter, it is possible that other E-box-binding proteins present in VZV-infected cells may be involved in IE62-dependent transactivation through this motif. However, no other E-box-binding cellular factors have yet been shown to be involved in this process.
Of interest, USF affects human immunodeficiency virus type 1 (HIV-1) transcription in a cell-specific manner, activating HIV-1 transcription in a T-cell line but acting as an inhibitor in epithelial cells (31). Since ORF10 is dispensable for VZV virulence in T-cell xenografts, disrupting the USF binding site in the OFR10 promoter would not be expected to alter VZV T-cell tropism (6). In contrast, our mutational analysis of the gI promoter permitted an assessment of the role of USF, because gI is required for VZV infection of differentiated human T cells (17, 29). These experiments showed that disrupting only the USF binding site in the gI promoter had no effect on VZV replication in T cells, although mutating both the USF and Sp1 sites reduced replication (17). Nevertheless, USF expression in T cells may influence the activation of the numerous other VZV promoters that are predicted to have this consensus binding site (43).
These ORF10 promoter experiments show that the cellular transcription factor USF is uniquely required as a cofactor with IE62 to produce the ORF10 protein in infected cells in vitro. Furthermore, we conclude that the cellular protein USF is a direct determinant of VZV virulence, since it is required to support normal replication of VZV in human epidermal cells within their usual microenvironment in vivo.
Acknowledgments
This work was supported by grants from the National Institute of Allergy and Infectious Diseases, AI53846 (to A.M.A.) and AI18449 (to W.T.R.).
We thank Leigh Zerboni, Jaya Rajamani, and Lee Wang of the A.M.A. laboratory for their valuable technical assistance. We thank Min Yang of the W.T.R. laboratory for preparation of the purified USF1 recombinant protein.
Footnotes
Published ahead of print on 24 January 2007.
REFERENCES
- 1.Arvin, A. M. 2001. Varicella-zoster virus, p. 2731-2767. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Baumeister, J., B. G. Klupp, and T. C. Mettenleiter. 1995. Pseudorabies virus and equine herpesvirus 1 share a nonessential gene which is absent in other herpesviruses and located adjacent to a highly conserved gene cluster. J. Virol. 69:5560-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bendall, A. S., and P. L. Molloy. 1994. Base preference for DNA binding by the bHLH-Zip protein USF: effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Res. 22:2801-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackwell, T. K., L. Kretzner, E. M. Blackwood, R. N. Eisenman, and H. Weintraub. 1990. Sequence-specific DNA binding by the c-Myc protein. Science 250:1149-1151. [DOI] [PubMed] [Google Scholar]
- 5.Blackwood, E. M., and R. N. Eisenman. 1991. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251:1211-1217. [DOI] [PubMed] [Google Scholar]
- 6.Che, X., L. Zerboni, M. H. Sommer, and A. M. Arvin. 2006. Varicella-zoster virus open reading frame 10 is a virulence determinant in skin cells but not in T cells in vivo. J. Virol. 80:3238-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, J. I., and S. E. Straus. 2001. Varicella-zoster virus and its replication, p. 2707-2730. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Cohen, J. I., and K. E. Seidel. 1994. Varicella-zoster virus (VZV) open reading frame 10 protein, the homolog of the essential herpes simplex virus protein VP16, is dispensable for VZV replication in vitro. J. Virol. 68:7850-7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohrs, R. J., M. Barbour, and D. H. Gilden. 1996. Varicella-zoster virus (VZV) transcription during latency in human ganglia: detection of transcripts mapping to genes 21, 29, 62, and 63 in a cDNA library enriched for VZV RNA. J. Virol. 70:2789-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.del Rio, T., H. C. Werner, and L. W. Enquist. 2002. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. J. Virol. 76:774-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Disney, G. H., T. A. McKee, C. M. Preston, and R. D. Everett. 1990. The product of varicella-zoster virus gene 62 autoregulates its own promoter. J. Gen. Virol. 71:2999-3003. [DOI] [PubMed] [Google Scholar]
- 12.Dorange, F., B. K. Tischer, J. F. Vautherot, and N. Osterrieder. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregor, P. D., M. Sawadogo, and R. G. Roeder. 1990. The adenovirus major late transcription factor USF is a member of the helix-loop-helix group of regulatory proteins and binds to DNA as a dimmer. Genes Dev. 4:1730-1740. [DOI] [PubMed] [Google Scholar]
- 14.Halazonetis, T. D., and A. N. Kandil. 1991. Determination of the c-MYC DNA-binding site. Proc. Natl. Acad. Sci. USA 88:6162-6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hay, J., and W. T. Ruyechan. 1992. Herpes simplex virus. Pathogenesis, immunobiology and control. Regulation of herpes simplex virus type 1 gene expression. Curr. Top. Microbiol. Immunol. 179:1-14. [PubMed] [Google Scholar]
- 16.Inchauspe, G., S. Nagpal, and J. M. Ostrove. 1989. Mapping of two varicella-zoster virus-encoded genes that activate the expression of viral early and late genes. Virology 173:700-709. [DOI] [PubMed] [Google Scholar]
- 17.Ito, H., M. H. Sommer, L. Zerboni, H. He, D. Boucaud, J. Hay, W. Ruyechan, and A. M. Arvin. 2003. Promoter sequences of varicella-zoster virus glycoprotein I targeted by cellular transactivating factors Sp1 and USF determine virulence in skin and T cells in SCIDhu mice in vivo. J. Virol. 77:489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones, J. O., M. H. Sommer, S. Stamatis, and A. M. Arvin. 2006. Mutational analysis of the varicella-zoster virus ORF62/63 intergenic region. J. Virol. 80:3116-3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaulen, H., P. Pognonec, P. D. Gregor, and R. G. Roeder. 1991. The Xenopus B1 factor is closely related to the mammalian activator USF and is implicated in the developmental regulation of TFIIIA gene expression. Mol. Cell. Biol. 11:412-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinchington, P. R., and J. I. Cohen. 2000. Viral proteins, p. 74-104. In A. M. Arvin and A. A. Gershon (ed.), Varicella zoster virus virology and clinical management. Cambridge University Press, Cambridge, United Kingdom.
- 21.Kinchington, P. R., J. P. Vergnes, P. Defechereux, J. Piette, and S. E. Turse. 1994. Transcriptional mapping of the varicella-zoster virus regulatory genes encoding open reading frames 4 and 63. J. Virol. 68:3570-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kost, R. G., H. Kupinsky, and S. E. Straus. 1995. Varicella-zoster virus gene 63: transcript mapping and regulatory activity. Virology 209:218-224. [DOI] [PubMed] [Google Scholar]
- 23.Kozlowski, M. T., L. Gan, J. M. Venuti, M. Sawadogo, and W. H. Klein. 1991. Sea urchin USF: a helix-loop-helix protein active in embryonic ectoderm cells. Dev. Biol. 148:625-630. [DOI] [PubMed] [Google Scholar]
- 24.McGeoch, D. J. 1992. Correlation between the HSV-1 DNA sequence and viral transcription maps, p. 29-47. In E. K. Wagner (ed.), Herpesvirus transcription and its regulation. CRC Press, Boca Raton, FL.
- 25.Meier, J. L., X. Luo, M. Sawadogo, and S. E. Straus. 1994. The cellular transcription factor USF cooperates with varicella-zoster virus immediate-early protein 62 to symmetrically activate a bidirectional viral promoter. Mol. Cell. Biol. 14:6896-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meier, J. L., and S. E. Straus. 1995. Interactions between varicella-zoster virus IE62 and cellular transcription factor USF in the coordinate activation of genes 28 and 29. Neurology 45:S30-S32. [DOI] [PubMed] [Google Scholar]
- 27.Michael, E., K. Kuck, and P. R. Kinchington. 1998. Anatomy of the varicella zoster virus open reading frame 4 promoter. J. Infect. Dis. 178:S27-S33. [DOI] [PubMed] [Google Scholar]
- 28.Moffat, J. F., L. Zerboni, P. R. Kinchington, C. Grose, H. Kaneshima, and A. M. Arvin. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J. Virol. 72:965-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moffat, J. F., H. Ito, M. Sommer, S. Taylor, and A. M. Arvin. 2002. Glyco-protein I of varicella-zoster virus is an essential virulence factor required for viral replication in skin and T cells. J. Virol. 76:8468-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriuchi, H., M. Moriuchi, and J. I. Cohen. 1995. Protein and cis-acting elements associated with transactivation of the varicella-zoster virus (VZV) immediately-early gene 62 promoter by VZV open reading frame 10 protein. J. Virol. 69:4693-4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naghavi, M. H., M. C. Estable, S. Schwartz, R. G. Roeder, and A. Vahlne. 2001. Upstream stimulating factor affects human immunodeficiency virus type 1 (HIV-1) long terminal repeat-directed transcription in a cell-specific manner, independently of the HIV-1 subtype and the core-negative regulatory element. J. Gen. Virol. 82:547-559. [DOI] [PubMed] [Google Scholar]
- 32.Peng, H., H. He, J. Hay, and W. T. Ruyechan. 2003. Interaction between the varicella zoster virus IE62 major transactivator and the cellular transcription factor Sp1. J. Biol. Chem. 278:38068-38075. [DOI] [PubMed] [Google Scholar]
- 33.Perera, L. P., J. D. Mosca, M. Sadeghi-Zadeh, W. T. Ruyechan, and J. Hay. 1992. The varicella-zoster virus immediate early protein, IE62, can positively regulate its cognate promoter. Virology 191:346-354. [DOI] [PubMed] [Google Scholar]
- 34.Perera, L. P. 2000. The TATA motif specifies the differential activation of minimal promoters by varicella zoster virus immediate-early regulatory protein IE62. J. Biol. Chem. 275:487-496. [DOI] [PubMed] [Google Scholar]
- 35.Pomeranz, L., and J. Blaho. 2000. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 74:10041-10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prendergast, G. C., and E. B. Ziff. 1991. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 251:186-189. [DOI] [PubMed] [Google Scholar]
- 37.Prendergast, G. C., D. Lawe, and E. B. Ziff. 1991. Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and Ras cotransformation. Cell 65:395-407. [DOI] [PubMed] [Google Scholar]
- 38.Rahaus, M., and M. H. Wolff. 1999. Influence of different cellular transcription factors on the regulation of varicella-zoster virus glycoproteins E (gE) and I (gI) UTR's activity. Virus Res. 62:77-88. [DOI] [PubMed] [Google Scholar]
- 39.Rahaus, M., N. Desloges, M. Yang, W. T. Ruyechan, and M. H. Wolff. 2003. Transcription factor USF, expressed during the entire phase of varicella-zoster virus infection, interacts physically with the major viral transactivator IE62 and plays a significant role in virus replication. J. Gen. Virol. 84:2957-2967. [DOI] [PubMed] [Google Scholar]
- 40.Roizman, B., and D. N. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. N. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 41.Ross, J., M. Williams, and J. I. Cohen. 1997. Disruption of the varicella-zoster virus dUTPase and the adjacent ORF9A gene results in impaired growth and reduced syncytia formation in vitro. Virology 234:186-195. [DOI] [PubMed] [Google Scholar]
- 42.Ruyechan, W. T., and J. Hay. 2000. DNA replication, p. 51-73. In A. M. Arvin and A. A. Gershon (ed.), Varicella zoster virus: virology and clonical management. Cambridge University Press, Cambridge, United Kingdom.
- 43.Ruyechan, W. T., H. Peng, M. Yang, and J. Hay. 2003. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 70:S90-S94. [DOI] [PubMed] [Google Scholar]
- 44.Sawadogo, M. 1988. Multiple forms of the human gene-specific transcription factor USF. II. DNA binding properties and transcriptional activity of the purified HeLa USF. J. Biol. Chem. 263:11994-12001. [PubMed] [Google Scholar]
- 45.Sawadogo, M., M. W. Van Dyke, P. D. Gregor, and R. G. Roeder. 1988. Multiple forms of the human gene-specific transcription factor USF. I. Complete purification and identification of USF from HeLa cell nuclei. J. Biol. Chem. 263:11985-11993. [PubMed] [Google Scholar]
- 46.Sirito, M., Q. Lin, T. Maity, and M. Sawadogo. 1994. Ubiquitous expression of the 43- and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res. 22:427-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan, X., P. Brunovskis, and L. F. Velicer. 2001. Transcriptional analysis of Marek's disease virus glycoprotein D, I, and E genes: gD expression is undetectable in cell culture. J. Virol. 75:2067-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner, E. K. 1985. Individual HSV-1 transcripts. Characterization of specific genes, p. 45-104. In B. Roizman (ed.), The herpesvirus, vol. 3. Plenum Press, New York, NY. [Google Scholar]
- 49.Wang, G., T. Suzutani, Y. Yamamoto, Y. Fukui, N. Nozawa, D. S. Schmid, I. Kurane, and N. Inoue. 2006. Generation of a reporter cell line for detection of infectious varicella-zoster virus and its application to antiviral studies. Antimicrob. Agents Chemother. 50:3142-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wingender, E., X. Chen, R. Hehl, H. Karas, I. Liebich, V. Matys, T. Meinhardt, M. Prüβ, I. Reuter, and F. Schacherer. 2000. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 28:316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang, M., J. Hay, and W. T. Ruyechan. 2004. The DNA element controlling expression of the varicella-zoster virus open reading frame 28 and 29 genes consists of two divergent unidirectional promoters which have a common USF site. J. Virol. 78:10939-10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, M., H. Peng, J. Hay, and W. T. Ruyechan. 2006. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J. Virol. 80:7339-7353. [DOI] [PMC free article] [PubMed] [Google Scholar]