

Abstract
JC virus (JCV)-specific CD8+ cytotoxic T lymphocytes (CTL) are associated with a favorable outcome in patients with progressive multifocal leukoencephalopathy (PML) and cross-recognize the polyomavirus BK virus (BKV). We sought to determine the frequency and phenotype in fresh blood of CD8+ T cells specific for two A*0201-restricted JCV epitopes, VP1p36 and VP1p100, and assess their impact on JC and BK viremia and viruria in 15 healthy subjects, eight human immunodeficiency virus-positive (HIV+) individuals, and nine HIV+ patients with PML (HIV+ PML patients) classified as survivors. After magnetic preenrichment of CD8+ T cells, epitope-specific cells ranged from 0.001% to 0.22% by tetramer staining, with no significant difference among the three study groups. By use of seven-color flow cytometry, there was no predominant differentiation phenotype subset among JCV-specific CD8+ T cells in healthy individuals, HIV+ subjects, or HIV+ PML patients. However, in one HIV+ PML patient studied in the acute phase, there was a majority of activated effector memory cells. BKV DNA was undetectable in all blood samples by quantitative PCR, while a low JC viral load was found in the blood of only one HIV+ and two HIV+ PML patients. JCV and BKV DNA were detected in 33.3% and 13.3% of all urine samples, respectively, independent of the presence of JCV-specific CTL. The detection of JCV DNA in the urine was associated with the presence of a JCV VP1p100 CTL response. Immunotherapies aiming at increasing the cellular immune response against JCV may be valuable in the treatment of HIV+ individuals with PML.
JC virus (JCV) is a polyomavirus that infects approximately 85% of the adult population worldwide (35). In most individuals, the virus is quiescent in the kidney or lymphoid organs, but in the setting of severe immunosuppression, such as in patients with AIDS or hematological malignancies or in organ transplant recipients, it can reactivate and spread to the central nervous system, causing a deadly demyelinating disease of the brain named progressive multifocal leukoencephalopathy (PML). The incidence of PML is 5.1% in patients with AIDS (26) and 3.3% in patients with hematological malignancies (12). Although there is no specific treatment for this condition, the immune reconstitution induced by highly active antiretroviral therapy (HAART) in human immunodeficiency virus (HIV)-infected patients has improved the survival from 10 to 50% in this population (17).
In previous studies, we have shown that the detection of peripheral blood CD8+ cytotoxic T lymphocytes (CTL) against HLA-A*0201-restricted JCV epitopes VP1p36 (8) and VP1p100 (19) after peptide stimulation and culture for 10 to 14 days is associated with a better outcome in PML patients. The presence of these cells early after PML onset had an 87% predictive value for disease stabilization (9). We have also detected JCV-specific CTL in the blood of most healthy individuals after in vitro stimulation, suggesting that these cells might play a role in the control of the viral replication and the protection against the development of PML in immunocompetent people (10). In these same studies, the frequency of JCV-specific CTL was estimated to be very low in fresh blood. We were able to detect JCV VP1p36 tetramer-positive CD8+ T cells in two HIV-infected patients with PML (HIV+ PML patients) and only in 2/10 healthy individuals. We therefore used a CTL sorting technique and showed that the frequency of these cells ranged from 1/2,494 to less than 1/100,000 peripheral blood mononuclear cells (PBMC). In one healthy individual, the frequency of JCV VP1p100 tetramer-positive CD8+ T cells was 1/75,200 PBMC. However, this technique required the use of large quantities of tetramer reagent and therefore was costly and not suitable for high-throughput testing.
The need for the development of a better method for detection of CTL in fresh blood was also spurred by our improved understanding of the cellular immune response against polyomaviruses. BK virus (BKV) is a human polyomavirus that has 75% sequence homology with JCV and causes nephropathy in kidney transplant recipients. Indeed, we and others have recently demonstrated that CTL specific for the HLA-A*0201-restricted JCV VP1p36 epitope cross-reacts with the BKV VP1p44 epitope and that the HLA-A*0201-restricted JCV VP1p100 epitope cross-reacts with the BKV VP1p108 epitope (5, 20, 30), suggesting that this immune response against both human polyomaviruses may be mediated by the same CTL populations.
To gain a better understanding of the dynamics of this cellular immune response, we sought to determine the differentiation profile of CD8+ T lymphocytes. In the case of JCV infection, the very low frequencies of virus-specific CTL make this a difficult task. Additionally, the determination of the phenotype is not reliable in antigen-stimulated CD8+ T lymphocytes after culture in vitro (13).
To address these challenges, we devised a more sensitive tetramer staining assay, using preenriched CD8+ T cells followed by seven-color flow cytometry. With this method, we determined the differentiation phenotype of CTL and measured the frequency of these cells in fresh blood samples of HIV-infected patients with and without PML and in samples from healthy control subjects. To better understand the role of the cellular immune response in the containment of both human polyomaviruses, we also correlated the presence of JCV VP1p36- and VP1p100-specific CD8+ T cells with detection of JCV and BKV DNA in urine and peripheral blood.
MATERIALS AND METHODS
Study subjects.
We enrolled a total of 32 HLA A*0201+ subjects in this study, including 15 healthy individuals aged 23 to 44 years (32.5 ± 6.3 years [mean ± standard deviation]), eight HIV-infected patients aged 38 to 68 years (51.3 ± 10.3 years), and nine HIV-infected patients with PML aged 36 to 47 years (41.7 ± 4.5 years). Eight PML patients were classified as survivors based on a recent consensus on terminology of PML (6) and were still alive an average of 7 years (2 to 12 years) after being diagnosed with PML. One patient was enrolled 3 weeks after the onset of PML, and samples of blood were collected prospectively over 19 weeks. He continues to improve 11 months after initial diagnosis. The mean CD4+ T-cell count was 358 (±254)/mm3 in the HIV+ group and 414 (±245)/mm3 in the HIV+ WPML group. We enrolled four HLA A*0201− healthy individuals as controls to assess the background of tetramer staining after magnetic sorting of CD8+ T cells and to determine the lower level of detection of this assay. All participants had detectable JCV and BKV antibodies in the blood. Informed consent was obtained according to institutional guidelines.
Major histocompatibility complex class I typing.
The major histocompatibility complex class I alleles expressed by the study subjects were determined using standard serologic tissue-typing procedures. Molecular analyses to determine HLA-A*02 subtypes were performed on all study subjects.
Hemagglutination inhibition assay.
Human serum antibody titers against BKV and JCV were determined by hemagglutination inhibition assay as described elsewhere (23).
PBMC preparation.
Between 2 × 107 and 2 × 108 PBMC were isolated from 30 to 40 ml of blood by centrifugation over a Ficoll-diatrizoate gradient. These cells were used for magnetic enrichment of CD8+ T cells or were cultured for 10 to 14 days in the presence of JCV CTL epitope peptides.
Magnetic enrichment of CD8+ T cells.
An enriched CD8+ T-cell population was obtained through negative selection using magnetic beads. Briefly, 2 × 107 to 8 × 107 PBMC were resuspended in cold MACS buffer (phosphate-buffered saline [PBS], 0.5% fetal calf serum [FCS], 2 mmol EDTA) and stained with a combination of biotin-labeled antibodies (CD8+ isolation kit II; Miltenyi Biotec) according to the manufacturer's instructions. Cells were incubated for 10 min at 4°C, and paramagnetic microbeads coated with monoclonal mouse antibiotin antibodies were added. After 15 min of incubation at 4°C, cells were washed and resuspended in 500 μl of MACS buffer. By use of this technique, 2 × 106 to 2 × 107 CD8+ T cells were negatively selected.
Stimulation of PBMC by CTL epitope peptides of JCV.
Three million PBMC were stimulated with HLA-A*0201-restricted nonamer epitope peptides of the JCV VP1 protein, VP1p36-SITEVECFL or VP1p100-ILMWEAVTL, at final concentrations of 2.5 μg/ml and 1 μg/ml, respectively. Cells were cultured in RPMI 1640-12% FCS in a well of a 24-well plate. After 72 h, an equal volume of medium containing 50 U of recombinant interleukin-2 was added to each culture well, and every 2 days thereafter, half of the medium was replaced. After 10 to 14 days, the frequency of JCV VP1p36 or JCV VP1p100 tetramer-positive cells was determined.
Staining and phenotypic analysis of JCV VP1p36- or JCV VP1p100-specific CD8+ T cells by seven-color flow cytometry.
The CD8+ T-cell-enriched fraction (2 × 106 to 2 × 107 cells) was washed in PBS with 2% FCS and centrifuged for 5 min at 300 × g. The pellet was resuspended in PBS using 100 μl/106 cells and stained with phycoerythrin-coupled tetrameric HLA-A*0201/VP1p36 or tetrameric HLA-A*0201/VP1p100. After 15 min of incubation at room temperature, the cells were stained with a combination of anti-CD3, -CD8α, -CCR7, -CD45RA, -CD38, and -HLA-DR and incubated for additional 15 min. Then, the lymphocyte samples were washed in PBS with 2% FCS and centrifuged for 5 min at 300 × g. The supernatants were decanted, and cells were resuspended in 0.5 ml of PBS containing 1.5% paraformaldehyde. Samples were analyzed on an LSR II flow cytometry system (BD Biosciences).
In addition, 5 × 105 PBMC cultured for 10 to 14 days were stained with the same tetramers. Flow cytometry analysis was performed as described above. Samples were analyzed on a FACSCalibur flow cytometry system (Becton Dickinson). Results were considered positive when tetramer-stained CD8+ T cells formed a distinct population and their number was ≥0.1%. Data presentation was performed using FlowJo software (Tree Star, Inc.).
Quantitative PCR detection for BKV and JCV DNA in the study subjects.
Quantitative real-time PCR (qPCR) was used to measure BKV and JCV viral loads in the plasma, PBMC, and urine of the study subjects. We used a QIAamp DNA blood mini kit (QIAGEN) to extract DNA from plasma, urine, or PBMC, following the instructions of the manufacturer and analyzing results in duplicate.
For JCV PCR, we used primers that represent nucleotides (nt) 4298 to 4320 (5′-AGAGTGTTGGGATCCTGTGTTTT-3′) and 4375 to 4352 (5′-GAGAAGTGGGATGAAGACCTGTTT-3′) of the large T gene sequence of JCV strain Mad 1, while for BKV PCR we used primers that represent nucleotides 2217 to 2241 (5′-TGCTAGGTATTTTGGGACTTTCACA-3′) and 2320 to 2302 (5′-GCCCCACACCCTGTTCATC-3′) of the VP1 gene sequence of BKV (Dunlop strain). Each 25-μl reaction mixture contained 400 nM of forward and reverse primers for JCV or 200 nM for BKV, 100 nM of JCV-specific probe (nt 4323 to 4350) (5′-6-carboxyfluorescein-TCATCACTGGCAAACATTTCTTCATGGC-6-carboxytetramethylrhodamine-3′) or BKV-specific probe (nt 2255 to 2283) (5′-6-carboxyfluorescein-TTCCCCCAGTACTTCATGTGACCAACACA-6-carboxytetramethylrhodamine-3′), and Rox reference dye and TaqMan universal PCR master mix (Applied Biosystems). Cycling conditions for both JCV and BKV qPCR were a first 50°C, 2-min step to decontaminate any possible carryover contamination and a 95°C, 10-min step to denature the template DNA and activate the polymerase, followed by 40 cycles consisting of 15 s at 95°C for denaturation and 1 min at 60°C for annealing and elongation. Plasmid DNA containing the BKV or JCV genome served to generate a standard curve against which samples were analyzed using 7300 system software (Applied Biosystems). All qPCR results were calculated as copy number per ml. The detection cutoff of the assay was 500 copies/ml of plasma and urine or 10 copies/μg of PBMC DNA.
Statistical analysis.
Categorical and numeric variables were compared using Fisher's test and the Mann-Whitney U test, respectively. All tests were two-tailed, and α of 0.05 was employed.
RESULTS
JCV VP1p36- and JCV VP1p100-specific CD8+ T cells are present in very low frequencies in the peripheral blood.
All study subjects were seropositive for JCV and BKV. Using preenrichment of a CD8+ T-cell fraction with paramagnetic beads followed by tetramer staining, we could detect a very low number of JCV-specific CTL in the fresh blood of the study subjects. To establish the background level of tetramer staining and identify the lower level of detection of this assay, we analyzed samples from four HLA-A*0201-negative individuals. In all, the frequency of tetramer-positive cells was lower than 0.001% CD8+ T cells by use of an LSR II flow cytometry analyzer (data not shown). Similarly, in most HLA-A*0201+ subjects, when the frequency of JCV VP1p36 or JCV VP1p100 tetramer-positive cells was lower than 0.001% in the unstimulated CD8+ T-cell-enriched fraction (Fig. 1A), negligible tetramer-positive cells were observed after subsequent in vitro peptide stimulation for 10 to 14 days (Fig. 1B). Conversely, when the frequency of tetramer-positive cells was greater than 0.001% in the unstimulated CD8+ T-cell-enriched fraction (Fig. 1C), a distinct population of tetramer-positive cells could always be detected after in vitro expansion (Fig. 1D). By use of this cutoff, a clear subpopulation of JCV-specific cells was identified in 29 of the CD8+ T-cell-enriched fresh blood samples of HLA A*0201+ study subjects by tetramer staining for either JCV VP1p36 or JCV VP1p100 (Table 1). After 10 to 14 days of peptide stimulation in vitro, we detected JCV-specific CTL in 38 PBMC samples for these epitopes (Table 1). Since in four of these samples, fresh blood tetramer staining could not be performed because of technical reasons, there were 34 tetramer-positive samples after in vitro stimulation for either JCV VP1p36 or JCV VP1p100 that were paired with fresh blood tetramer staining for the corresponding epitopes. Therefore, the sensitivity of the tetramer staining assay performed on CD8+ T-cell-enriched fresh blood samples was 85.2% (29/34). Since all positive fresh blood samples also had detectable JCV-specific CTL after in vitro culture, the specificity of this test in fresh samples was 100% (Table 1). Altogether, peptide stimulation allowed for an average 3.1 log in vitro expansion (range, 1.24 to 3.93) of tetramer staining cells. A JCV VP1p36- and/or JCV VP1p100-specific CTL population was detected in the peripheral blood of 9/15 (60%) healthy individuals, 4/8 HIV-infected subjects (50%), and 7/9 (77.7%) HIV+ PML patients. JCV VP1p36-specific CTL were found in 16/32 (50%) study subjects, and their frequency ranged from 0.001 to 0.022% of total CD8+ T-cell population, while JCV VP1p100 CTL were found in 13/24 (54.2%) study subjects tested, and their frequency ranged from 0.0014 to 0.015%. We did not find any significant difference between frequencies of either JCV VP1p36- or JCV VP1p100-specific CD8+ cells among the three study groups (Fig. 2).
FIG. 1.

Threshold of detection of JCV-specific CD8+ T lymphocytes by tetramer staining of the CD8+-enriched fraction of fresh blood samples from HLA A*0201+ study subjects. When the frequency of tetramer staining cells was lower than 0.001% in the CD8+-enriched fraction (A), no expansion was observed after peptide stimulation (stim.) and culture for 10 to 14 days (<0.1%) (B). If the frequency of JCV-specific cells was greater than 0.001% in the CD8+-enriched fraction (C), a clear population could always be identified after stimulation (D). The percentage of all CD8+ T cells that bind the tetramer is indicated in each panel.
TABLE 1.
Frequencies of JCV VP1p36- and VP1p100-specific CD8+ T-cell-enriched fresh blood samples and cultured PBMC after peptide stimulation
| Subject | % JCV VP1p36-specific CD8+ T cells
|
% JCV VP1p100-specific CD8+ T cells
|
||
|---|---|---|---|---|
| Fresh blood | After peptide stimulation | Fresh blood | After peptide stimulation | |
| Healthy control no. | ||||
| 1 | 0.0026 | 2.6 | —a | 47.1 |
| 2 | — | — | 0.0014 | 7.1 |
| 3 | — | — | NAb | — |
| 4 | — | — | 0.0028 | 0.8 |
| 5 | 0.0048 | 3.8 | 0.0075 | 9.3 |
| 6 | — | — | NA | — |
| 7 | 0.0062 | 12.2 | — | — |
| 8 | 0.003 | 25.4 | NA | 19.3 |
| 9 | 0.0098 | 4.7 | 0.0056 | 1.7 |
| 10 | — | — | — | 7.8 |
| 11 | — | — | — | — |
| 12 | — | — | — | — |
| 13 | — | — | 0.0057 | 4.2 |
| 14 | — | — | — | — |
| 15 | 0.007 | 20.9 | — | — |
| HIV+ patient no. | ||||
| 1 | 0.013 | 21.9 | 0.0017 | 1.6 |
| 2 | 0.0068 | 12 | NA | 2.8 |
| 3 | 0.001 | 0.1 | 0.0053 | 25.4 |
| 4 | — | 1 | NA | NA |
| 5 | — | — | — | — |
| 6 | — | — | NA | — |
| 7 | 0.0027 | 5.3 | 0.0075 | 0.1 |
| 8 | — | — | — | — |
| HIV+ PML patient no. | ||||
| 1 | 0.0013 | 0.6 | NA | 5.1 |
| 2 | 0.0043 | 3.9 | NA | 26.5 |
| 3 | 0.022 | 3.4 | 0.015 | 2.1 |
| 4 | 0.009 | 22.3 | 0.002 | 1.3 |
| 5 | — | — | 0.0035 | 10 |
| 6 | — | — | — | — |
| 7 | 0.0014 | 7.2 | 0.0091 | 29.1 |
| 8 | — | 0.2 | — | 29.4 |
| 9 | 0.011 | 37.9 | 0.011 | 18.2 |
—, undetectable.
NA, not available.
FIG. 2.
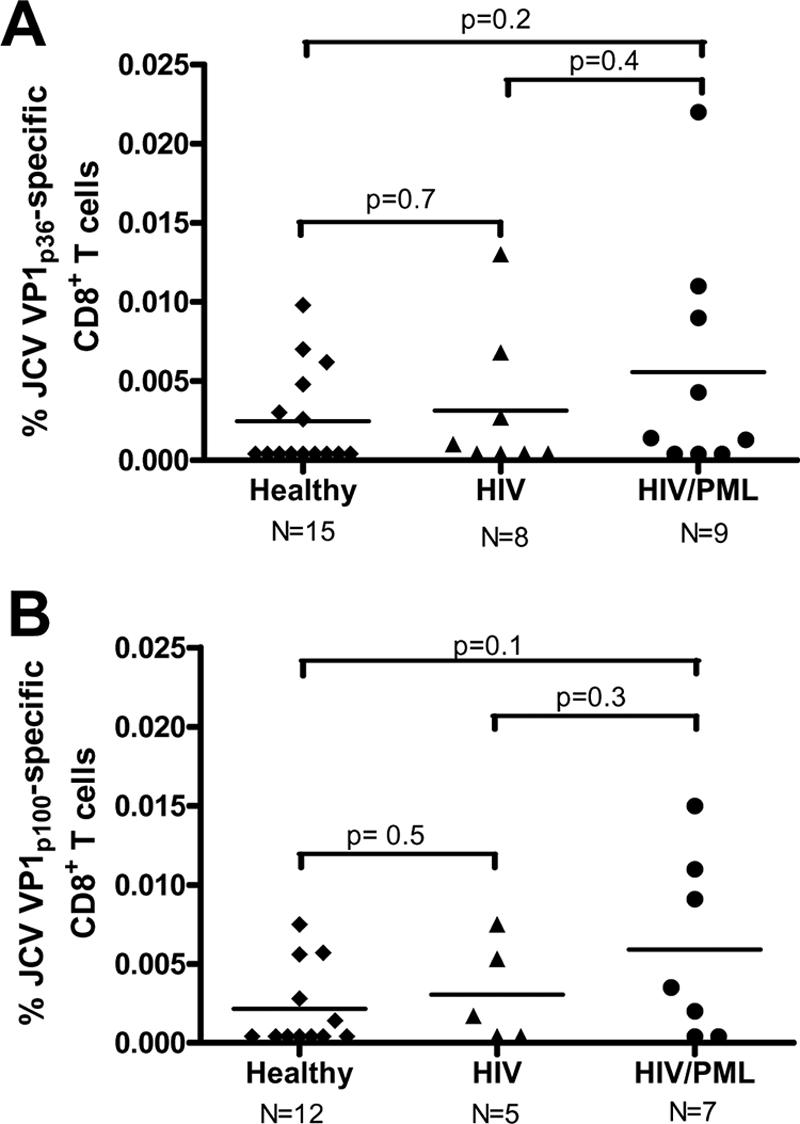
Frequencies of JCV VP1p36-specific (A) and JCV VP1p100-specific (B) CD8+ T cells in healthy control subjects, HIV+ individuals, and HIV+ PML patients. There was no significant difference in results among the three groups.
Phenotypic analysis of JCV-specific CD8+ T-cell populations in peripheral blood.
On the basis of expression of the cell surface markers CCR7 and CD45RA, four subsets of JCV-specific CD8+ T cells have been defined: CCR7+ CD45RA+ naïve T cells (TN cells), CCR7+ CD45RA− central memory T cells (TCM cells), CCR7− CD45RA− effector memory T cells (TEM cells), and CCR7− CD45RA+ effector or effector memory RA+ T cells (TEMRA cells) (29). In healthy subjects or HIV-infected patients without PML, there was not a predominant subset of JCV-specific CD8+ T cells in the peripheral blood. CCR7+ CD45RA+, CCR7+ CD45RA−, and CCR7− CD45RA− T cells were present in similar frequencies in both groups (Fig. 3). However, in HIV+ PML patients with long-term disease, CCR7− CD45RA− TEM cells accounted for a large proportion of JCV VP1p36-specific (49.8%) and VP1p100-specific (45.5%) cells, although there was no significant difference compared to results for the other study subject groups (data not shown). CCR7− CD45RA+ TEMRA cells were present only in very low numbers (<10%) in all three groups. Expression of activation marker HLA-DR or CD38 was not observed in JCV-specific CD8+ T cells in the three study populations, except for a PML patient enrolled 3 weeks after the onset of symptoms, as described below.
FIG. 3.

Phenotypic analysis of JCV VP1p36-specific (A) and JCV VP1p100-specific (B) CD8+ T-cell subsets. There was no predominant phenotype in the healthy control subjects, HIV+ individuals, or HIV+ PML patients. Error bars represent standard errors of the means.
JCV VP1p36-specific CD8+ T cells from an HIV+ patient with PML during the early phase of disease display a CCR7− CD45RA− phenotype and express activation markers.
To study the initial dynamics of the CD8+ T-cell response against JCV, we determined the frequency and phenotype of JCV VP1p36-specific CD8+ T cells prospectively in one HIV-infected individual with PML during the early phase of disease. This patient was a 38-year-old gentleman who presented with a subacute history of short-term memory problems and right hemiparesis 7 months after being diagnosed with HIV. His initial CD4+ T-cell count was 1/mm3, and his HIV RNA level was 7,830 copies/ml. He was treated with a combination of zidovudine-lamivudine (Combivir) and lopinavir-ritonavir (Kaletra), and his viral load declined to <50 copies/ml. At PML onset, his CD4+ T-cell count was 84 cell/mm3 and the plasma HIV RNA level was still undetectable. A magnetic resonance imaging scan of the brain showed multiple hyperintense lesions in T2-weighted and FLAIR sequences located in the white matter of both hemispheres. JCV DNA was detected in the cerebrospinal fluid, and a diagnosis of PML was made.
The frequency of JCV VP1p36 tetramer-positive cells in this patient's blood was the highest among those of all study subjects. Three weeks after the onset of symptoms, the frequency was 0.022% of CD8+ T cells. JCV DNA was detected in his plasma at 4.6 × 103 copies/ml. Two weeks later, the frequency peaked at 0.044% and declined thereafter (Fig. 4A). The patient continued on HAART and was started on the 5HT2A receptor antagonist mirtazapine at 15 mg/day, since it has been shown that this receptor is involved in the infection of glial cells by JCV (11). This patient made an excellent recovery. By 19 months after disease onset, the CD4+ T-cell count rose to 237 cells/mm3 and the HIV viral load remained undetectable. His neurological exam showed only a mild right-leg weakness.
FIG. 4.

(A) Longitudinal study of the frequency of JCV VP1 p36-specific CD8+ T cells in fresh blood of an HIV+ patient recently diagnosed with PML. (B) The CCR7− CD45RA− effector memory (TEM) phenotype of CD8+ T cells was predominant in the early phase of disease (top row). After 3 weeks from the onset of symptoms, we observed a strong expression of activation markers CD38 and HLA-DR, which gradually disappeared over the next few months. Percentages of CD8+ cells stained with the various markers are indicated in the panels.
Interestingly, the great majority (93.7%) of his JCV VP1p36 tetramer-positive CD8+ T cells were CCR7− and CD45RA− (Fig. 4B). This phenotype is usually associated with effector memory T cells. At the first evaluation, JCV VP1p36-specific CD8+ T cells expressed activation marker CD38 and HLA-DR molecules on their surface, suggesting that these cells were highly activated during JCV reactivation (34). Over the first 4 months of the disease, the expression of both molecules gradually disappeared (Fig. 4B), concomitant with clinical improvement.
Relationship between JCV and BKV replication in blood and urine and presence of JCV VP1p36- or JCV VP1p100-specific CD8+ cells.
We sought to investigate a possible correlation between the presence of JCV-specific CD8+ T cells and viral replication in blood and urine. Using a quantitative assay, we detected JCV DNA in urine samples of 4/15 healthy individuals (range, 7.18 × 102 to 1.48 × 106 copies/ml), 3/6 HIV-infected subjects (range, 5.72 × 103 to 5.72 × 107 copies/ml), and 3/9 PML patients (range, 5.3 × 102 to 6.58 × 104 copies/ml) (Table 2). Urine samples were not available for two HIV-infected patients. Nine out of 10 patients (90%) with urinary JCV excretion had a detectable JCV VP1p100-specific cellular immune response in their blood, while these CTL could be found in only 10/20 (50%) subjects with no JCV viruria (P = 0.049). There was no significant difference in the presence of JCV VP1p36-specific CTL between those subjects with JCV excretion in urine (6/10) and those without (10/20) (P = 0.7).
TABLE 2.
JCV and BKV urinary excretion in different study groups
| Study group | No. of patients with viruria/total no. of patients (%)
|
|
|---|---|---|
| JCV DNA in urine | BKV DNA in urine | |
| Healthy controls | 4/15 (26.6) | 2/15 (13.3) |
| HIV+ patients | 3/6 (50) | 1/6 (16.6) |
| HIV+ PML patients | 3/9 (33.3) | 1/9 (11.1) |
| Total | 10/30 (33.3) | 4/30 (13.3) |
BKV DNA was found in the urine samples of 2/15 (12.9%) healthy, 1/6 (16.6%) HIV-infected, and 1/9 (11.1%) HIV-infected PML subjects (Table 2). BKV viral load ranged from 1.4 × 103 to 5 × 104 copies/ml. Concomitant JCV and BKV urinary excretion was not observed with any of the individuals. Two out of four subjects with urinary BKV excretion had a detectable cellular immune response against both HLA A*0201-restricted JCV epitopes. No particular patterns of CD8+ T-cell differentiation or presence of activation markers were noticed in these two subjects.
Finally, JCV DNA was found in the plasma of three of the study subjects. In one PML patient, 4.6 × 103 copies/ml were detected, whereas very low copy levels were found in the PBMC samples of an HIV-infected individual (22.7 copies/μg DNA) and another PML patient (39.6 copies/μg DNA). All three subjects had JCV VP1p36- and/or JCV VP1p100-specific cells detected in the peripheral blood. BKV DNA was not found in the plasma or PBMC samples of any of the study subjects.
DISCUSSION
Since phenotypic changes are commonly observed to occur in T cells after culture, we devised a method to study the dynamics of the immune response against JCV directly in peripheral blood. Preenrichment of the CD8+ T-cell population via immunomagnetic beads followed by tetramer staining had a high sensitivity (85.2%) and specificity (100%) compared to results from the tetramer staining of PBMC after peptide stimulation and expansion of these cells in vitro for 10 to 14 days. This method, along with seven-color flow cytometry, allowed us to identify JCV-specific CD8+ T cells and characterize their differentiation profile in most study subjects.
Interestingly, compared to results for other persistent viral infections, JCV-specific CTL were found in considerably low frequencies. Indeed, whereas CD8+ T cells against Epstein-Barr virus (32), cytomegalovirus (31), and HIV (14) are present in frequencies ranging from 0.1 to 5.8% of total CD8+ T lymphocytes, the percentage of JCV-specific cells was much lower. This low magnitude of the CD8+ T-cell response against JCV could be explained by the pattern of viral replication and the anatomical sites of viral latency. In most individuals, primary infection happens during childhood and the virus subsequently remains latent or replicates intermittently in the kidney or the lymphoid organs (25). Since viremia is not usually found in immunocompetent individuals (18), low-level antigen synthesis or inefficient antigen presentation at these sites may lead to a limited CD8+ T-cell response. However, since JCV is not very virulent and causes PML only in the setting of severe immunosuppression, a small virus-specific memory cell population may be sufficient to prevent widespread viral replication in healthy individuals and most HIV-infected patients. During viral reactivation, based on the response observed to occur in a patient recently diagnosed with PML, there may be a marked increase of this CD8+ T-cell pool and a switch to an effector memory phenotype, leading to disease stabilization and marked clinical improvement.
We did not find differences among the frequencies of JCV-specific CD8+ T cells in fresh blood samples in the three groups studied. Although HIV infection is the major predisposing condition for the development of PML (36), in our study, the HIV-infected group had only a modest decrease of CD4+ T-cell counts and a relatively preserved immune system, whereas all of the PML patients had a good outcome and most of them had inactive disease. However, it is also possible that PML patients had a redistribution of CD8+ T cells in their central nervous system, since we have found these cells in cerebrospinal fluid (7) and brain (38) in previous studies.
Recently, different subsets of memory CD8+ T cells have been identified based on the expression of CCR7, a chemokine receptor that controls homing to secondary lymphoid organs, and CD45RA, a transmembrane phosphatase that regulates signaling through the T-cell receptor-CD3 complex (34). CCR7+ CD45RA+ TN cells are naïve T cells that have not yet been antigen stimulated, CCR7+ CD45RA− TCM cells are central memory T cells that home to the secondary lymphoid organs and readily proliferate and differentiate into effector cells in response to antigenic stimulation, CCR7− CD45RA− TEM cells are effector memory T cells that migrate to inflamed peripheral tissues and can display immediate effector functions, and CCR7− CD45RA+ TEMRA cells are effector memory cells that express the highest levels of perforin and correspond to terminally differentiated effector cells (28, 29).
The analysis of the CD8+ T-cell differentiation profile in the healthy individuals and HIV-infected patient populations did not reveal a predominant cell subset. CCR7− CD45RA− TEM, CCR7+ CD45RA− TCM, and CCR7+ CD45RA+ TN cells were seen in similar numbers among the three groups. Although the CCR7+ CD45RA+ pool traditionally corresponds to a TN phenotype, it might actually include antigen-experienced cells. Geginat et al. have observed that cytokine-stimulated TCM cells acquire CD45RA expression without losing CCR7, adopting an apparent naïve phenotype (13). Interestingly, in one PML patient with early disease, the TCM subset corresponded to more than 90% of the JCV-specific CTL. Although we cannot make firm conclusions based on this single case, this dramatic increase might suggest that these cells play a role in the acute fight against JCV; however, this needs to be confirmed in other individuals. Indeed, this patient has made a remarkable recovery, and 19 months after disease onset, the patient has only minimal neurological sequelae.
There is considerable heterogeneity in the extents of differentiation of the CD8+ T-cell populations in response to viral infections (2, 37). While circulating cytomegalovirus-specific CD8+ T cells are terminally differentiated TEMRA cells, HIV-specific CD8+ T cells are largely CCR7− CD45RA− TEM cells, suggesting that HIV may interfere with T-cell differentiation and an effective immune response (4). In our study, we found a profile similar to that of HIV-specific CD8+ T-cell infection, with low percentages of TEMRA cells, even in a patient soon after the diagnosis. Whether this represents a problem in the T-cell differentiation or may reflect a particular differentiation state associated with JCV infection remains to be determined. Since we did not study JCV-specific CD8+ T-cell differentiation at the sites of active viral replication, such as in the brain of PML patients, we also cannot exclude the possibility of the presence of terminally differentiated CCR7− CD45RA+ TEMRA cells in the central nervous system.
We and others have demonstrated a cross-reactivity between the CTL epitopes JCV VP1p36 and JCV VP1p100 and BKV VP1p44 and BKV VP1p108 (5, 20, 30), respectively, indicating that the same population of CD8+ T cell could be functionally active against these two viruses. In the present study, we had the opportunity to follow prospectively in the early phase of disease one PML patient who had the highest JCV-specific CTL response among all study subjects and whose virus-specific CD8+ T cells expressed activation markers HLA-DR and CD38, which are usually seen at the time of viral replication (34). Since this patient had active PML and low-level JC viremia and since BKV DNA was not detected in his blood and urine samples, this suggests that the VP1p36-specific CD8+ T lymphocytes were directed against JCV only and might have had a role in the stabilization of the disease. In addition, our results differ from a recent report by Sharma et al., who attempted to characterize the phenotype of the cross-reactive BKV-specific CTL in healthy individuals (30). Indeed, these authors tested cells after peptide stimulation and culture in vitro, which is bound to alter their differentiation markers, whereas we used unstimulated fresh blood samples, as necessary for characterization of the phenotype of these cells ex vivo.
Asymptomatic JC viruria occurs in up to one-third of healthy individuals, while JC viremia is found mainly in immunosuppressed individuals who are at risk of developing PML (18). Therefore, one of the goals of this study was to determine the effect of the cellular immune response on the level of polyomavirus expression in blood and urine. While BKV DNA was not found in any of the plasma or PBMC samples, a low JC viral load was detected in PBMC samples of one HIV-infected individual and one PML patient and in the plasma of another PML patient. Since JCV viremia has been associated with profound immunosuppression in HIV-infected people (18), it is not surprising that viremia was detected in only three individuals, as the patients with HIV infection had a relatively preserved CD4+ T-cell count.
The presence of virus-specific CD8+ T cells did not prevent the excretion of JCV or BKV in the urine of some study subjects, regardless of their immune status. Interestingly, however, JC replication in the kidney may induce a distinct CTL response. In our study, JCV excretion in the urine was associated with the presence of VP1p100-specific CD8+ T cells in the blood. Recently, our group had shown that A*0201+ kidney transplant recipients with polyomavirus nephropathy, a condition related to active BKV replication in the kidney, have a predominant response against the cross-reactive BKV VP1p108 epitope, compared to healthy individuals, who have a predominant BKV VP1p44 response, which cross-reacts with JCV VP1p36 (5). This suggests that the cross-reacting JCV VP1p100 and BKV VP1p108 epitopes may be presented preferentially to CTL in association with the HLA A*0201 molecule in the renal tubular cells during human polyomavirus infections.
PML remains an important disease since its incidence in HIV-infected patients has not decreased significantly in the HAART era (1) and because of the growing number of organ transplant recipients worldwide who may be at risk for PML. In addition, PML has been reported to occur in multiple sclerosis and Crohn's patients taking natalizumab (3, 16, 21, 33), a monoclonal antibody against the α4β1-integrin, and this class of medications, called selective adhesion molecule inhibitors, are attractive candidates for treatment of a variety of autoimmune diseases (27). There is recent evidence that natalizumab treatment significantly modulated proliferative responses of immune cells, although various cell subsets were differentially affected. Further studies will be necessary to determine the fate of JCV-specific T cells during natalizumab therapy. This may help clarify why these patients are susceptible to PML but not to other viral infections (15, 24). Prospective follow-up of the frequency and phenotype of JCV-specific CD8+ T-cell response in these populations at risk would also provide valuable information on the dynamic interaction between the virus and the host.
In summary, we were able to detect very low frequency JCV-specific CD8+ T-cell populations in the peripheral blood. There was not a predominant differentiation phenotype subset among JCV VP1p36- or JCV VP1p100-specific CTL in healthy individuals, HIV-infected subjects, or PML survivors. Prospective analysis of one PML patient revealed that JCV VP1p36-specific CTL were activated early in the course of disease, indicating that these cells were indeed responding to JCV and not to BKV. The cellular immune response against polyomaviruses was associated with the absence of viremia, but not viruria, and the detection of JCV DNA in the urine was associated with the presence of a JCV VP1p100 CTL response. Immunotherapies aiming at increasing the cellular immune response against JCV may prove to be a valuable option for the treatment of PML.
Acknowledgments
This work was supported in part by Public Health Service grants R01 NS/AI 041198 and NS 047029, the Harvard Medical School Center for AIDS Research (CFAR), an NIH-funded program (P30 AI60354), and the Ellen R. Cavallo Research Fund to I. J. Koralnik. M. A. Lima is the recipient of a fellowship from the National Scientific and Technological Development Council, CNPq, Brazil.
Footnotes
Published ahead of print on 17 January 2007.
REFERENCES
- 1.Antinori, A., A. Ammassari, M. L. Giancola, A. Cingolani, S. Grisetti, R. Murri, L. Alba, B. Ciancio, F. Soldani, D. Larussa, G. Ippolito, and A. De Luca. 2001. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J. Neurovirol. 7:323-328. [DOI] [PubMed] [Google Scholar]
- 2.Appay, V., P. R. Dunbar, M. Callan, P. Klenerman, G. M. Gillespie, L. Papagno, G. S. Ogg, A. King, F. Lechner, C. A. Spina, S. Little, D. V. Havlir, D. D. Richman, N. Gruener, G. Pape, A. Waters, P. Easterbrook, M. Salio, V. Cerundolo, A. J. McMichael, and S. L. Rowland-Jones. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379-385. [DOI] [PubMed] [Google Scholar]
- 3.Berger, J. R., and I. J. Koralnik. 2005. Progressive multifocal leukoencephalopathy and natalizumab—unforeseen consequences. N. Engl. J. Med. 353:414-416. [DOI] [PubMed] [Google Scholar]
- 4.Champagne, P., G. S. Ogg, A. S. King, C. Knabenhans, K. Ellefsen, M. Nobile, V. Appay, G. P. Rizzardi, S. Fleury, M. Lipp, R. Forster, S. Rowland-Jones, R. P. Sekaly, A. J. McMichael, and G. Pantaleo. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410:106-111. [DOI] [PubMed] [Google Scholar]
- 5.Chen, Y., J. Trofe, J. Gordon, R. A. Du Pasquier, P. Roy-Chaudhury, M. J. Kuroda, E. S. Woodle, K. Khalili, and I. J. Koralnik. 2006. Interplay of cellular and humoral immune responses against BK virus in kidney transplant recipients with polyomavirus nephropathy. J. Virol. 80:3495-3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cinque, P., I. J. Koralnik, and D. B. Clifford. 2003. The evolving face of human immunodeficiency virus-related progressive multifocal leukoencephalopathy: defining a consensus terminology. J. Neurovirol. 9(Suppl. 1):88-92. [DOI] [PubMed] [Google Scholar]
- 7.Du Pasquier, R. A., P. Autissier, Y. Zheng, J. Jean-Jacques, and I. J. Koralnik. 2005. Presence of JC virus-specific CTL in the cerebrospinal fluid of PML patients: rationale for immune-based therapeutic strategies. AIDS 19:2069-2076. [DOI] [PubMed] [Google Scholar]
- 8.Du Pasquier, R. A., M. J. Kuroda, J. E. Schmitz, Y. Zheng, K. Martin, F. W. Peyerl, M. Lifton, D. Gorgone, P. Autissier, N. L. Letvin, and I. J. Koralnik. 2003. Low frequency of cytotoxic T lymphocytes against the novel HLA-A*0201-restricted JC virus epitope VP1p36 in patients with proven or possible progressive multifocal leukoencephalopathy. J. Virol. 77:11918-11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Pasquier, R. A., M. J. Kuroda, Y. Zheng, J. Jean-Jacques, N. L. Letvin, and I. J. Koralnik. 2004. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain 127:1970-1978. [DOI] [PubMed] [Google Scholar]
- 10.Du Pasquier, R. A., J. E. Schmitz, J. Jean-Jacques, Y. Zheng, J. Gordon, K. Khalili, N. L. Letvin, and I. J. Koralnik. 2004. Detection of JC virus-specific cytotoxic T lymphocytes in healthy individuals. J. Virol. 78:10206-10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elphick, G. F., W. Querbes, J. A. Jordan, G. V. Gee, S. Eash, K. Manley, A. Dugan, M. Stanifer, A. Bhatnagar, W. K. Kroeze, B. L. Roth, and W. J. Atwood. 2004. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380-1383. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Suarez, J., D. de Miguel, I. Krsnik, H. Banas, I. Arribas, and C. Burgaleta. 2005. Changes in the natural history of progressive multifocal leukoencephalopathy in HIV-negative lymphoproliferative disorders: impact of novel therapies. Am. J. Hematol. 80:271-281. [DOI] [PubMed] [Google Scholar]
- 13.Geginat, J., A. Lanzavecchia, and F. Sallusto. 2003. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood 101:4260-4266. [DOI] [PubMed] [Google Scholar]
- 14.Gray, C. M., J. Lawrence, J. M. Schapiro, J. D. Altman, M. A. Winters, M. Crompton, M. Loi, S. K. Kundu, M. M. Davis, and T. C. Merigan. 1999. Frequency of class I HLA-restricted anti-HIV CD8+ T cells in individuals receiving highly active antiretroviral therapy (HAART). J. Immunol. 162:1780-1788. [PubMed] [Google Scholar]
- 15.Hauser, S. L., and H. L. Weiner. 2006. Natalizumab: immune effects and implications for therapy. Ann. Neurol. 59:731-732. [DOI] [PubMed] [Google Scholar]
- 16.Kleinschmidt-DeMasters, B. K., and K. L. Tyler. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 353:369-374. [DOI] [PubMed] [Google Scholar]
- 17.Koralnik, I. J. 2006. Progressive multifocal leukoencephalopathy revisited: has the disease outgrown its name? Ann. Neurol. 60:162-173. [DOI] [PubMed] [Google Scholar]
- 18.Koralnik, I. J., D. Boden, V. X. Mai, C. I. Lord, and N. L. Letvin. 1999. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology 52:253-260. [DOI] [PubMed] [Google Scholar]
- 19.Koralnik, I. J., R. A. Du Pasquier, M. J. Kuroda, J. E. Schmitz, X. Dang, Y. Zheng, M. Lifton, and N. L. Letvin. 2002. Association of prolonged survival in HLA-A2+ progressive multifocal leukoencephalopathy patients with a CTL response specific for a commonly recognized JC virus epitope. J. Immunol. 168:499-504. [DOI] [PubMed] [Google Scholar]
- 20.Krymskaya, L., M. C. Sharma, J. Martinez, W. Haq, E. C. Huang, A. P. Limaye, D. J. Diamond, and S. F. Lacey. 2005. Cross-reactivity of T lymphocytes recognizing a human cytotoxic T-lymphocyte epitope within BK and JC virus VP1 polypeptides. J. Virol. 79:11170-11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langer-Gould, A., S. W. Atlas, A. J. Green, A. W. Bollen, and D. Pelletier. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 353:375-381. [DOI] [PubMed] [Google Scholar]
- 22.Reference deleted.
- 23.Miller, N. R., E. O. Major, and W. C. Wallen. 1983. Transfection of human fetal glial cells with molecularly cloned JCV DNA. Prog. Clin. Biol. Res. 105:29-40. [PubMed] [Google Scholar]
- 24.Niino, M., C. Bodner, M. L. Simard, S. Alatab, D. Gano, H. J. Kim, M. Trigueiro, D. Racicot, C. Guerette, J. P. Antel, A. Fournier, F. Grand'maison, and A. Bar-Or. 2006. Natalizumab effects on immune cell responses in multiple sclerosis. Ann. Neurol. 59:748-754. [DOI] [PubMed] [Google Scholar]
- 25.Polo, C., J. L. Perez, A. Mielnichuck, C. G. Fedele, J. Niubo, and A. Tenorio. 2004. Prevalence and patterns of polyomavirus urinary excretion in immunocompetent adults and children. Clin. Microbiol. Infect. 10:640-644. [DOI] [PubMed] [Google Scholar]
- 26.Power, C., J. G. Gladden, W. Halliday, M. R. Del Bigio, A. Nath, W. Ni, E. O. Major, J. Blanchard, and M. Mowat. 2000. AIDS- and non-AIDS-related PML association with distinct p53 polymorphism. Neurology 54:743-746. [DOI] [PubMed] [Google Scholar]
- 27.Rice, G. P., H. P. Hartung, and P. A. Calabresi. 2005. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64:1336-1342. [DOI] [PubMed] [Google Scholar]
- 28.Sallusto, F., J. Geginat, and A. Lanzavecchia. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22:745-763. [DOI] [PubMed] [Google Scholar]
- 29.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708-712. [DOI] [PubMed] [Google Scholar]
- 30.Sharma, M. C., W. Zhou, J. Martinez, L. Krymskaya, T. Srivastava, W. Haq, D. J. Diamond, and S. F. Lacey. 2006. Cross-reactive CTL recognizing two HLA-A*02-restricted epitopes within the BK virus and JC virus VP1 polypeptides are frequent in immunocompetent individuals. Virology 350:128-136. [DOI] [PubMed] [Google Scholar]
- 31.Singhal, S., J. C. Shaw, J. Ainsworth, M. Hathaway, G. M. Gillespie, H. Paris, K. Ward, D. Pillay, P. A. Moss, and D. J. Mutimer. 2000. Direct visualization and quantitation of cytomegalovirus-specific CD8+ cytotoxic T-lymphocytes in liver transplant patients. Transplantation 69:2251-2259. [DOI] [PubMed] [Google Scholar]
- 32.Tan, L. C., N. Gudgeon, N. E. Annels, P. Hansasuta, C. A. O'Callaghan, S. Rowland-Jones, A. J. McMichael, A. B. Rickinson, and M. F. Callan. 1999. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 162:1827-1835. [PubMed] [Google Scholar]
- 33.Van Assche, G., M. Van Ranst, R. Sciot, B. Dubois, S. Vermeire, M. Noman, J. Verbeeck, K. Geboes, W. Robberecht, and P. Rutgeerts. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353:362-368. [DOI] [PubMed] [Google Scholar]
- 34.van Lier, R. A., I. J. ten Berge, and L. E. Gamadia. 2003. Human CD8(+) T-cell differentiation in response to viruses. Nat. Rev. Immunol. 3:931-939. [DOI] [PubMed] [Google Scholar]
- 35.Weber, T., C. Trebst, S. Frye, P. Cinque, L. Vago, C. J. Sindic, W. J. Schulz-Schaeffer, H. A. Kretzschmar, W. Enzensberger, G. Hunsmann, and W. Luke. 1997. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J. Infect. Dis. 176:250-254. [DOI] [PubMed] [Google Scholar]
- 36.Weber, T., F. Weber, H. Petry, and W. Luke. 2001. Immune response in progressive multifocal leukoencephalopathy: an overview. J. Neurovirol. 7:311-317. [DOI] [PubMed] [Google Scholar]
- 37.Wherry, E. J., and R. Ahmed. 2004. Memory CD8 T-cell differentiation during viral infection. J. Virol. 78:5535-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wuthrich, C., S. Kesari, W. K. Kim, K. Williams, R. Gelman, D. Elmeric, U. De Girolami, J. T. Joseph, T. Hedley-Whyte, and I. J. Koralnik. 2006. Characterization of lymphocytic infiltrates in progressive multifocal leukoencephalopathy: co-localization of CD8(+) T cells with JCV-infected glial cells. J. Neurovirol. 12:116-128. [DOI] [PubMed] [Google Scholar]