

Abstract
Egress of herpes simplex virus type 1 (HSV-1) from the nucleus of the infected cell to extracellular spaces involves a number of distinct steps, including primary envelopment by budding into the perinuclear space, de-envelopment into the cytoplasm, cytoplasmic reenvelopment, and translocation of enveloped virions to extracellular spaces. UL20/gK-null viruses are blocked in cytoplasmic virion envelopment and egress, as indicated by an accumulation of unenveloped or partially enveloped capsids in the cytoplasm. Similarly, UL11-null mutants accumulate unenveloped capsids in the cytoplasm. To assess whether UL11 and UL20/gK function independently or synergistically in cytoplasmic envelopment, recombinant viruses having either the UL20 or UL11 gene deleted were generated. In addition, a recombinant virus containing a deletion of both UL20 and UL11 genes was constructed using the HSV-1(F) genome cloned into a bacterial artificial chromosome. Ultrastructural examination of virus-infected cells showed that both UL20- and UL11-null viruses accumulated unenveloped capsids in the cytoplasm. However, the morphology and distribution of the accumulated capsids appeared to be distinct, with the UL11-null virions forming aggregates of capsids having diffuse tegument-derived material and the UL20-null virus producing individual capsids in close juxtaposition to cytoplasmic membranes. The UL20/UL11 double-null virions appeared morphologically similar to the UL20-null viruses. Experiments on the kinetics of viral replication revealed that the UL20/UL11 double-null virus replicated in a manner similar to the UL20-null virus. Additional experiments revealed that transiently expressed UL11 localized to the trans-Golgi network (TGN) independently of either gK or UL20. Furthermore, virus infection with the UL11/UL20 double-null virus did not alter the TGN localization of transiently expressed UL11 or UL20 proteins, indicating that these proteins did not interact. Taken together, these results show that the intracellular transport and TGN localization of UL11 is independent of UL20/gK functions, and that UL20/gK are required and function prior to UL11 protein in virion cytoplasmic envelopment.
Herpes simplex virus type 1 (HSV-1) morphogenesis occurs in multiple stages within infected cells. Initially, the virion capsid assembles within the nucleus and the virion acquires an initial envelope by budding into the perinuclear spaces (39). Subsequently, these enveloped virions fuse with the outer nuclear lamellae, leading to the accumulation of unenveloped capsids in the cytoplasm. Within the cytoplasm, a number of additional tegument proteins attach to the capsid and the fully tegumented capsids bud into cytoplasmic vesicles, which mostly likely originate from the trans-Golgi network (TGN). Enveloped virions are ultimately secreted to extracellular spaces through the utilization of cellular vesicular trafficking systems (7, 19, 20, 33, 40, 43). The process by which the tegumented cytoplasmic capsids bud into TGN-derived vesicles is not well understood. The prevalent model calls for specific interactions among viral tegument proteins and membrane proteins and glycoproteins embedded within TGN membranes as key factors that drive cytoplasmic virion envelopment. This model is supported by evidence that specific mutations within tegument proteins and multiple membrane proteins and glycoproteins inhibit cytoplasmic envelopment (32, 34). Apparently, multiple glycoproteins may be concurrently involved in cytoplasmic virion envelopment. The simultaneous absence of both gM and gE or of gM and the gE cytoplasmic tail results in inhibition of cytoplasmic envelopment for pseudorabies virus (PRV) (5, 6); however, deletion of gM or gE does not appear to affect HSV-1 cytoplasmic envelopment. In contrast, deletion of both HSV-1 gD and gE causes accumulation of capsids in the cytoplasm of infected cells, presumably due to loss of contacts with tegument proteins (32, 34). These results suggest that PRV and HSV-1 cytoplasmic envelopment may rely on different repertoires of protein-protein interactions to drive cytoplasmic virion envelopment.
Of particular interest to these investigations are the membrane proteins UL11 (2, 23, 24), UL20 (3, 14, 18, 30), and UL53 (gK) (13, 21, 22), which are known to be important determinants of cytoplasmic envelopment for both PRV and HSV-1. The UL11 gene encodes a 96-amino-acid tegument protein, which is N-terminally myristylated (28) and palmitylated (25). The UL11 protein localizes to nuclear and TGN-derived membranes in infected cells (1) but only to the TGN-derived membranes in noninfected cells (4). UL11 was shown to specifically interact with the UL16 tegument protein, providing a potential docking mechanism for tegumented capsids onto TGN-derived membranes(26, 42). An HSV UL11-null mutant obtained by deletion of most of the UL11-coding region accumulated capsids in the nucleus and unenveloped capsids in the cytoplasm (2), while a PRV null virus with the entire UL11 gene deleted showed accumulation of unenveloped capsids in the cytoplasm of infected cells embedded in tegument-like material (24).
The UL20 and UL53 (gK) genes encode multipass transmembrane proteins of 222 and 338 amino acids, respectively, and are conserved in all alphaherpesviruses (9, 29, 38). UL20p and gK localize to TGN membranes after endocytosis from cell surfaces (14). UL20p and gK are essential for cytoplasmic virion morphogenesis, since mutant viruses lacking either gK or UL20p accumulate capsids within the cytoplasm that are unable to acquire envelopes by budding into TGN-associated membranes (11, 12, 14, 18, 30). Furthermore, UL20p is essential for virus-induced cell fusion caused by either gB or gK syncytial mutations, and it is necessary for gK cell surface expression. Recently, our laboratory has shown that gK and UL20 interact and that this interaction is essential for their cotransport and membrane fusion and virion morphogenesis functions (15, 31).
The purpose of the present investigations was twofold: (i) to revisit the role of UL11 in virion morphogenesis and egress by constructing a new recombinant UL11-null virus in the HSV-1(F) genetic background that could be directly compared to our previously constructed gK-null and UL20-null viruses and (ii) to investigate whether UL11 and the gK/UL20 heterodimer functioned synergistically or independently of each other in the late stages of cytoplasmic virion morphogenesis. The bacterial artificial chromosome (BAC)-cloned HSV-1(F) viral genome was used to generated UL11 single-null and UL11/UL20 double-null viruses. Characterization of the replication and ultrastructural characteristics of these recombinant viruses revealed that UL11 played an important role in cytoplasmic virion envelopment. Furthermore, although UL11 and UL20 were independently transported and localized at the TGN, UL20 functions preceded and were required for UL11 functions in cytoplasmic virion envelopment.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics. The UL19- and UL20-complementing cell line G5 was a gift of P. Desai (Johns Hopkins Medical Center) (10). A plasmid encoding UL11-green fluorescent protein (GFP) was a gift of J. Wills (26). Construction of both a UL20-3xFLAG plasmid and a gK-V5 plasmid was described previously (15). Cell line Fd20-1 constitutively expressing UL20 was constructed in this laboratory (30).
Construction of HSV-1 mutants with deletions of the UL11 and/or UL20 genes (pYEbac102, pYEbac102ΔUL11, pYEbac102ΔUL20, and pYEBac102ΔUL11ΔUL20.).
Insertion-deletion mutagenesis of pYEbac102 DNA was accomplished in Escherichia coli with the λ gam recE recT (GET) recombination system (36, 37) as described previously for mutagenesis of the Kaposi's sarcoma-associated herpesvirus genome (27). Electrocompetent YEbac102 Escherichia coli DH10B cells were transformed with plasmid pGETrec, which contains the genes recE, recT, and bacteriophage λ gam, and grown on plates containing chloramphenicol (12.5 μg/ml) and ampicillin (100 μg/ml). Individual colonies were picked and grown overnight in Luria-Bertani (LB) medium containing chloramphenicol and ampicillin. The next day, the culture was inoculated into 250 ml of LB medium containing chloramphenicol and ampicillin until an optical density at 600 nm of 0.4 was reached. Addition of l-arabinose to a final concentration of 0.2% (wt/vol) and further incubation for 40 min induced expression of the recE, recT, and λ gam genes from plasmid pGETrec. The cells were then harvested and made electrocompetent.
For the ΔUL11-Kan mutation, a PCR fragment containing a kanamycin resistance (Kan) gene cassette flanked by ∼50 bp of viral sequences on both sides was used for recombination to construct pYEbacΔUL11, containing the Kan gene cassette within the targeted UL11 genomic region. Specifically, the Kan gene cassette was inserted 94 nucleotides downstream of the UL11 ATG codon to avoid interruption of the UL12 open reading frame (ORF), which overlaps with the 5′ terminus of the UL11 gene by 87 nucleotides. The remaining coding sequence of the UL11 gene was deleted up to three nucleotides past the TAA termination codon. The construction of the ΔUL20-GFP-Zeo mutation was described previously (31). For the ΔUL11ΔUL20 mutation, a PCR fragment containing either a Kan gene cassette or a GFP-zeocin resistance cassette flanked by ∼50 bp of viral sequences on both sides was used for recombination to construct pYEbac102ΔUL11ΔUL20. Briefly, 40 μl of electrocompetent DH10B cells harboring both pYEbac102 and pGETrec were electroporated with 200 ng of each PCR product to delete the target gene(s) (UL11 or UL20) with standard electroporation parameters (1.8 kV/cm, 200 Ω, and 25 μF). Following electroporation, cells were grown in 1 ml of LB medium for 60 min and subsequently streaked onto LB agar plates containing chloramphenicol (12.5 μg/ml) as well as either kanamycin (50 μg/ml) or zeocin (25 μg/ml). Mutant pYEbac102 DNA containing a deletion in the UL11 or UL20 gene was isolated from bacterial colonies, and a second round of electroporation was performed to remove plasmid pGETrec. Following electroporation, cells were grown on agar plates containing chloramphenicol as well as either kanamycin or zeocin.
Confirmation of the targeted mutations in pYEbac102 DNA.
HSV-1 BAC DNAs (pYEbac102, pYEbac102ΔUL11, and pYEbac102ΔUL11ΔUL20) were purified from 500 ml of BAC cultures with the QIAGEN large-construct kit (QIAGEN, Valencia, CA). PCR primers that lie outside both UL11 and UL20 were designed. A PCR test was performed to verify that the desired mutations were present. All mutations were sequenced to verify the presence of the desired mutations in the BACs. To verify that no spurious mutations were introduced into the viral genome of the individual BAC mutants during mutagenesis or during the transfection procedure, each null mutation was complemented by expression vectors containing the appropriate gene or genes. Each complemented virus was verified to have the plaque phenotype characteristics of the wild-type BAC YEbac102.
Transfection of HSV-1 BAC DNAs.
Transient transfection of cells with BAC DNAs was performed with Lipofectamine 2000 (Invitrogen). Vero cells (pYEbacΔUL11) and Fd20-1 cells (pYEbacΔUL11ΔUL20) were grown to 95% confluence in six-well plates. Cells were transfected with BAC DNA mixed with Lipofectamine 2000 in Opti-MEM medium as recommended by the manufacturer (Invitrogen). After 6 h of incubation at 37°C, the medium was removed from the transfected cells, the cells were washed with phosphate-buffered saline, and fresh Dulbecco's modified Eagle's medium with 10% fetal calf serum was added. At 72 h posttransfection, virus stocks were collected.
One-step growth kinetics of YEbac102 mutants.
Analysis of one-step growth kinetics was as described previously (12, 16). Briefly, each virus at a multiplicity of infection (MOI) of 2 was adsorbed to approximately 6 × 105 Vero cells at 4°C for 1 h. Thereafter, warm medium was added, and virus was allowed to penetrate for 2 h at 37°C. Any remaining extracellular virus was inactivated by low-pH treatment (0.1 M glycine, pH 3.0). Cells and supernatants were harvested immediately thereafter (0 h) or after 4, 8, 12, 18, 24, or 36 h of incubation at 37°C. Virus titers were determined by end point titration of virus stocks on Vero cells for pYEbacΔUL11 or on G5 cells for pYEbac102ΔUL11ΔUL20.
Electron microscopy.
Cell monolayers were infected with the indicated virus at an MOI of 3. All cells were prepared for transmission electron microscopy examination 24 h postinfection (hpi). Infected cells were fixed in a mixture of 2% paraformaldehyde and 1.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.3. Following treatment with 1% OsO4 and dehydration in an ethanol series, the samples were embedded in Epon-Araldyte resin and polymerized at 70°C. Thin sections were made on an MTXL ultratome (RMC Products), stained with 5% uranyl acetate and citrate-nitrate-acetate lead, and observed with a Zeiss 10 transmission electron microscope as described previously (14, 30).
Confocal microscopy.
To determine intracellular localization of UL20 and gK in the presence or absence of UL11, Vero cells were grown on coverslips in six-well plates to 95% confluence and then transfected with UL11, UL11 and UL20, UL11 and gK, or UL11, UL20, and gK by using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. After 6 h of incubation at 37°C, the medium was removed from the transfected cells, cells were washed with phosphate-buffered saline, and fresh Dulbecco's modified Eagle's medium with 10% fetal calf serum was added. At 48 h posttransfection cells were washed with Tris-buffered saline (TBS), fixed with electron microscopy grade 3% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) for 15 min, washed twice with PBS-50 mM glycine, and permeabilized with 1.0% Triton X-100. Monolayers were subsequently blocked for 1 h with 7% normal goat serum and 7% bovine serum albumin in TBS (TBS blocking buffer) before incubation for 2 h with either anti-GFP (Immunology Consultants Laboratory, Newberg, OR) for detection of UL11, anti-FLAG (Sigma Chemical) for detection of UL20, anti-V5 (Invitrogen, Carlsbad, CA) for detection of gK, anti-TGN 46 (Serotec, Raleigh, NC) for detection of the trans-Golgi network, or a combination of these antibodies as indicated. Alternatively, to determine the intracellular localization of UL11 and UL20 during an active virus infection, 143 TK− cells were prepared as described above with the exception that 24 h posttransfection the cells were infected with YEbac102ΔUL11ΔUL20 at an MOI of 2. At 48 h postinfection, the cells were prepared as described above.
To visualize specific fluorescence, secondary antibodies were added to the cells. Briefly, cells were washed thoroughly and Alexafluor 488, Alexafluor 594, and Alexafluor 647 at a 1:750 dilution were added and incubated for 1 h. Cells were washed again and mounted on slides. The fluorescence was then visualized using a Leica TCS SP2 laser-scanning confocal microscope (Leica Microsystems, Exton, PA) fitted with a CS APO 63× Leica objective (numerical aperture, 1.4). Individual optical sections in the z axis, averaged six times, were collected at the zoom indicated in the figure legends in series in the different channels at a resolution of 1,024 × 1,024 pixels as described previously (14-17). Images were compiled and rendered in Adobe Photoshop.
RESULTS
Construction of the HSV-1 BACs pYEbac102ΔUL11 and pYEbac102ΔUL11ΔUL20.
The complete HSV-1(F) genome has been cloned into a BAC (pYEbac102), enabling the genetic manipulation of the HSV-1 genome in E. coli (41). Previously, we utilized the BAC-based GET homologous recombination system to construct deletions within the gB, UL20, and gK genes in E. coli (31). A similar strategy was utilized to construct an HSV-1 BAC with most of the UL11 ORF deleted. Deletion of the UL11 gene was accomplished using specific oligonucleotide primers (Table 1). Specifically, the UL11 deletion encompassed 194 bp of the UL11 ORF, while 97 bp of the 5′ UL11 ORF remained intact. This deletion was engineered to ensure that the UL12 ORF, which overlaps with the UL11 ORF by 86 bp, was unaffected (see Materials Methods). A similar deletion of the UL11 gene was constructed in the pYEbac102ΔUL20 genetic background.
TABLE 1.
Oligonucleotide primers used for GET recombination
| Primer designation | Primer name | Sequencea | Purpose (product size, bp) |
|---|---|---|---|
| A | 3′ UL11-Kan | 5′-GGGTTTTTTAAAAACGACACGCGTGCGACCGTATACAGAAATTGTTTTGGcggttgatgagagctttgttgtaggtggac-3′ | UL11 GET recombination (1,208) |
| B | 5′ UL11-Kan | 5′-AACGTCCTCATCACCGACGACGGGGAGGTCGTCTCGCTGACCGCCCACGACTagccacgttgtgtctcaaaatctctgatgtta-3′ | |
| C | 3′ UL20-GFPZeo | 5′-CACGGACATCCCCCAAAACACGGGCGCCGACAACGGCAGACGATCCCTCTTGATGTTagacatgataagatacattgatgagtttgg-3′ | UL20 GET recombination (2,025) |
| D | 5′ UL20 GFPZeo | 5′-CTGACGTAAGCGACCCTTTGCGGTTTCGGTCTCCCCACCTCCACCGCACACCCCgtccgttacataacttacggtaaatgg-3′ | |
| a | 3′ UL11-Rev | 5′-GCACCAGCGCGGAGGAGGGC-3′ | Diagnostic PCR for wild-type |
| b | 5′UL11-For | 5′-ATTGTACGCCCAAGATACAACACCG-3′ | UL11 (794) or ΔUL11-Kan (1,701) |
| c | 3′ UL20-Rev | 5′-ACGGCCTTATCAAAACACTCGCCTC-3′ | Diagnostic PCR for wild-type |
| d | 5′ UL20-For | 5′-GAAGGCTGCGGCCTCGTTCCAG-3′ | UL20 (909) or ΔUL20-GFPZeo (2,469) |
HSV-1-homologous regions are denoted by uppercase letters; lowercase letters signify sequences which bind to the marker gene.
For construction of the ΔUL11-Kan mutation, primers A and B were used for homologous recombination and positioned to remove a 198-bp region of the UL11 gene upon insertion of the kanamycin cassette extending from nucleotide 96 of UL11 to 3 bases past the UL11 stop codon (Table 1; Fig. 1). The UL11 deletion was constructed so that it does not disrupt the UL12 ORF, which overlaps the 5′ end of UL11 by 87 bases (see Materials and Methods). An identical UL11 insertion-deletion was constructed on both the wild-type pYEbac102 genetic background and the pYEbac102ΔUL20 genetic background. A similar methodology was previously used to construct a recombinant virus carrying a deletion of the UL20 gene after insertion of a GFP-zeocin gene cassette (ΔUL20-GFPZeo) (14). This construction deleted a 353-bp region of the UL20 gene extending from the UL20 ATG to the HpaI site located within the UL20 ORF (Table 1; Fig. 1). Each pYEbac102 mutant construct contains one or more of the insertion-deletion mutations mentioned above: pYEbac102ΔUL11 contains the ΔUL11-Kan mutation, pYEbacΔUL20 contains the ΔUL20-GFPZeo mutation, and pYEbac102ΔUL11ΔUL20 contains both the ΔUL11-Kan and the ΔUL20-GFPZeo mutations (Fig. 1).
FIG. 1.

Schematic of the strategy for the construction of pYEbac102 mutant BACs. (A) The top line represents the prototypic arrangement of the HSV-1 genome, with the unique long (UL) and unique short (US) regions flanked by the terminal repeat (TR) and internal repeat (IR) regions. (B) Shown are the expanded genomic regions of the UL11 and UL20 ORFs, the approximate locations of the genomic sites to which insertion of the marker genes was targeted, and the primers used in diagnostic PCR to confirm the presence of each mutation. (C) PCR fragments containing the kanamycin resistance or GFP-zeocin resistance gene cassette flanked by approximately 50 bp of viral sequences on both sides were used for targeted GET recombination in E. coli to construct pYEbac102 mutant BACs with insertion-deletion mutations in the UL11 and/or UL20 ORFs, respectively. The approximate locations of the primers used in amplification of each PCR fragment are also shown. CMV, cytomegalovirus.
PCR-based confirmation of the pYEbac102ΔUL11 and pYEbac102ΔUL11ΔUL20 genotypes.
The YEbac102-based genomic constructs were tested for the presence of the engineered insertion-deletion mutations via diagnostic PCR. Primers a and b (Table 1; Fig. 1), located outside and bracketing the UL11 gene, amplified the predicted 794-bp UL11 DNA fragments from pYEbac102 and pYEbac102ΔUL20 (Fig. 2, lanes 2 and 3). Primers c and d (Table 1; Fig. 1), located outside and bracketing the UL20 gene, amplified the predicted 909-bp UL20 DNA fragments from pYEbac102 and pYEbac102ΔUL11 (Fig. 2, lanes 7 and 9). In contrast, diagnostic PCR against the pYEbac102ΔUL11 and pYEbac102ΔUL11ΔUL20 DNAs using primers a and b (Table 1; Fig. 1) produced a PCR-amplified DNA fragment of 1,608 bp, as predicted due to the insertion of the kanamycin gene cassette (Fig. 2, lanes 4 and 5). Primers c and d (Table 1; Fig. 1), located outside and bracketing the UL20 gene, amplified the predicted 2,469-bp DNA fragment from pYEbac102ΔUL20 and pYEbac102ΔUL11ΔUL20 due to the insertion of the GFP-zeocin gene cassette (Fig. 2, lanes 8 and 10).
FIG. 2.

PCR-based diagnostic analysis of pYEbac102ΔUL11 and pYEbac102ΔUL20 mutants. Oligonucleotide primers a and b (Table 1) were utilized to amplify DNA fragments containing the inserted Kan gene cassette. (i) Amplification with primers a and b produced the predicted 1,701-bp DNA fragment for the pYEbac102ΔUL11 and pYEbac102ΔUL11ΔUL20 genomes, consistent with the insertion of the Kan gene cassette, and the predicted 794-bp fragment for the pYEbac102 and pYEbac102ΔUL20 controls. (ii) Amplification with primers c and d (Table 1) produced the predicted 2,469-bp DNA fragment for the pYEbac102ΔUL20 and pYEbac102ΔUL11ΔUL20, consistent with insertion of the GZ gene cassette, and the 999-bp predicted DNA fragment for the pYEbac102 and pYEbac102ΔUL11ΔUL20 controls.
Production of infectious virus from pYEbac102-based constructs.
To generate virus stocks from the mutant pYEbac102 constructs, transient transfection of individual BAC DNAs was performed into Vero cells or the Fd20-1 cell line, which is transformed with the UL20 gene and was shown to efficiently complement UL20-null viruses (30). Specifically, pYEbac102 and pYEbac102ΔUL11 were transfected into Vero cells, while pYEbac102ΔUL20 and pYEbac102ΔUL11ΔUL20 were transfected into the UL20-complementing Fd20-1 cells (30). For all transfection experiments, virus plaques became visible at 72 h posttransfection, and virus stocks were collected at appropriate points when exhibiting maximum cytopathic effects.
Plaque morphology and replication kinetics of HSV-1 YEbac102 mutants.
As we have noted previously (31), construction of mutant HSV-1 viruses using the YEbac102 plasmid allows for the rapid generation of recombinant viruses carrying desired mutations without the need for extensive plaque purification, which is normally needed when recombinant viruses are produced via classical homologous recombination in cell culture. To assess and compare the effects of the deletion of the UL11 and UL20 genes in the context of the same viral HSV-1(F) genome on cell-to-cell spread, the plaque morphologies of the YEbac102, YEbac102ΔUL11, YEbacΔUL20, and YEbac102ΔUL11ΔUL20 viruses were examined in Vero cells in the presence or absence of complementation by either the UL11 or UL20 protein provided in trans via transient expression (Fig. 3). Complementation experiments were performed by transfection of expression plasmids for UL20, UL11, or both UL20 and UL11 followed by infection with either the ΔUL20, ΔUL11, or ΔUL11ΔUL20 virus at 24 h posttransfection. As expected, the wild-type YEbac102 produced large plaques on all infected cells (Fig. 3A and B). Infection of noncomplementing cells with either YEbac102ΔUL20 or YEbac102ΔUL20ΔUL11 resulted in very small plaques containing on average one or two cells (Fig. 3E and G), while infection of noncomplementing cells with the pYEbac102ΔUL11 virus resulted in plaques which were approximately one-third the average size of those of the wild-type virus (Fig. 3C). Complementation of YEbac102ΔUL11, YEbac102ΔUL20, or YEbac102ΔUL11/UL20 by UL11, UL20, or UL20 plus UL11, respectively, produced viral plaques similar in size to those of the parental wild-type virus (Fig. 3D, F, and H). Furthermore, complementation of YEbacΔUL20ΔUL11 with the UL20 alone produced a viral plaque similar in size to that of the YEbacΔUL11 virus (not shown). Complementation of the mutant viruses to wild-type plaque phenotypes was observed at a rate of 60 to 70% for all mutant viruses, indicating the absence of any secondary mutations that could account for the observed mutant phenotypes.
FIG. 3.

Plaque phenotypes of UL11-null, UL20-null, and UL11/UL20 double-null viruses under complementing and noncomplementing conditions. Vero cell monolayers were either mock transfected or transfected with plasmids expressing UL11, UL20, or both UL11 and UL20. Transfected cells were infected at 24 h posttransfection with the corresponding viruses, pYEbac102, pYEbac102ΔUL11, pYEbac102ΔUL20, and pYEbac102ΔUL11ΔUL20. Individual viral plaques were visualized at 24 h postinfection by immunohistochemistry.
To examine the effects of the various mutations on virus replication, Vero cells were infected at an MOI of 2 with either the wild-type or each mutant virus. Virus stocks were prepared at 0, 4, 8, 12, 18, 24, and 36 hpi and titrated in triplicate onto complementing cells (Fig. 4). The kinetics of both YEbac102ΔUL20 and YEbac102ΔUL11ΔUL20 were similar to each other and substantially slower than that of the YEbac102 virus, with maximum titers reduced more than 3 logs in comparison to the YEbac102 at 36 hpi. The replication kinetics of the YEbac102ΔUL11 virus was reduced in comparison to that of YEbac102, with maximum titers reduced by more than 1 log at 36 hpi.
FIG. 4.
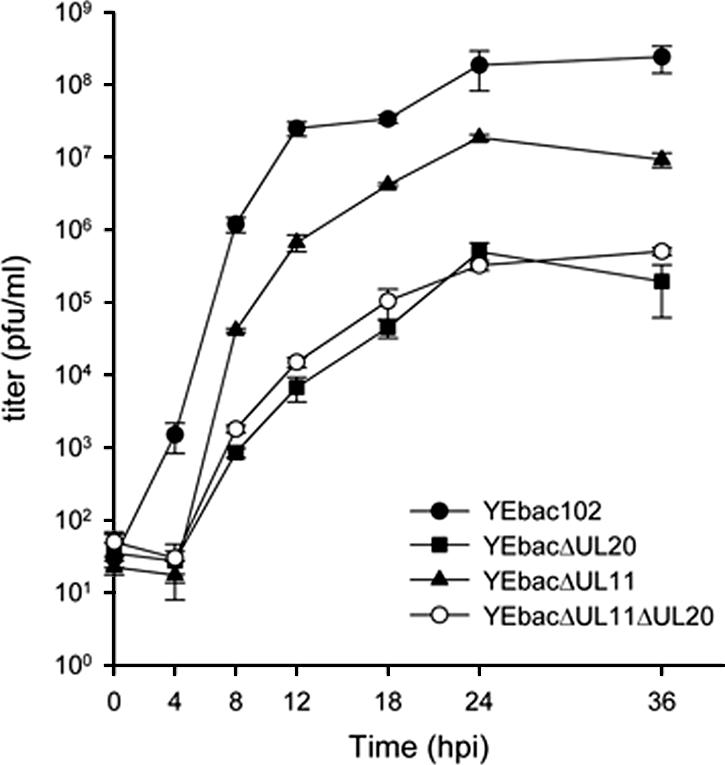
Viral replication kinetics. A comparison of the viral replication characteristics of YEbac102 (•), YEbac102ΔUL20 (▪), YEbac102ΔUL11 (▴), and YEbac102ΔUL11ΔUL20 (○) on Vero cells is shown. One-step growth kinetics of infections virus production were calculated after infection at an MOI of 2 followed by incubation at 37°C. Error bars indicate standard deviations.
Ultrastructural characterization of the YEbac102ΔUL11 and YEbac102ΔUL11ΔUL20 mutant viruses.
The ultrastructural phenotypes of the YEbac102ΔUL20, YEbac102ΔUL11, and YEbac102ΔUL11ΔUL20 viruses relative to the YEbac102 parental virus were investigated utilizing transmission electron microscopy at 24 hpi. As expected, the YEbac102 virus exhibited no apparent defects in virion egress, as exemplified by the presence of fully enveloped virions extracellularly as well as the presence of fully enveloped virions intracellularly (31) (not shown). Unlike the case for the wild-type virus, ultrastructural visualization of YEbac102ΔUL11-, YEbac102ΔUL20-, and YEbac102ΔUL11ΔUL20-infected Vero cells revealed cytoplasmic defects in virion envelopment. The YEbac102ΔUL11 mutant produced largely unenveloped capsids in the cytoplasm embedded within morphologically darker-stained areas that may be caused by the accumulation of tegument proteins (Fig. 5a). This ultrastructural phenotype appeared to be dissimilar to that of a previously constructed HSV-1(F) null virus (2) inasmuch as there was no noticeable accumulation of capsids within nuclei of infected cells. In contrast, the YEbac102ΔUL11 ultrastructural morphology was similar to that produced by a recombinant PRV that lacked the UL11-homologous gene, although the tegument-like staining material surrounding the unenveloped capsids did not appear to be as concentrated as in the PRV case (24). As reported previously (31), the YEbac102ΔUL20 mutant virus produced unenveloped capsids in the cytoplasm, as well as aberrantly enveloped virions (Fig. 5b). Ultrastructural examination of Vero cells infected with the YEbac102ΔUL11ΔUL20 double-null virus revealed virion morphogenetic defects that were quite similar to those of the UL20-null virus and different from those of the UL11-null virus (Fig. 5c).
FIG. 5.


Ultrastructural morphologies of the YEbac102ΔUL11 (a), YEbac102ΔUL20 (b), and YEbac102ΔUL11ΔUL20 (c) viruses. Confluent cell monolayers were infected with the indicated virus at an MOI of 2, incubated for 24 h at 37°C, and prepared for transmission electron microscopy. Panels A, low magnification of an infected cell; panels B and C, higher magnifications of the cells shown in panels A. Nuclear (N), cytoplasmic (C), and extracellular (E) spaces are marked; bars show the relative magnification scale.
UL11 and UL20 are independently transported to the TGN.
It has been previously reported that UL11 localizes to the TGN (25). In agreement with these findings, transient expression of UL11 in Vero cells resulted in localization of the UL11 protein in the TGN (Fig. 6).
FIG. 6.

Digital images of confocal micrographs showing UL11 localization. Vero cell monolayers were transfected with the UL11-GFP-expressing plasmid. At 24 h posttransfection, cells were washed thoroughly, fixed, and stained with the anti-GFP antibody (A1) or with the Golgi specific marker TGN46 (A2) and fluorescence was visualized by confocal microscopy. A3, overlay. Magnification, ×63; zoom, ×2.
Previously, we showed that gK or UL20 alone remained at the rough endoplasmic reticulum when expressed alone. However, coexpression of gK and UL20 resulted in the TGN localization of both proteins, strongly suggesting that these two proteins interact and that this interaction is necessary for the coordinate transport to the TGN (14). To determine whether either UL20 or gK expression was able to alter the TGN localization of the UL11 protein, the UL11 gene was transiently expressed in Vero cells concurrently with either the UL20 or gK gene alone or with both the gK and UL20 genes. UL11 localized to the TGN in the presence of either UL20, gK, or both UL20 and gK (Fig. 7). Additional experimentation was performed to ascertain whether the presence or absence of the UL20 or gK gene could affect the TGN localization of the UL11 protein in the context of other viral proteins. In these experiments, 143 TK− cells were transfected with either the UL11, UL20, or both UL11 and UL20, and 24 h posttransfection, cells were infected with the YEbac102ΔUL11ΔUL20 virus. Again, the UL11 protein localized efficiently to the TGN (Fig. 8).
FIG. 7.

Comparison of UL11 intracellular localization in the presence or absence of gK/UL20. Vero cells were transfected with a combination of plasmids expressing UL11-GFP, UL20, and/or gK. At 24 h posttransfection, cells were washed thoroughly, fixed, and prepared for confocal microscopy after staining with the appropriate antibodies under the appropriate conditions. UL11 was stained with the anti-GFP antibody, UL20 with the anti-FLAG antibody, and gK with the anti-V5 antibody. Magnification, ×63; zoom ×2.
FIG. 8.

Intracellular localization of the UL11 and UL20 proteins in virus-infected cells. Vero cells were transfected with plasmids expressing UL11-GFP, UL20, or both. At 24 h posttransfection, cells were infected at an MOI of 2 with YEbac102ΔUL11ΔUL20. At 24 h postinfection, cells were washed thoroughly, fixed, stained with appropriate antibodies, and prepared for confocal microscopy. Magnification, ×63; zoom ×2.
DISCUSSION
HSV-1 cytoplasmic virion envelopment is thought to be mediated by complex interactions among tegument proteins and viral glycoproteins embedded in the TGN membranes. A number of viral glycoproteins have been shown to either interact with tegument proteins or otherwise affect cytoplasmic envelopment. In this study, we examined potential physical and functional relationships between the UL11 protein and the gK/UL20 heterodimer in cytoplasmic virion envelopment. Specifically, it was of interest to compare the ultrastructural phenotype of a newly constructed UL11-null virus in the HSV-1(F) background with those of the gK-null and UL20-null viruses and to determine whether UL11 and the gK/UL20 heterodimer functioned in a dependent manner or independently of each other. The salient features of our results are as follows: (i) the HSV-1 UL11 protein functioned exclusively in cytoplasmic envelopment as evidenced by the accumulation of cytoplasmic capsids in cells infected with the HSV-1(F) UL11-null virus in a manner similar to the previously described PRV UL11-null virus (24); (ii) the UL20-null and UL11-null plaque morphologies, as well as their ultrastructural phenotypes, were substantially different; (iii) a UL20/UL11 double-null virus produced plaques and ultrastructural morphologies closely resembling those of the UL20-null viral phenotype; (iv) the UL11/UL20 double-null virus replicated with replication kinetics similar to that of the UL20-null virus; and (v) UL11 and UL20 were transported independently of each other in transient-transfection experiments. These results led to the conclusion that UL20 and UL11 transport and localize at the TGN independently of each other; however UL20 must function at a virion morphogenetic step occurring prior to and required for UL11 function.
Recently, we constructed and characterized deletion mutations of the gK, UL20, and gB genes by utilizing the HSV-1(F) genome cloned into a BAC system (31). The HSV-1 gK-null and UL20-null viruses produced smaller plaques than the original gK-null and UL20-null viruses, which were constructed via classical recombination/deletion methodologies in the HSV-1(KOS) genetic background (14, 22). This apparent very-small-plaque phenotype could be due to the difference in viral strains. Alternatively, we favor the hypothesis that the BAC-assisted construction of mutant viruses allowed the rapid generation of mutant genomes in E. coli, substantially lowering the possibility that compensatory mutations that increase plaque size may be selected after serial passage of the KOS gK- or UL20-null viruses. Appreciably, the very-small-plaque phenotype of the UL20/gK-null viruses exemplify that the gK/UL20 proteins are essential for virus spread.
To compare the role of UL11 in virion morphogenesis, the YEbac102ΔUL11 (ΔUL11) virus was produced using our previously described BAC-assisted mutagenesis procedure (27, 31). The ΔUL11 virus was constructed by deleting most of the UL11 ORF, taking care not to interrupt the UL12 ORF, which overlaps with the UL11 ORF by 87 bp. Previously, a similar UL11-null virus, R7219, carrying a UL11 deletion of 176 bp was constructed using classical recombination experiments (2). In comparison, the ΔUL11 virus carried an additional deletion of 18 bp. Overall, the new ΔUL11virus confirmed previous findings with the R7219 virus, since both viruses exhibited a significant reduction in virus replication. However, the R7219 virus exhibited a major defect in capsid envelopment via budding through the nuclear membrane, resulting in an unusual accumulation of intranuclear capsids as well as a reduced number of enveloped capsids in the cytoplasm (2). In contrast to these findings, the ΔUL11virus accumulated capsids in the cytoplasm of infected cells that failed to acquire cytoplasmic envelopes. There was no apparent defect in nuclear egress as evidenced by the lack of accumulation of capsids in the nucleus or incomplete nuclear budding processes. Importantly, the ΔUL11 virus accumulated capsids in the cytoplasm of infected cells in large aggregates surrounded by electron-dense material, which may be derived by tegument proteins. A similar ultrastructural phenotype was observed for the PRV UL11-null virus, with the exception that capsid aggregates were embedded within strongly electron-dense structures of uniform density attributed to the accumulation of capsid proteins (24). Therefore, the HSV-1(F) UL11 gene functions in cytoplasmic envelopment in a manner similar to that of the PRV UL11 gene.
The UL11 protein is anchored to TGN membranes through an N-terminal myristylate anchor. In addition, the UL11 protein contains a dileucine and an acidic amino acid motif, which are known to be involved in physical interactions with the UL16 protein, as well as in recycling from the plasma membranes to the TGN. Deletion of either motif abrogates UL16 interaction and plasma membrane TGN recycling (25, 26). The ΔUL11 virus is predicted to code for the first 32 amino acids of the UL11 protein, effectively retaining the dileucine motif while deleting the entire acidic motif. In contrast, the R7219 virus is predicted to code for the first 39 amino acids of the UL11 protein, effectively retaining the dileucine motif as well as three of the seven amino acids of the acidic acid motif. Thus, it can be predicted that both UL11 peptides encoded by the ΔUL11 and R7219 viruses could be myristylated and anchored to TGN membranes. However, the ΔUL11 protein could not interact with the UL16 protein, since the entire acidic motif is deleted. In contrast, it is possible that the R7219 UL11 protein may be able to interact with the UL16 protein, since the dileucine motif is intact and three of the seven acidic amino acids remain with the protein. It has been reported that the UL16 tegument protein is present on intranuclear assemblons and a structural component of mature virions (35). Therefore, these subtle differences in the UL11 amino-terminal peptides may account for the reported delay of nuclear egress for the R7219 virus, which may be caused by an aberrant interaction of the UL11 amino-terminal peptide encoded by the R7219 virus with UL16 at the nuclear membrane.
The ΔUL11 ultrastructural phenotype is similar to that of the PRV UL11-null virus, with the exception that the PRV UL11-null virus formed capsids within a highly uniform electron-dense material, which appeared to be derived from tegument proteins. In contrast, the ΔUL11 mutant appeared to have a diffuse electron-dense material surrounding the unenveloped capsids. The 32-amino-acid UL11 peptide encoded by the UL11-null virus retains the site of myristylation at its N terminus and thus could be anchored to the TGN membranes. The PRV UL11-null virus does not code for any UL11-derived peptide, since the UL11 initiation codon was altered by site-directed mutagenesis (24). Therefore, the ΔUL11 amino-terminal peptide within TGN membranes as well as in the cytoplasm may interact with other tegument proteins, preventing the more pronounced aggregation of tegument proteins produced by the PRV UL11-null virus.
To address potential synergistic effects between the UL11 and UL20 genes in cytoplasmic envelopment, we constructed the ΔUL20/UL11 double-null virus. Surprisingly, the ΔUL20/UL11 double-null virus exhibited replication characteristics very similar to those of the UL20-null virus. Similarly, the ultrastructural phenotype of the double-null virus was largely similar to that of the UL20-null virus, without any apparent contribution by the UL11 null mutation. Additional studies with transient-expression systems revealed that UL11 and UL20/gK were transported to the TGN independently of each other, indicating that they did not physically or otherwise functionally interact either in cells expressing only the UL11 and UL20/gK genes or in cells infected with virus. Taken together, these results suggest that the UL11 functions in cytoplasmic envelopment are totally dependent on UL20 expression. One way to interpret this apparent dependence of UL11 on UL20 is that the UL20 protein functions in an earlier cytoplasmic envelopment step with UL11, which is required for UL11 function. However, this prediction is contrary to the observation that the UL11-null virions accumulate capsids with aberrant tegument-derived structures, apparently distal to TGN membranes. One interpretation of these results is that in the absence of UL20 protein there is an irreversible attachment to TGN membranes that cannot be overcome by the UL11 null mutation. In contrast, the UL11 null mutation may allow initial binding of tegumented capsids to membranes followed by capsid release, forming the observed aggregates of capsids embedded in tegument-like material. In this instance, the known physical interaction of UL11 with UL16 may in part be responsible for the observed defect in tegument protein accumulation in the cytoplasm, since in the absence of UL11, UL16 as well as other tegument proteins that may interact with UL11 would not bind to TGN membranes but would remain free in the cytoplasm. An alternative scenario is based on the observation that the cellular intracytoplasmic membranes seemed to be restructured in the absence of the PRV UL11, indicating that UL11 may play some stabilization role in Golgi-TGN membranes in infected cells (24). In this regard, it is possible that gK/UL20 may play a similar stabilization role for TGN membranes and specifically at capsid budding sites. In Vero cells Golgi stacks are largely fragmented during infection (8), which would argue against overall Golgi-TGN stabilization roles for either gK or UL20. However, concurrent action of gK/UL20 and UL11 at TGN budding sites may occur, with gK/UL20 playing a dominant role over UL11.
The UL20/gK heterodimer is of paramount importance in cytoplasmic envelopment, since deletion of either gene causes profound accumulation of cytoplasmic capsids and reduction of viral titers by more than 3 logs, while viral plaques are greatly diminished in size. It is important to determine if the UL20/gK functions precedes those of other viral glycoproteins that are known to function in cytoplasmic virion envelopment, such as gE/gI and gM. In this regard, an investigation of the phenotypes of double mutants carrying deletions in more than one gene may shed some light on the sequence of events that control cytoplasmic envelopment.
Acknowledgments
P.A.F. was supported by a Louisiana Board of Regents Fellowship. The work was supported by NIH/NIAID grant R01 AI43000 to K.G.K.
Footnotes
Published ahead of print on 10 January 2007.
REFERENCES
- 1.Baines, J. D., R. J. Jacob, L. Simmerman, and B. Roizman. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines, J. D., and B. Roizman. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines, J. D., P. L. Ward, G. Campadelli-Fiume, and B. Roizman. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowzard, J. B., R. J. Visalli, C. B. Wilson, J. S. Loomis, E. M. Callahan, R. J. Courtney, and J. W. Wills. 2000. Membrane targeting properties of a herpesvirus tegument protein-retrovirus Gag chimera. J. Virol. 74:8692-8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brack, A. R., J. M. Dijkstra, H. Granzow, B. G. Klupp, and T. C. Mettenleiter. 1999. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 73:5364-5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brack, A. R., B. G. Klupp, H. Granzow, R. Tirabassi, L. W. Enquist, and T. C. Mettenleiter. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 74:4004-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Browne, H., S. Bell, T. Minson, and D. W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campadelli, G., R. Brandimarti, L. C. Di, P. L. Ward, B. Roizman, and M. R. Torrisi. 1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 90:2798-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debroy, C., N. Pederson, and S. Person. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36-48. [DOI] [PubMed] [Google Scholar]
- 10.Desai, P., N. A. DeLuca, J. C. Glorioso, and S. Person. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 67:1357-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietz, P., B. G. Klupp, W. Fuchs, B. Kollner, E. Weiland, and T. C. Mettenleiter. 2000. Pseudorabies virus glycoprotein K requires the UL20 gene product for processing. J. Virol. 74:5083-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster, T. P., X. Alvarez, and K. G. Kousoulas. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 77:499-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster, T. P., and K. G. Kousoulas. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster, T. P., J. M. Melancon, J. D. Baines, and K. G. Kousoulas. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 78:5347-5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster, T. P., J. M. Melancon, T. L. Olivier, and K. G. Kousoulas. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 78:13262-13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster, T. P., G. V. Rybachuk, and K. G. Kousoulas. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 75:12431-12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs, W., B. G. Klupp, H. Granzow, C. Hengartner, A. Brack, A. Mundt, L. W. Enquist, and T. C. Mettenleiter. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 76:8208-8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs, W., B. G. Klupp, H. Granzow, and T. C. Mettenleiter. 1997. The UL20 gene product of pseudorabies virus functions in virus egress. J. Virol. 71:5639-5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harley, C. A., A. Dasgupta, and D. W. Wilson. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 75:1236-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchinson, L., and D. C. Johnson. 1995. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 69:5401-5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayachandra, S., A. Baghian, and K. G. Kousoulas. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 71:5012-5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopp, M., H. Granzow, W. Fuchs, B. Klupp, and T. C. Mettenleiter. 2004. Simultaneous deletion of pseudorabies virus tegument protein UL11 and glycoprotein M severely impairs secondary envelopment. J. Virol. 78:3024-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopp, M., H. Granzow, W. Fuchs, B. G. Klupp, E. Mundt, A. Karger, and T. C. Mettenleiter. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J. Virol. 77:5339-5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loomis, J. S., R. J. Courtney, and J. W. Wills. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417-11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luna, R. E., F. Zhou, A. Baghian, V. Chouljenko, B. Forghani, S. J. Gao, and K. G. Kousoulas. 2004. Kaposi's sarcoma-associated herpesvirus glycoprotein K8.1 is dispensable for virus entry. J. Virol. 78:6389-6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacLean, C. A., B. Clark, and D. J. McGeoch. 1989. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 70:3147-3157. [DOI] [PubMed] [Google Scholar]
- 29.MacLean, C. A., S. Efstathiou, M. L. Elliott, F. E. Jamieson, and D. J. McGeoch. 1991. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J. Gen. Virol. 72:897-906. [DOI] [PubMed] [Google Scholar]
- 30.Melancon, J. M., T. P. Foster, and K. G. Kousoulas. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 78:7329-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melancon, J. M., R. E. Luna, T. P. Foster, and K. G. Kousoulas. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167-180. [DOI] [PubMed] [Google Scholar]
- 33.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mettenleiter, T. C. 2006. Intriguing interplay between viral proteins during herpesvirus assembly or: the herpesvirus assembly puzzle. Vet. Microbiol. 113:163-169. [DOI] [PubMed] [Google Scholar]
- 35.Nalwanga, D., S. Rempel, B. Roizman, and J. D. Baines. 1996. The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236-242. [DOI] [PubMed] [Google Scholar]
- 36.Narayanan, K., R. Williamson, Y. Zhang, A. F. Stewart, and P. A. Ioannou. 1999. Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 6:442-447. [DOI] [PubMed] [Google Scholar]
- 37.Orford, M., M. Nefedov, J. Vadolas, F. Zaibak, R. Williamson, and P. A. Ioannou. 2000. Engineering EGFP reporter constructs into a 200 kb human beta-globin BAC clone using GET recombination. Nucleic Acids Res. 28:E84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramaswamy, R., and T. C. Holland. 1992. In vitro characterization of the HSV-1 UL53 gene product. Virology 186:579-587. [DOI] [PubMed] [Google Scholar]
- 39.Roizman, B., and A. E. Sears. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 40.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]