

Abstract
CCR5-using human immunodeficiency virus type 1 (HIV-1) isolates typically gain CXCR4 use via multiple mutations in V3 and often V1/V2 regions of envelope, and patterns of mutations are distinct for each isolate. Here, we report that multiple CXCR4-using variants of a parental CCR5-using HIV-1 isolate, SF162, obtained by either target cell selection or CCR5 inhibition have a common mutation pattern characterized by the same two V3 mutations and that these mutations preexisted in some of the SF162 stocks. These results imply that SF162 has a single pathway for acquiring CXCR4 use and that prolonged culture is sufficient to select for R5X4 variants.
Human immunodeficiency virus type 1 (HIV-1) isolated from infected individuals uses CCR5 and/or CXCR4 coreceptors for cell entry (27). Infection is transmitted by CCR5-using HIV-1 variants (R5) that often later evolve into variants using CXCR4 exclusively or together with CCR5 (X4 or R5X4), and this evolution is associated with accelerating progression to AIDS (5, 6, 12, 39). R5 or X4 phenotype is determined by the amino acid sequence of HIV-1 gp120, particularly of V3 and V1/V2, and less frequently of other regions (4, 7, 8, 10, 11, 17, 21, 23, 30, 34-36). The cause and mechanisms of the evolution towards CXCR4 usage are not fully understood. Target cell selection and the abundance of natural ligands for CCR5 and CXCR4 are thought to be two important selection factors (24, 25, 29), which have been modeled in vitro by propagation of R5 variants in cell lines expressing CXCR4 only (13, 18, 30, 32) or in the presence of CCR5 ligands (1, 15, 26, 28, 37). Evolution towards CXCR4 usage in vivo (38, 39) and in vitro seems to go along multiple pathways, and most R5X4 variants have diverse mutation patterns, although some common features (e.g., charged amino acids at positions 11 and 25 of the V3 loop) have been observed (20, 22, 32).
Earlier, it was reported that CXCR4-using variants of the R5 HIV-1 SF162 isolate (9) selected by viral propagation on CXCR4-expressing T-cell lines acquired two consistent mutations in V3, I309R and A316V (13, 18). Here, we have independently selected for CXCR4-using variants by propagating SF162 in cells expressing CXCR4 but not CCR5 (see reference 32) or in peripheral blood mononuclear cells (PBMC) in the presence of escalating concentrations of RANTES. We found that the same two V3 mutations occurred in seven independently selected CXCR4-using variants of SF162 and that the I309R mutation precedes the A316V mutation under both modes of selection. Moreover, even without RANTES, serial passage of SF162 in PBMC also selected for the same two V3 mutations, albeit more slowly than in T-cell lines or in high RANTES concentrations. Interestingly, one of the SF162 stocks obtained by culture in PBMC of HIV-1 from the AIDS Research and Reference Reagent Program already contained a minor population (11%) harboring the I309R mutation and an even smaller population (<2%) with both the I309R and A316V mutations.
In the first set of experiments virus was selected in the course of five weekly passages by serial dilution of U87-CD4-CCR5 cells by U87-CD4-CXCR4 cells (see reference 32). The SF162 stock used for these experiments was initially derived from transfection of 293T cells with a molecular clone and subsequently had been propagated in PBMC. Seventy-three independent envelope (Env) clones were PCR amplified from the SF162 virus stock used for these experiments and sequenced, and all were found to match the canonical SF162 sequence in the database (Table 1).
TABLE 1.
SF162 Env mutations in R5X4 variants
| Virus namec | No. of clones
|
||||
|---|---|---|---|---|---|
| No. with mutations/total no. sequenced | No. with specific mutation(s) in V3/total no. with mutations
|
No. with other mutation in gp160; mutation (region) or reference | |||
| I309 only | A316V only | I309R + A316V | |||
| DEM SF162 lab stock | 0/73 | 0 | 0 | 0 | 0 |
| A-5 | 4/4 | 4/4 | 0 | 0 | 0 |
| A-10 | 4/4 | 0 | 0 | 4/4 | 0 |
| B-5 | 4/4 | 4/4 | 0 | 0 | 3/4; T132I (V1) |
| B-10 | 4/4 | 0 | 0 | 4/4 | 4/4; T132I (V1) |
| C-10 | 4/4 | 0 | 0 | 4/4 | 4/4; V255M (C2) |
| D-10 | 4/4 | 0 | 0 | 4/4 | 4/4; I269N (gp41) |
| LBM SF162 lab stock | 9/73 | 8 | 0 | 1 | 11; Table 2 |
| C-2 | 0/36 | 0 | 0 | 0 | NDb |
| R50-2a | 0/21 | 0 | 0 | 0 | ND |
| R90-2a | 0/18 | 0 | 0 | 0 | ND |
| C-3 | 4/10 | 4 | 0 | 0 | ND |
| R50-3 | 3/9 | 1 | 1 | 1 | ND |
| R90-3 | 3/9 | 3 | 0 | 0 | ND |
| C-4 | 7/10 | 6 | 0 | 0 | ND |
| R50-4 | 6/10 | 0 | 0 | 6 | ND |
| R90-4 | 6/9 | 5 | 0 | 1 | ND |
| C-5 | 5/8 | 3 | 0 | 0 | ND |
| R50-5 | 7/10 | 0 | 0 | 7 | ND |
| R90-5 | 5/7 | 3 | 0 | 2 | ND |
| C-6 | 5/10 | 2 | 0 | 3 | ND |
| R50-6 | 6/9 | 1 | 0 | 5 | ND |
| R90-6 | 4/10 | 2 | 0 | 2 | ND |
| C-7 | 2/7 | 1 | 0 | 1 | ND |
| R50-7 | 5/9 | 0 | 0 | 5 | ND |
| R90-7 | 5/8 | 3 | 0 | 2 | ND |
| C-13 | 10/15 | 0 | 0 | 10 | 11; Table 2 |
| R50-13 | 15/15 | 0 | 0 | 15 | 9; Table 2 |
| R90-13 | 15/15 | 0 | 0 | 15 | 9; Table 2 |
| MT-4 C-13 | 15/15 | 0 | 0 | 15 | 11; Table 2 |
| MT-4 R90-13 | 14/14 | 0 | 0 | 14 | 11; Table 2 |
R50 and R90 are SF162 variants isolated from PBMC cultured with RANTES increased every second weekly passage from 0.5 nM at passage 1 to 240 nM at passage 10 or from 50 nM at passage 1 to 2,400 nM at passage 10, respectively. The concentration of RANTES was kept constant from passages 10 to 13. C, control variants selected in parallel but without RANTES. Numbers indicate at what passage the viruses were isolated.
ND, only sequence data for C2-V3-C3 (Table 2).
DEM, Donald E. Mosier; LBM, Leonid B. Margolis.
SF162 variants capable of replication on pure U87-CD4-CXCR4 cells were further selected by five weekly passages in MT-2 cells. Four independent selection experiments were performed, and the selected CXCR4-using variants were designated SF162A, -B, -C, and -D, with numbers indicating 5 or 10 weeks of selection (Table 1). The replication capacities and sensitivities of the four selected variants to the CCR5 inhibitor PSC-RANTES (19, 31) and the CXCR4 inhibitor AMD3100 (14) are shown in Fig. 1. Note that the I309R mutation alone in SF162A-5 and SF162B-5 decreased replication capacity on both CCR5- and CXCR4-expressing target cells, a loss of fitness previously described for coreceptor switch intermediates (30, 32). Table 1 shows the sequence changes associated with the R5X4 phenotype, with the same I309R + A316V mutations shared by all four final variants. Variant SF162A-10 has only the two V3 mutations, but variants B-, C-, and D-10 each have one additional unique mutation that impacts replication capacity and sensitivity to coreceptor inhibitors (e.g., the I269V change in gp41 in D-10 [Fig. 1]).
FIG. 1.

Replication and coreceptor inhibitor sensitivity of SF162 envelope mutants. A. Mean replication of SF162 and mutated variants A, B, C, and D after 5 or 10 weeks of selection (Table 1 shows sequence changes) assayed on U87-CD4-CCR5 target cells (R5 p24) or U87-CD4-CXCR4 target cells (X4 p24) measured by capsid p24 antigen enzyme-linked immunosorbent assay. Data are means ± standard errors of the means for triplicate assays. The stock SF162 gave <10 pg/ml p24 on CXCR4 target cells. B. Inhibition of virus replication on U87-CD4-CCR5 target cells by the CCR5 inhibitor PSC-RANTES. C. Inhibition of virus replication on U87-CD4-CXCR4 cells by the CXCR4 inhibitor AMD3100.
In the second set of experiments we selected SF162 variants by infecting 20 million phytohemagglutinin-activated PBMC with 104 50% tissue culture infective doses of uncloned virus and applying selective pressure by increasing RANTES concentrations weekly. The amount of RANTES was increased every second weekly passage either from 0.5 nM at passage 1 to 240 nM at passage 10 or from 50 nM at passage 1 to 2,400 nM at passage 10. The concentration of RANTES was kept constant from passages 10 to 13. Variants isolated under these two protocols were designated as R50 and R90, respectively, and control variants selected in parallel without RANTES were designated as C, followed by the passage number at isolation.
Virus samples from passages 2 to 7 and 13 were collected, and multiple independent Env clones were sequenced (Table 1). No mutations in SF162 Env were detected at R50-2, R90-2, or C-2, even though a minor population (8/73 clones, 11%) of the starting stock harbored the I309R substitution, and 1/73 clones had both the I309R and A316V mutations. This apparent loss (which was soon reversed) of the I309R mutation may be related to lower replication fitness in cell lines (Fig. 1). However, by week 3 of culture, 40% of control Env clones had the I309R mutation, and 33% of Env clones of R50 variants had both the I309R and the A316V mutations. These trends continued during weeks 4 to 7 of selection, and by 13 weeks, both the I309R and A316V mutations were found in all Env clones except for one out of seven sequences in C-13. Further culture of these variants in MT-4 cells selected for both V3 mutations without exception whether or not they were growing under the pressure of RANTES.
The rare presence of Env variants in the SF162 stock harboring both V3 mutations led us to examine the entire gp160 (gp120 + gp41) sequence of the starting SF162 stock and the passage 13 isolates (Table 2). We found up to 10 variable sites in gp120 and gp41 in the SF162 stock, with two invariant changes in gp41 that had previously been observed in SF162 obtained from the same source (3). In addition to the two V3 mutations, RANTES selection appeared to select for the M434K change in the bridging sheet of gp120 and E137K in HR2 of gp41 (Table 2), changes that may impact the kinetics of virus entry (33).
TABLE 2.
Sequence changes in SF162 gp120 and gp41 before and after selection
| Protein, region, and mutation | No. of mutated Env clones/total no. of clones for virusa:
|
|||||
|---|---|---|---|---|---|---|
| SF162 stock from LBMb | C-13 | R50-13 | R90-13 | MT-4 C-13 | MT-4 R90-13 | |
| gp120 | ||||||
| C1 | ||||||
| K46R | 0/7 | 0/10 | 0/9 | 0/9 | 1/9 | 1/9 |
| V1 | ||||||
| R151K | 3/7 | 4/10 | 0/9 | 0/9 | 5/9 | 0/9 |
| G152K | 0/7 | 0/10 | 0/9 | 0/9 | 0/9 | 2/9 |
| V2 | ||||||
| I165V | 2/7 | 0/10 | 0/9 | 0/9 | 0/9 | 0/9 |
| Y177H | 0/7 | 6/10 | 7/9 | 7/9 | 5/9 | 9/9 |
| C2 | ||||||
| N197K | 0/7 | 0/10 | 0/9 | 0/9 | 2/9 | 0/9 |
| D230N | 0/16 | 3/19 | 0/18 | 0/18 | 4/18 | 0/18 |
| V3 | ||||||
| I309R | 8/73 | 10/15 | 15/15 | 15/15 | 15/15 | 14/14 |
| A316V | 1/73 | 10/15 | 15/15 | 15/15 | 15/15 | 14/14 |
| β20-21 | ||||||
| Q428K | 0/15 | 0/15 | 11/15 | 0/15 | 0/15 | 0/15 |
| E429K | 0/15 | 1/15 | 0/15 | 0/11 | 0/15 | 0/15 |
| M434K | 0/15 | 3/15 | 14/17 | 10/11 | 9/17 | 18/18 |
| gp41 | ||||||
| C5 | ||||||
| L494V | 7/9 | 10/10 | 8/8 | 3/5 | 6/8 | 9/9 |
| FP | ||||||
| A14T | 2/9 | 3/10 | 0/9 | 0/9 | 0/9 | 2/9 |
| HR1 | ||||||
| R46K | 4/9 | 1/10 | 0/9 | 0/9 | 0/9 | 0/9 |
| HR2 | ||||||
| N129S | 19/19 | 18/18 | 15/16 | 13/13 | 18/18 | 18/18 |
| E137K | 1/18 | 0/10 | 4/10 | 5/8 | 0/10 | 5/8 |
| TM | ||||||
| V182I | 3/9 | 4/10 | 4/10 | 5/8 | 4/10 | 0/8 |
| T189A | 9/9 | 10/10 | 10/10 | 8/8 | 10/10 | 8/8 |
| CP | ||||||
| R196K | 0/9 | 0/10 | 0/10 | 5/8 | 0/10 | 4/8 |
| L274Q | 6/9 | 10/10 | 10/10 | 8/8 | 10/10 | 8/8 |
Bold roman numbers indicate an increase in mutation frequencies (positive selection) relative to SF162 stock sequence, and bold italicized numbers indicate a decrease in mutation frequencies (negative selection).
LBM, Leonid B. Margolis.
To investigate whether the mutations evolving in SF162 passaged in PBMC reflected biological properties of the mutated viruses, we evaluated viral replication in MT-4 cells (CXCR4+ CCR5−) as well as in PBMC and in explants of human tonsillar tissue (16) in the presence of CXCR4 ligand JM-2987 (functionally equivalent hydrobromide salt of AMD3100). Mutants isolated at different passages along with control SF162 were inoculated into MT-4 cells (2.5 ng p24 per 106 cells), and viral replication was monitored for 5 days (Fig. 2). Parental SF162 as well as R90-2 and C-2 variants replicated in these cells to a very low level. SF162 variants isolated from later passages, R90-4, R90-7, and R90-13 as well as C-4, C-7, and C-13, replicated in MT-4 much more efficiently and in a passage-dependent manner. MT-4 cells infected with the R90-13 isolate produced >50,000 times more p24 than cells infected with the R90-2 isolate, and >10 times more than with the C-13 isolate (Fig. 2). A similar pattern of infection was observed for R50 variants. The ability of SF162 isolates to infect MT-4 cells strongly correlated with the level of I309R/A316V quasispecies (R = 0.99), although a role for other mutations cannot be excluded.
FIG. 2.
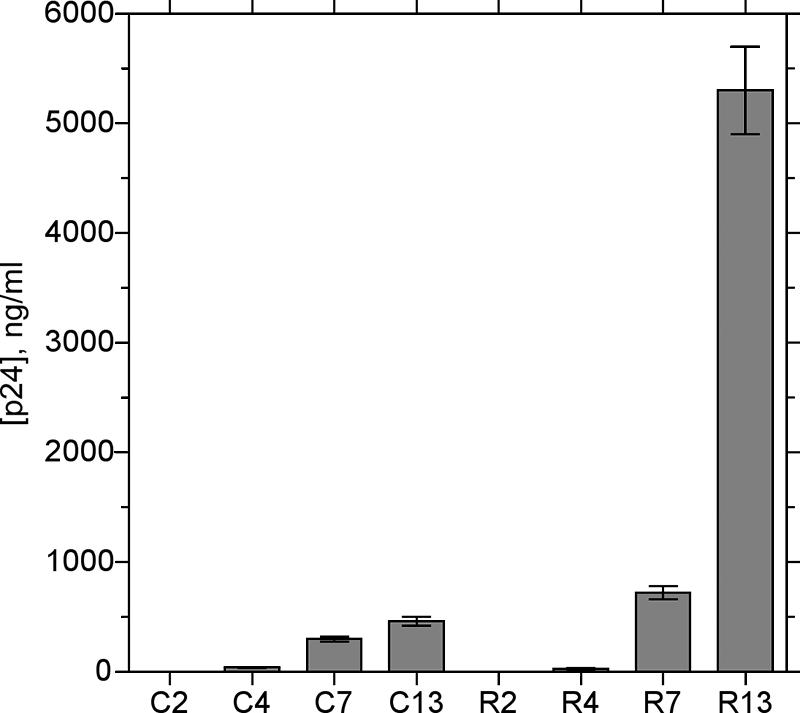
Replication in MT-4 cell line of SF162 variants propagated in PBMC with and without RANTES. SF162 was propagated in PBMC in the presence of escalating concentrations of RANTES and passaged as described in the text. R, R90 variants isolated from PBMC in the presence of RANTES; C, control variants isolated from PBMC. Numbers indicate the passages at which viruses have been isolated (Table 1). Presented are average cumulative amounts (± standard errors of the means) of HIV-1 p24 capsid antigen released by infected MT-4 cells over 5 days of infection.
Replication of the parental stock of SF162 in PBMC was almost completely inhibited by RANTES, whereas infection of PBMC by the C-13 isolate was only partially inhibited and infection by the R90-13 isolate was RANTES insensitive. By contrast, R90-13 replication in PBMC was almost completely (by 98.3% ± 0.3%) inhibited by 100 nM JM-2987, whereas, under similar conditions, R90-2 was inhibited by only 7.2% ± 1.6%. Furthermore, in tonsillar explants (16) inoculated with R90-13 or R50-13, viral replication was consistently inhibited by 100 to 1,000 nM JM-2987 (data not shown).
These results suggest that under all tested conditions SF162 has a single pathway of V3 mutations leading to CXCR4 use and that prolonged replication in activated PBMC is sufficient to select for these two recurring mutations. The presence of CCR5-binding chemokines accelerates this selection, but the SF162 evolutionary pathway remains the same. The unique pathway of SF162 evolution may be related to its site of isolation (9). We speculate that when the precursor of SF162 spread from plasma to the brain, it lost the I309R + A316V mutations and reverted from R5X4 to R5 HIV-1. We observed the same codon changes in all V3 mutations, which appears to reflect the codon bias imposed by the A-rich HIV-1 genome (2) and may facilitate forward or reverse mutation of these two sites. Our results suggest that, for HIV-1 variants with an intrinsic tendency to mutate towards CXCR4 usage or that already have acquired important mutations on this pathway, the high level of CC chemokines in HIV-infected patients or the use of CCR5 inhibitors may accelerate the pace of HIV evolution from a less-pathogenic R5 towards more-pathogenic (5, 12) R5X4 or X4 variants.
Nucleotide sequence accession numbers.
The Env sequences reported here have been deposited with GenBank under accession numbers EF208071 through EF208086.
Acknowledgments
This work was supported by NIH grant AI-52778 to D.E.M. The work of Y.K. and L.B.M. was supported by the NICHD Intramural Program.
We are grateful to M. R. Santi and the staff of the Children's National Medical Center for providing tonsillar tissues.
This is publication 18549-IMM from The Scripps Research Institute.
Footnotes
Published ahead of print on 3 January 2007.
REFERENCES
- 1.Aarons, E. J., S. Beddows, T. Willingham, L. Wu, and R. A. Koup. 2001. Adaptation to blockade of human immunodeficiency virus type 1 entry imposed by the anti-CCR5 monoclonal antibody 2D7. Virology 287:382-390. [DOI] [PubMed] [Google Scholar]
- 2.Berkhout, B., A. Grigoriev, M. Bakker, and V. V. Lukashov. 2002. Codon and amino acid usage in retroviral genomes is consistent with virus-specific nucleotide pressure. AIDS Res. Hum. Retrovir. 18:133-141. [DOI] [PubMed] [Google Scholar]
- 3.Binley, J. M., T. Wrin, B. Korber, M. B. Zwick, M. Wang, C. Chappey, G. Stiegler, R. Kunert, S. Zolla-Pazner, H. Katinger, C. J. Petropoulos, and D. R. Burton. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78:13232-13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjorndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyo. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bozzette, S., J. McCutchan, S. Spector, B. Wright, and D. Richman. 1993. A cross-sectional comparison of persons with syncytium- and non-syncytium-inducing human immunodeficiency virus. J. Infect. Dis. 168:1374-1379. [DOI] [PubMed] [Google Scholar]
- 6.Brumme, Z. L., J. Goodrich, H. B. Mayer, C. J. Brumme, B. M. Henrick, B. Wynhoven, J. J. Asselin, P. K. Cheung, R. S. Hogg, J. S. Montaner, and P. R. Harrigan. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis. 192:466-474. [DOI] [PubMed] [Google Scholar]
- 7.Carrillo, A., and L. Ratner. 1996. Cooperative effects of the human immunodeficiency virus type 1 envelope variable loops V1 and V3 in mediating infectivity for T cells. J. Virol. 70:1310-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan, S. Y., R. F. Speck, C. Power, S. L. Gaffen, B. Chesebro, and M. A. Goldsmith. 1999. V3 recombinants indicate a central role for CCR5 as a coreceptor in tissue infection by human immunodeficiency virus type 1. J. Virol. 73:2350-2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng-Mayer, C., and J. A. Levy. 1988. Distinct biological and serological properties of human immunodeficiency viruses from the brain. Ann. Neurol. 23(Suppl.):S58-S61. [DOI] [PubMed] [Google Scholar]
- 10.Cheng-Mayer, C., M. Quiroga, J. W. Tung, D. Dina, and J. A. Levy. 1990. Viral determinants of human immunodeficiency virus type 1 T-cell or macrophage tropism, cytopathogenicity, and CD4 antigen modulation. J. Virol. 64:4390-4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocchi, F., A. L. DeVico, A. Garzino-Demo, A. Cara, R. C. Gallo, and P. Lusso. 1996. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 2:1244-1247. [DOI] [PubMed] [Google Scholar]
- 12.Connor, R. I., K. E. Sheridan, D. Ceradini, S. Choe, and N. R. Landau. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185:621-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dejucq, N., G. Simmons, and P. R. Clapham. 2000. T-cell line adaptation of human immunodeficiency virus type 1 strain SF162: effects on envelope, vpu and macrophage-tropism. J. Gen. Virol. 81:2899-2904. [DOI] [PubMed] [Google Scholar]
- 14.Donzella, G. A., D. Schols, S. W. Lin, J. A. Este, K. A. Nagashima, P. J. Maddon, G. P. Allaway, T. P. Sakmar, G. Henson, E. De Clercq, and J. P. Moore. 1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 4:72-77. [DOI] [PubMed] [Google Scholar]
- 15.Este, J. A., C. Cabrera, J. Blanco, A. Gutierrez, G. Bridger, G. Henson, B. Clotet, D. Schols, and E. De Clercq. 1999. Shift of clinical human immunodeficiency virus type 1 isolates from X4 to R5 and prevention of emergence of the syncytium-inducing phenotype by blockade of CXCR4. J. Virol. 73:5577-5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glushakova, S., B. Baibakov, J. Zimmerberg, and L. B. Margolis. 1997. Experimental HIV infection of human lymphoid tissue: correlation of CD4+ T cell depletion and virus syncytium-inducing/non-syncytium-inducing phenotype in histocultures inoculated with laboratory strains and patient isolates of HIV type 1. AIDS Res. Hum. Retrovir. 13:461-471. [DOI] [PubMed] [Google Scholar]
- 17.Groenink, M., R. A. Fouchier, S. Broersen, C. H. Baker, M. Koot, A. B. van't Wout, H. G. Huisman, F. Miedema, M. Tersmette, and H. Schuitemaker. 1993. Relation of phenotype evolution of HIV-1 to envelope V2 configuration. Science 260:1513-1516. [DOI] [PubMed] [Google Scholar]
- 18.Harrowe, G., and C. Cheng-Mayer. 1995. Amino acid substitutions in the V3 loop are responsible for adaptation to growth in transformed T-cell lines of a primary human immunodeficiency virus type 1. Virology 210:490-494. [DOI] [PubMed] [Google Scholar]
- 19.Hartley, O., H. Gaertner, J. Wilken, D. Thompson, R. Fish, A. Ramos, C. Pastore, B. Dufour, F. Cerini, A. Melotti, N. Heveker, L. Picard, M. Alizon, D. Mosier, S. Kent, and R. Offord. 2004. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. USA 101:16460-16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman, N. G., F. Seillier-Moiseiwitsch, J. Ahn, J. M. Walker, and R. Swanstrom. 2002. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J. Virol. 76:3852-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang, S. S., T. J. Boyle, H. K. Lyerly, and B. R. Cullen. 1991. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 253:71-74. [DOI] [PubMed] [Google Scholar]
- 22.Jensen, M. A., F. S. Li, A. B. van 't Wout, D. C. Nickle, D. Shriner, H. X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77:13376-13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozak, S. L., E. J. Platt, N. Madani, F. E. Ferro, Jr., K. Peden, and D. Kabat. 1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol. 71:873-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margolis, L., and R. Shattock. 2006. Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper’ problem resolved? Nat. Rev. Microbiol. 4:312-317. [DOI] [PubMed] [Google Scholar]
- 25.Margolis, L. B., S. Glushakova, J. C. Grivel, and P. M. Murphy. 1998. Blockade of CC chemokine receptor 5 (CCR5)-tropic human immunodeficiency virus-1 replication in human lymphoid tissue by CC chemokines. J. Clin. Investig. 101:1876-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marozsan, A. J., S. E. Kuhmann, T. Morgan, C. Herrera, E. Rivera-Troche, S. Xu, B. M. Baroudy, J. Strizki, and J. P. Moore. 2005. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology 338:182-199. [DOI] [PubMed] [Google Scholar]
- 27.Moore, J. P., S. G. Kitchen, P. Pugach, and J. A. Zack. 2004. The CCR5 and CXCR4 coreceptors—central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retrovir. 20:111-126. [DOI] [PubMed] [Google Scholar]
- 28.Mosier, D. E., G. R. Picchio, R. J. Gulizia, R. Sabbe, P. Poignard, L. Picard, R. E. Offord, D. A. Thompson, and J. Wilken. 1999. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J. Virol. 73:3544-3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overbaugh, J., and C. R. Bangham. 2001. Selection forces and constraints on retroviral sequence variation. Science 292:1106-1109. [DOI] [PubMed] [Google Scholar]
- 30.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastore, C., G. R. Picchio, F. Galimi, R. Fish, O. Hartley, R. E. Offord, and D. E. Mosier. 2003. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 47:509-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastore, C., A. Ramos, and D. E. Mosier. 2004. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol. 78:7565-7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reeves, J. D., J. L. Miamidian, M. J. Biscone, F. H. Lee, N. Ahmad, T. C. Pierson, and R. W. Doms. 2004. Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and entry inhibitor sensitivity. J. Virol. 78:5476-5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. de Goede, R. P. van Steenwijk, J. M. Lange, J. K. Schattenkerk, F. Miedema, and M. Tersmette. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66:1354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shioda, T., J. A. Levy, and C. Cheng-Mayer. 1992. Small amino acid changes in the V3 hypervariable region of gp120 can affect the T-cell-line and macrophage tropism of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 89:9434-9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71:7136-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trkola, A., S. E. Kuhmann, J. M. Strizki, E. Maxwell, T. Ketas, T. Morgan, P. Pugach, S. Xu, L. Wojcik, J. Tagat, A. Palani, S. Shapiro, J. W. Clader, S. McCombie, G. R. Reyes, B. M. Baroudy, and J. P. Moore. 2002. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. USA 99:395-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Rij, R. P., H. Blaak, J. A. Visser, M. Brouwer, R. Rientsma, S. Broersen, A. M. de Roda Husman, and H. Schuitemaker. 2000. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J. Clin. Investig. 106:1039-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van 't Wout, A. B., H. Blaak, L. J. Ran, M. Brouwer, C. Kuiken, and H. Schuitemaker. 1998. Evolution of syncytium-inducing and non-syncytium-inducing biological virus clones in relation to replication kinetics during the course of human immunodeficiency virus type 1 infection. J. Virol. 72:5099-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]