

Abstract
Infection by Kaposi's sarcoma-associated herpesvirus (KSHV) is required for the development of Kaposi's sarcoma (KS), a highly inflammatory angiogenic tumor of endothelial cells commonly found in untreated AIDS patients. Angiopoietin 2 (Ang-2) modulates the vasculature during inflammation and angiogenesis, but the mechanism by which KSHV regulates Ang-2 expression has not been investigated. Here, we show that KSHV infection of primary human umbilical vein endothelial cells induced the expression and release of Ang-2, which in turn was required for KSHV-induced paracrine-dependent angiogenesis in vivo. Ang-2 was strongly expressed in small vessels and spindle tumor cells in KS tumors. Mechanistically, KSHV activated the Ang-2 promoter via AP-1 and Ets1 transcriptional factors, which were mediated by ERK, JNK, and p38 mitogen-activated protein kinase (MAPK) pathways. Our findings demonstrate the importance of Ang-2 in KS angiogenesis and define a novel role for AP-1 and MAPK pathways in regulating angiogenesis. This study also illustrates a distinct mechanism by which a tumor virus modulates vasculature to promote tumorigenesis and exemplifies the convergence of oncogenesis and angiogenesis pathways in tumor development.
Two families of endothelium-specific angiogenic factors, vascular endothelial growth factors (VEGFs) and angiopoietins, regulate vascular development and angiogenesis in a complementary and coordinated fashion (35, 70). While VEGFs enhance angiogenesis by promoting the proliferation of endothelial cells (ECs), angiopoietins are necessary for blood vessel remodeling, sprouting, and maturation (35). Angiopoietin 1 (Ang-1) is an agonist of the Tie-2 receptor and stabilizes blood vessels by promoting adhesive interactions between ECs and mural cells (33, 67). Conversely, Ang-2 is an antagonist of the Tie-2 receptor and induces the detachment of mural cells and destabilizes the existing blood vessels, a process that is essential for new blood vessel formation (26, 44). Ang-2 has limited expression in normal tissues but is strongly expressed in remodeling vasculature during embryogenesis, inflammation, and carcinogenesis (26, 42, 44). Thus, Ang-2 is implicated as an essential factor in tumor-driven angiogenesis (20, 68).
The expression of Ang-2 is upregulated by cytokines, growth factors, and environmental conditions, which include VEGF, angiotensin II, leptin, estrogen, HER2, tumor necrosis factor alpha (TNF-α), and hypoxia (11, 14, 25, 37, 45, 54, 56, 77). Examination of the promoter of Ang-2 revealed Ets1 as an important transcriptional factor that regulates the expression of Ang-2 (32, 34). A glucose-responsive GC box in the Ang-2 promoter mediates increased Ang-2 expression during glycolysis by decreasing mSin3A-Sp3 complex binding to the promoter as a result of the coordinated methylglyoxal and O-linked N-acetylglucosamine modifications of mSin3A and Sp3, respectively (76). Despite these studies, the mechanism controlling the expression of Ang-2, particularly in tumor-driven angiogenesis, remains unclear.
Kaposi's sarcoma (KS) is the dominant malignancy in patients with AIDS. Because human immunodeficiency virus type 1 infection is common in sub-Saharan Africa, KS is now one of the most frequent neoplasms in the region, and in some countries, it accounts for up to 30% of all cancers (55). Infection by Kaposi's sarcoma-associated herpesvirus (KSHV) is necessary for the development of all clinical forms of KS, including classical KS; transplantation or iatrogenic KS; African or endemic KS; and AIDS-related or epidemic KS (17). KS is a highly angiogenic vascular spindle tumor of proliferating ECs infiltrated with inflammatory cells. While early-stage KS resembles hyperplasia, the late stage is either clonal or oligoclonal containing genetic instability, and thus is considered a true cancer (65). Despite intensive studies, the mechanism by which KSHV induces malignant transformation and the development of KS is incompletely understood. While KSHV infection of human primary ECs induces chromosome instability (58), the extensive angiogenesis and inflammation in KS lesions can also contribute to cellular transformation and development of malignant tumors (18). A number of KSHV proteins, including viral interleukin 6 (vIL-6) (open reading frame [ORF]-K2), vGPCR (ORF74), vCCL-1 (ORF-K6), and vCCL-II (ORF-K4), promote angiogenesis by regulating various cellular signaling pathways (5, 7, 43, 50, 59, 63, 64). KSHV infection also directly induces several angiogenic and inflammatory cytokines, including VEGF, bFGF, IL-6, IL-8, GRO-α, TNF-β, and ephrin B2, most of which are expressed in KS lesions (46, 47, 52, 72, 75). The mRNAs of both Ang-1 and Ang-2 have been detected at higher levels in KS tumors than in the adjacent normal tissues (9), but the protein expression levels of these angiogenic factors in KS tumors remain unknown. Furthermore, the mechanism by which KSHV induces Ang-1 and Ang-2 has not been determined. In this study, we showed that Ang-2 protein was highly expressed in KS tumors and that KSHV infection of primary human umbilical vein ECs (HUVEC) induced Ang-2, which was required for KSHV-induced angiogenesis. In addition, we found that KSHV induced Ang-2 expression by activating AP-1 and Ets1 via the ERK, JNK, and p38 mitogen-activated protein kinase (MAPK) pathways. These results demonstrate an important role for Ang-2 in KS tumorigenesis and define novel pathways controlling the expression of Ang-2.
MATERIALS AND METHODS
Virus preparation.
Concentrated virus was prepared from BCBL-1 cells or 293 and BCBL-1 cells infected with a recombinant KSHV, BAC36 (57). Briefly, supernatant from O-tetradecanoylphorbol 13-acetate (Sigma, St. Louis, MO)-induced cells was first centrifuged twice at 5,000 × g for 10 min to eliminate cell debris and then at 100,000 × g for 1 h with 20% sucrose as a cushion. The final pellet was dissolved in basic culture medium without growth factors and other supplements overnight. Fresh virus preparations were used in the experiments.
Cell culture and KSHV infection.
HUVEC and human dermal microvascular ECs (DMVEC) obtained from Clonetics (Walkersville, MD) were grown in EBM-2 medium (Clonetics). KSHV infection was previously described (27, 81). For all the experiments, HUVEC cultures were infected with BAC36 KSHV at a 40 to 50% efficiency based on the percentage of cells expressing green fluorescent protein (GFP) at 2 days postinfection (dpi). DMVEC were infected with KSHV prepared from BCBL-1 cells. To inhibit a MAPK pathway, a specific inhibitor was added to the culture 30 min prior to infection: U0126 (10 μM) for the MEK pathway, SB203580 (50 μM) for the p38 pathway, and JNK inhibitor II (50 μM) for the JNK pathway, all of which were purchased from Calbiochem (Oakland, CA).
Western blot analysis.
To detect Ang-2 secretion, protein preparations from supernatants of mock- and KSHV-infected HUVEC cultures were separated in sodium dodecyl sulfate-polyacrylamide electrophoresis gels under reducing conditions and transferred to nitrocellulose membranes as previously described (28). The membrane was incubated first with a purified goat anti-Ang-2 immunoglobulin G (IgG) (Santa Cruz, Santa Cruz, CA) and then with a donkey anti-goat IgG-horseradish peroxidase conjugate (Sigma). Specific signals were revealed with chemiluminescence substrates and recorded on films or a Cyclone Phosphorimager (Perkin-Elmer, Shelton, CT). For the detection of Ets1 and β-actin, protein preparations were directly prepared from mock- and KSHV-infected HUVEC. Purified IgG of a rabbit polyclonal antibody to Ets1 (Santa Cruz) was used to detect the Ets1 protein. A goat anti-rabbit IgG-horseradish peroxidase conjugate (Sigma) was used to reveal the signal. β-Actin was detected with a mouse IgG monoclonal antibody (Santa Cruz) and revealed with a goat anti-mouse IgG-horseradish peroxidase conjugate (Sigma).
RNA extraction and RT-PCR.
Total RNA from HUVEC was purified using an RNA isolation kit (Promega, Madison, WI), which included an RNase-free DNase 1 treatment step to remove any possible genomic-DNA contamination. Total RNA was then converted into cDNA using Superscript H− reverse transcriptase (Invitrogen, Carlsbad, CA) with a poly(dT)18 primer. Reverse transcription (RT)-PCR was conducted to quantify individual mRNA transcripts. The Ang-2 primers were 5′TGGAAGCTGGAGGAGGCGGGTGG3′ and 5′ATGTGGTGGAAGAGGACACAGTG3′. The amplified 638-bp fragment is within the C terminus of the Ang-2 gene coding region. β-Actin was used for normalization. The β-actin primers were 5′CTGGAACGGTGAAGGTGACA3′ and 5′AAGGGACTTCCTGTAACAATGCA3′, which amplified a fragment of 140 bp. The PCR products were analyzed on a 1.2% agarose gel by electrophoresis. PCR was also carried out on the RNA samples without RT to exclude any possible genomic-DNA contamination.
To monitor the KSHV replication program in the infected cultures, we carried out RT-PCR for a set of KSHV genes that included two lytic genes, RTA (ORF50) and ORF-K8.1, and two latent genes, major latent nuclear antigen (LANA or LNA, ORF73) and vFLIP (ORF-K13). The RTA primers were 5′CACAAAAATGGCGCAAGATGA3′ (forward) and 5′TGGTAGAGTTGGGCCTTCAGTT3′ (reverse), which amplified a product of 98 bp. The ORF-K8.1 primers were 5′AAAGCGTCCAGGCCACCACAGA3′ (forward) and 5′GGCAGAAAATGGCACACGGTTAC3′ (reverse), which amplified a product of 160 bp. The LANA primers were 5′GCAGACACTGAAACGCTGAA3′ (forward) and 5′AGGTGAGCCACCAGGACTTA3′ (reverse), which amplified a product of 101 bp. The vFLIP primers were 5′GGATGCCCTAATGTCAATGC3′ (forward) and 5′GGCGATAGTGTTGGGAGTGT3′ (reverse), which amplified a product of 113 bp.
Immunocytochemistry.
After microwave pretreatment (pH 7.2) for epitope retrieval, 3- or 4-μm serial sections from paraffin-embedded, formalin-fixed KS lesions were stained with an affinity-purified goat polyclonal antibody to Ang-2 (R&D Systems, Abingdon, United Kingdom) at 1:100 dilution or to an isotype control antibody (R&D Systems). The primary antibodies were then revealed with an anti-goat-horseradish peroxidase conjugate (R&D Systems), and the signal intensity was increased by the tyramine amplification technique, as described previously (51, 71). To detect LANA, a rat monoclonal antibody to LANA (Novocastra Laboratories, Newcastle Upon Tyne, United Kingdom) was used at 1:750 dilution, and the signals were revealed with an anti-rat secondary antibody-horseradish peroxidase conjugate (R&D Systems). KS specimens were obtained from the San Antonio Cancer Institute Pathology Core Laboratory, in accordance with U.S. regulations, and from the archives of the Institute for Pathology, Medizinische Hochschule Hannover, and the Krankenhaus Nordstadt, where specimens were acquired in accordance with guidelines of the local ethics committees.
In vivo Matrigel angiogenesis assay.
C57/B6 mice (4 weeks old) were subcutaneously injected with supernatants from mock- or KSHV-infected HUVEC cultures (Supmock or SupKSHV, respectively) mixed with Matrigel (Becton Dickinson, Franklin Lakes, NJ). Supernatants were obtained by infecting HUVEC with KSHV virions in basic EBM-2 without supplements (SupKSHV) or by adding basic EBM-2 without supplements to the cells (Supmock) for 12 h. Cellular debris was then removed by centrifugation at 12,000 rpm for 30 min. Six groups of animals were used, including those injected with SupKSHV, Supmock, SupKSHV plus 1 μg of purified goat anti-Ang-2 IgG (Santa Cruz) or purified control IgG from normal goat serum (Santa Cruz), or Supmock plus 100 ng recombinant Ang-2 (Calbiochem) with or without the anti-Ang-2 antibody. For each group, four mice, each with two sites, were injected. Each site was injected with 200 μl of a corresponding culture supernatant and 350 μl Matrigel. To observe angiogenesis, the Matrigel plugs were surgically removed 10 days after injection and immediately photographed to evaluate the growth of blood vessels. Histological sections were stained with Masson's trichrome (Sigma, St. Louis, MO) for ECs/vessels (red) and Matrigel (blue) and an antibody to VE-Cadherin (Santa Cruz). The hemoglobin content was assayed using the Drabkin reagent (Sigma) after the Matrigel specimens were homogenized in 50 mM EDTA. The Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio approved all animal housing and surgical procedures in accordance with the University of Texas Health Science Center at San Antonio guidelines.
Reporter constructs and reporter assay.
A 3.2-kb genomic fragment containing the full-length human Ang-2 promoter was PCR cloned into the pGL3-basic vector (Promega) to obtain the reporter plasmid pEBS1. There are 10 putative Ets1-binding sites (EBSs), annotated as E1 to E10, and 8 putative AP-1-binding sites (ABSs), annotated as A1 to A8, in the Ang-2 promoter (see Fig. 4A). Based on the positions of the EBSs and ABSs, consecutive deletion constructs pEBS1 to -10 containing different truncations of the Ang-2 promoter regions, designated D1 to D10, were generated by PCR amplification (see Fig. 4A). Mutant promoter reporters were generated using the pEBS7 reporter as a template by site-directed mutagenesis to ablate the ABSs or EBSs using the QuikChange site-directed mutagenesis kit according to the instructions of the manufacturer (Stratagene, San Diego, CA). The correct mutations of the sites were verified by DNA sequencing (Table 1) . Other plasmids used were expression plasmids of AP-1 members, pRSV-c-Fos and pRSV-c-Jun, provided by B. E. Sawaya (Temple University). The dominant-negative mutant (DN) of the c-Fos expression plasmid (termed A-Fos) was from Charles Vinson (National Institutes of Health). The c-Jun DN expression plasmid pCMV-TAM67 was from Bradford W. Ozanne (Beatson Institute). The p38 DN (pcDNA3-p38/AF) was from Jiahua Han (The Scripps Institute). The JNK DN (HA-JNK1 [APF]) was from Lin Mantell (New York University School of Medicine). The ERK DN (pCEP4L-HA-ERK1K71R) was from Melanie Cobb (University of Texas Southwestern Medical Center). The Ets1 DN, which lacked a transcription activation domain corresponding to amino acids 306 to 441, was obtained by PCR cloning into the pCMV-myc vector (BD Biosciences, Palo Alto, CA) using primers 5′CTATGTGCGGGACCGTGCTGACC3′ and 5′TCACTCGTCGGCATCTGGCTTGAC3′.
FIG. 4.

Identification of ABS and EBS as the KSHV-responsive cis elements in the Ang-2 promoter. (A) Schematic illustration of the Ang-2 promoter deletion constructs (pEBS1 to -10), with D1 to D10 corresponding to the respective deletion regions. ABS1 to ABS8 and EBS1 to EBS10 are abbreviated as A1 to A8 and E1 to E10, respectively. (B) KSHV activation of the full-length Ang-2 promoter-reporter pEBS1 at 12 and 52 hpi. Activation was calculated by standardizing the reporter activity of the mock-infected cells as 1. (C) KSHV activation of the Ang-2 promoter-reporter deletion constructs at 52 hpi. Activation was calculated by standardizing the control vector pGL3-basic reporter activity in mock-infected cells as 1. The numbers above the bars denote the increase (n-fold) in KSHV-infected cells compared to mock-infected cells. (D) KSHV activation of the reporter pEBS7 and its mutant constructs with mutation of either a single site (pMutE7, pMutE8, pMutA7, and pMutA8) or dual sites (pMutE7A7 and pMutA7E8) at 12 and 52 hpi. Activation was calculated by standardizing the reporter activity of the mock-infected cells as 1. In all the experiments, cells transfected with either deletion or mutation constructs of the Ang-2 promoter for 12 h were infected with KSHV and assayed for luciferase at either 12 or 52 hpi. The results represent averages with standard deviations.
TABLE 1.
Mutagenesis analysis of putative ABSs and EBSs in the Ang-2 promoter
| Putative site | Sequencea
|
|
|---|---|---|
| Wild type | Mutant | |
| E7 | 5′-ATTTTTCCTGT-3′ | 5′-ATTTAGCATGT-3′ |
| E8 | 5′-ACAGGAAGATA-3′ | 5′-ACAAGTCGATA-3′ |
| A7 | 5′-AGTGACCCCC-3′ | 5′-AGAGTCCGCC-3′ |
| A8 | 5′-GCTGACACAG-3′ | 5′-GCAGTCAGAG-3′ |
Mutated sites are underlined.
Transfection of HUVEC was carried out using Cytopure Transfection Reagent according to the instructions of the manufacturer (MP Biomedicals, Irvine, CA). In all the experiments, we estimated the transfection efficiency to be around 30 to 40% using the GFP expression vector pEGFP (Clontech Laboratories, Inc., Mountain View, CA) as a control. The β-galactosidase vector pSV-β-Gal was included to control the transfection efficiency. Luciferase activities were measured with a specific assay kit (Promega). To identify the cis elements that were responsive to KSHV infection, cells transfected with deletion or mutation constructs of the Ang-2 promoter for 12 h were infected with KSHV and assayed for luciferase at either 12 or 52 hours postinfection (hpi). To determine the effect of an expression plasmid on the Ang-2 promoter, reporter pEBS1 was cotransfected with the expression plasmid.
Electrophoretic mobility shift assay (EMSA).
Nuclear extracts were prepared from mock- or KSHV-infected HUVEC as previously described (73). Annealed double-stranded consensus AP-1 (5′CGGTGACTCACAGCT3′) and Ets1 (5′CCCCTACAGGAAGATAACGG3′) oligonucleotides were labeled with [γ-32P] ATP. For the gel shift assay, 4 μg of nuclear extract was incubated for 20 min at room temperature with 5 × 105 cpm DNA probe in a total volume of 20 μl binding buffer containing 10 mM Tris-HCl at pH 7.6, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 5% glycerol, 1% bovine serum albumin, 2 μg of poly(dI-dC). For the competition assay, cold AP-1 wild-type probe and its corresponding mutant probe (5′CGGAAATTGACAGCT3′) or cold Ets1 probe and its corresponding mutant probe (5′CCCCTACATTCCGATAACGG3′) at 100-fold were added to the respective reaction mixtures. All the mutated sites are underlined. For the supershift assay, 1 μg purified rabbit polyclonal anti-c-Fos and anti-c-Jun IgG, or IgG of a negative control antibody (Santa Cruz), was added to the reaction mixture. Samples were loaded onto a 6% nondenaturing polyacrylamide gel for electrophoresis. Images were captured with the Cyclone Phosphorimager.
Chromatin immunoprecipitation (ChIP) assay.
Briefly, 4 ×105 HUVEC were washed with phosphate-buffered saline (PBS) and incubated at 37°C for 10 min with culture medium containing 1% formaldehyde. The cells were then washed twice with ice-cold PBS and harvested with a scraper in PBS. After centrifugation, the cell pellets were resuspended in the above-mentioned lysis buffer used for Western blotting lysate preparation, left on ice for 10 min, and sonicated for 10 s six to eight times. This process generated a smear with average DNA sizes of 0.6 to 0.8 kb. After centrifugation at 10,000 × g for 10 min at 4°C, the supernatants were diluted 10-fold in PBS and immunoprecipitated with the antibody to Ets1 or c-Jun at 4°C overnight. Purified IgG from a rabbit polyclonal antibody to Tie2 (Santa Cruz) was used as a negative control antibody. The chromatin-antibody complexes were then pulled down with protein G-agarose beads (Sigma), which were then sequentially washed four times with PBS-Tween and once with TE (10 mM Tris at pH 7.5, 1 mM EDTA). The immunoprecipitates were resuspended in TES (10 mM Tris at pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.05% sodium dodecyl sulfate, and 100 μg/ml proteinase K) and incubated at 50°C for 6 h, followed by phenol-chloroform extraction and ethanol precipitation. The DNA was dissolved in 20 μl of sterile water. PCR was carried out to amplify the immunoprecipitated Ang-2 promoter fragment using primers 5′CTAGCTAGCTATTTTGCCAGCTTAGCAC3′ (forward) and 5′AACTTAACTTGAGGCAAACACAC3′, which amplified a product of 641 bp. Initial DNA inputs were also amplified to estimate the recovery yields. As an additional control, a PCR was carried out to amplify the Ang-2 exon 9 outside the Ang-2 promoter region using primers 5′TGTGGTCCTTCCAACTTGAACGGAATG3′ and 5′ATGTGGTGGAAGAGGACACAGTG3′, which amplified a product of 246 bp.
RESULTS
KSHV infection induces the expression and release of Ang-2.
KSHV infection of primary HUVEC transforms them into spindle shapes, a phenotype reminiscent of the endothelial tumor cells in KS lesions (27). Since Ang-2 is the main factor involved in destabilizing the existing vasculature that is required for initiating angiogenesis, we hypothesized that induction of Ang-2 by KSHV might be one of the early events leading to KS angiogenesis. Indeed, KSHV infection of HUVEC induced secretion of Ang-2 as early as 12 hpi (Fig. 1A). KSHV infection also upregulated Ang-2 mRNA expression, which peaked at 52 hpi (Fig. 1C). In contrast, we did not detect Ang-2 secretion or mRNA expression in the mock-infected cultures (Fig. 1A and C). Since we calibrated the signals with the β-actin gene, the detected upregulation of Ang-2 mRNA by KSHV infection could not be due to cell doubling (Fig. 1C). In samples that were not subjected to RT, we did not detect any signals in both mock- and KSHV-infected cells (Fig. 1C), indicating that the amplified signals were from Ang-2 mRNA rather than any contaminated genomic DNA. To determine whether KSHV induction of Ang-2 expression could be sustained after longer infection, we examined the mRNA expression of Ang-2 at 144 hpi (6 dpi). As shown in Fig. 1C, the expression of Ang-2 mRNA was low but remained upregulated in KSHV-infected HUVEC even at 6 dpi.
FIG. 1.

KSHV infection of HUVEC induces Ang-2 expression and release. (A) KSHV infection induced Ang-2 release in HUVEC. Culture supernatants (50 μl) from mock- or KSHV-infected HUVEC at 12 or 52 hpi were examined for Ang-2 protein by Western blotting. A recombinant Ang-2 was used as a control. β-Actin expression was measured and used to normalize cell numbers. (B) KSHV infection induced Ang-2 release in DMVEC. Supernatants from mock- or KSHV-infected DMVEC at 7 dpi were examined for Ang-2 protein by Western blotting. (C) RT-PCR detection of Ang-2 and β-actin transcripts, as well as KSHV transcripts, for RTA, K8.1, LANA, and vFLIP genes in mock- or KSHV-infected HUVEC at different hpi. PCR was also carried out for Ang-2 and β-actin without RT in the RNA samples to exclude any possible genomic-DNA contamination.
We have previously shown that KSHV infection of HUVEC is productive at the early stage of infection (27, 78). To correlate the status of KSHV replication with Ang-2 upregulation, we examined the expression of a set of KSHV genes, including two lytic genes, the RTA and K8.1 genes, and two latent genes, the LANA and vFLIP genes. As we had previously reported, KSHV lytic genes were expressed at all the time points examined (Fig. 1C). These results further confirm that KSHV infection of HUVEC is productive. Since both viral latent and lytic genes are expressed in this system (78), KSHV upregulation of Ang-2 could be mediated by both latent and lytic genes.
In contrast to the HUVEC system, previous studies have shown that the default replication program in KSHV infection of DMVEC is latency (40). KSHV immediately establishes latent infection following infection of DMVEC. To determine whether KSHV also upregulates Ang-2 expression in latently infected ECs, we examined the protein expression of Ang-2 in DMVEC at 7 dpi, at which time KSHV had established latent infection in the culture (40). As shown in Fig. 1B, the expression of Ang-2 protein was significantly upregulated in KSHV-infected DMVEC compared to mock-infected cells. Analysis of mRNA expression in three separate microarray experiments using Affymetrix U133A 2.0 arrays showed that the expression of Ang-2 mRNA was upregulated 2.5 (±0.5)-fold in KSHV-infected DMVEC compared to mock-infected cells. These results indicate that KSHV upregulates the expression of Ang-2 in both default latent and productive infection systems.
Protein expression of Ang-2 in KS lesions.
To extend our observations to clinical samples, we performed immunohistochemical staining for Ang-2 protein expression in KS lesions. Consistent with the in vitro results, we found Ang-2 protein expression in 23 of 27 KS lesions, 20 of which had strong staining in intratumor vessels while 16 had focal staining in tumor cells (Fig. 2A and B). Of the 23 Ang-2-positive lesions, the majority of the tumor cells were also positive for KSHV LANA (Fig. 2C), confirming KSHV infection. Of the four Ang-2-negative lesions, two were also LANA negative. These results are consistent with the detection of high Ang-2 mRNA levels in KS lesions (9) and suggest an essential role for Ang-2 in KS angiogenesis.
FIG. 2.

Representative illustration of immunohistochemical detection of Ang-2 protein in sequential sections of a KS tumor. (A and B) Ang-2 staining showing strong expression of Ang-2 in small intratumor vessels (black arrows) and cytoplasmic staining in polygonal tumor cells (white arrows). Both panels A and B were from the same field, with B at a higher magnification. (C) Strong nuclear staining of LANA was seen in the majority of the tumor cells. (D) A section incubated with an isotype control antibody instead of either anti-Ang-2 or anti-LANA antibody was negative. The star identifies the position of the tumor in each section (panels A, C, and D).
Ang-2 is necessary for KSHV-induced paracrine-dependent angiogenesis.
To determine the role of Ang-2 in KS angiogenesis, we examined the angiogenic effects of KSHV-induced Ang-2 in a tumor-independent model involving subcutaneous implantation of reconstituted basement membrane (Matrigel) into mice and subsequent surgical removal of the pellets for angiogenesis assessment. Matrigel specimens containing SupKSHV were reddish with visible blood vessels (Fig. 3A, i). Histological examination revealed a large number of both micro- and macrovessels (Fig. 3B, i) that stained positive for VE-cadherin, a marker for ECs (Fig. 3C, i). In contrast, Matrigel specimens containing Supmock were white, with many fewer blood vessels (Fig. 3A, ii). Histological examination also revealed very few vessels (Fig. 3B, ii, and C, ii). To verify the functionality of the vessels, we determined the relative hemoglobin contents in the Matrigel pellets (Fig. 3D). Matrigel specimens containing SupKSHV had 30% higher levels of hemoglobin than those containing Supmock. These results indicated that KSHV infection of HUVEC could induce angiogenesis by a paracrine mechanism. To examine whether Ang-2 is essential for KSHV-induced paracrine-dependent angiogenesis, we used a specific antibody to inhibit the function of Ang-2 in this model. Addition of the anti-Ang-2 antibody to the Matrigel pellets abolished the angiogenic effect of SupKSHV, while a control antibody had no effect (Fig. 3A-C, iii and iv, respectively). Further evidence that Ang-2 was capable of inducing angiogenesis in this assay was provided by analysis of the Matrigel specimens containing Supmock supplemented with human recombinant Ang-2. They also had a strong reddish color and were rich in blood vessels (Fig. 3A to C, v). The anti-Ang-2 antibody abolished these angiogenic effects (Fig. 3A to C, vi). Examination of the relative hemoglobin contents showed that, similar to the Matrigel specimens containing SupKSHV, the specimens containing SupKSHV plus control antibody and Supmock plus recombinant Ang-2 had high levels of hemoglobin, while those containing SupKSHV plus anti-Ang-2 antibody and Supmock plus recombinant Ang-2 and anti-Ang-2 antibody had low levels, resembling that of Supmock (Fig. 3D). Taken together, these results indicated that KSHV-induced Ang-2 was necessary for the induction of angiogenesis and that angiogenesis could be recapitulated by treatment of Matrigel with mock-infected HUVEC culture supernatants supplemented with Ang-2 alone.
FIG. 3.

Ang-2 is required for KSHV-induced paracrine-dependent angiogenesis. (A) Supernatants from KSHV- and mock-infected HUVEC cultures were assayed for their abilities to induce angiogenesis in an in vivo angiogenesis assay. Matrigel pellets containing supernatants from HUVEC cultures infected by KSHV for 12 h (SupKSHV) had a large number of new blood vessels (i), while fewer blood vessels were present in those containing supernatants from mock-infected HUVEC cultures (Supmock) (ii). Addition of 1 μg/ml of IgG of an anti-Ang-2 antibody, but not a control antibody, to the Matrigel pellets inhibited the formation of blood vessels (iii and iv, respectively). Addition of 100 ng/ml of recombinant Ang-2 to the Matrigel pellets was sufficient to induce the formation of a large number of new blood vessels (v), which was inhibited by the anti-Ang-2 antibody (vi). (B) Histological staining of Matrigel pellet sections from panel A with Masson's trichrome showing promotion of both macro- and microvessels by KSHV-induced Ang-2 (the arrowheads show the borders of macrovessels, while the arrows show microvessels). (C) VE-cadherin staining of Matrigel pellet sections from panel A showing that the vessels were formed from ECs (the arrowheads show the borders of macrovessels, while the arrows show microvessels). (D) Hemoglobin contents of the Matrigel specimens from panel A. In all experiments, four mice, each injected at two sites, were used for each group. Representative illustrations are shown in panels A to C. In the hemoglobin content assay, the results are averages with standard deviations (D).
KSHV induces Ang-2 by activating AP-1 and Ets1.
Since we observed KSHV upregulation of Ang-2 mRNA, we determined possible KSHV transcriptional activation of the Ang-2 promoter. KSHV infection indeed activated the full-length Ang-2 promoter reporter pEBS1 by 2.88- and 10.51-fold at 12 and 52 hpi, respectively (Fig. 4B), which was consistent with the induction of Ang-2 mRNA by KSHV (Fig. 1B). Because KSHV activated the Ang-2 promoter to a much higher level at 52 hpi than at 12 hpi, subsequent promoter deletion analyses were conducted at the later time point. Previous studies identified 10 putative EBSs (E1 to E10) in the Ang-2 promoter (Fig. 4A) (32, 34). Our detailed examination of the Ang-2 promoter identified eight putative ABSs (A1 to A8) (Fig. 4A). Deletion analyses indicated that the promoter regions D8, D7, D5, D2, and D1 contributed positively to the Ang-2 promoter activity during KSHV infection (Fig. 4C). Compared to the luciferase vector control, reporters pEBS8, -7, -5, -2, and -1 had 14.12-, 46.13-, 55.32-, 44.96-, and 78.92-fold higher luciferase activities, respectively. Deletion of the D8, D7, D5, D2, and D1 regions reduced the luciferase activity by 2.62-, 3.27-, 1.63-, 1.38-, and 1.76-fold, respectively. Based on these results, we concluded that D8 containing the E8 site and D7 containing the E7 and A7 sites were the most important promoter regions contributing to the Ang-2 promoter activity during KSHV infection. KSHV infection increased the pEBS8 and pEBS7 reporters 3.61- and 4.33-fold at 52 hpi, respectively.
We further performed mutagenesis analyses using the pEBS7 reporter as a template to determine the contributions of the individual EBS and ABS sites in these regions to the Ang-2 promoter activity in response to KSHV infection at both 12 and 52 hpi (Fig. 4D). Multiple mutations were introduced into each E7, E8, A7, or A8 site to abolish the corresponding EBS or ABS (Table 1). KSHV infection activated the wild-type pEBS7 reporter 1.85-fold at 12 hpi, which was abolished by mutating the A7 site alone or together with other sites (Fig. 4D). Mutation of the E7, E8, or A8 site had no effect on KSHV activation of the pEBS7 reporter at that time point. KSHV infection activated the pEBS7 reporter 4.57-fold at 52 hpi, which was abolished by mutating either the E7 or A7 site alone or together with the other sites (Fig. 4D). Mutation of the E8 site reduced KSHV activation of the pEBS7 reporter 2.34-fold, while mutation of the A8 site alone had no effect at this time point. These results indicated that KSHV activation of the Ang-2 promoter at 12 hpi involved the A7 site and at 52 hpi involved the A7, E7, and E8 sites.
The above-mentioned results indicated that both AP-1 and Ets1 were involved in KSHV induction of Ang-2. Indeed, the DNs of c-Fos and c-Jun abolished KSHV activation of the Ang-2 promoter at 12 hpi (Fig. 5A), while DNs of c-Fos, c-Jun, and Ets1 abolished KSHV activation of the Ang-2 promoter at 52 hpi (Fig. 5B). The inhibition effects of the DNs on KSHV induction of the Ang-2 promoter were dose dependent (Fig. 5A, b and c, and B, b to d). Based on these results, we concluded that AP-1 was responsible for KSHV early induction of Ang-2 while both AP-1 and Ets1 were responsible for KSHV late induction of Ang-2.
FIG. 5.

KSHV induction of Ang-2 is mediated by AP-1 and Ets1. (A) Activity of the full-length Ang-2 promoter reporter pEBS1 cotransfected with the DNs at 12 hpi. (a) DN of c-Fos, c-Jun, or Ets-1. (b) Dose response to DN of c-Fos. (c) Dose response to DN of c-Jun. In all the experiments, cells transfected with DN for 12 h were infected with KSHV and assayed for luciferase at 12 hpi. (B) Activity of the full-length Ang-2 promoter reporter pEBS1 cotransfected with the DNs at 52 hpi. (a) DN of c-Fos, c-Jun, or Ets-1. (b) Dose response to DN of Ets1. (c) Dose response to DN of c-Fos. (d) Dose response to DN of c-Jun. In all the experiments, cells transfected with the DNs for 12 h were infected with KSHV and assayed for luciferase at 52 hpi. (C) RT-PCR detection of Ets1 and β-actin transcripts in KSHV-infected HUVEC at different hpi. (D) Mock- or KSHV-infected HUVEC were examined for the expression of Ets1 and β-actin at 52 hpi by Western blotting. (E) KSHV-induced AP-1 binding to ABS at 12 and 52 hpi detected by EMSA. Nuclear extract was prepared from mock (M)- or KSHV (K)-infected HUVEC at 12 or 52 hpi. Cold wild-type (W) or mutant (M) probe at 100× was used as a competitor. An antibody to either c-Fos or c-Jun or a control antibody was used for the supershift assay. (F) KSHV-induced Ets1 binding to EBS at 52 hpi, but not at 12 hpi, as detected by EMSA. Nuclear extract was prepared from mock (M)- or KSHV (K)-infected HUVEC at 12 or 52 hpi. Cold wild-type (W) or mutant (M) probe at 100× was used as a competitor. (G) ChIP assay using an antibody to c-Jun, Ets1, or Tie2 (control) to pull down a 641-bp DNA fragment containing the ABSs and EBSs in the Ang-2 promoter from mock- or KSHV-infected HUVEC at 12 or 52 hpi. No DNA in exon 9 of Ang-2, lying outside of the Ang-2 promoter, was pulled down. Input DNA was also amplified to estimate the recovery yields of ChIP. (H) Reporter activity of the full-length Ang-2 promoter reporter pEBS1 cotransfected with the expression plasmids of c-Fos, c-Jun, c-Fos plus c-Jun, or vector pCMV-myc. Luciferase activity was assayed at 48 h posttransfection. In all the reporter assays, results represent averages with standard deviations.
If AP-1 and Ets1 were involved in KSHV transcriptional induction of Ang-2, their activation by KSHV should precede the induction of Ang-2. Indeed, KSHV induced AP-1 binding to its consensus element as early as 2 hpi and peaked at 6 hpi (75). Similarly, we observed peak KSHV induction of Ets1 mRNA at 52 hpi (Fig. 5C), and Ets1 protein was also significantly induced by KSHV at 52 hpi (Fig. 5D). A gel shift assay detected strong AP-1 binding to the ABSs at both 12 and 52 hpi (Fig. 5E), which was inhibited by a wild-type cold probe but not the corresponding mutant cold probe and supershifted by an antibody to c-Fos or c-Jun but not IgG of a negative control antibody. Similarly, strong Ets1 binding to the EBSs was observed at 52 hpi, which was inhibited by wild-type cold probe but not the corresponding mutant cold probe, while no Ets1 band was visible at 12 hpi (Fig. 5F).
We next performed a ChIP assay to determine whether KSHV-activated AP-1 and Ets1 bound to the Ang-2 promoter in vivo (Fig. 5G). A 641-bp DNA fragment containing the ABS and EBSs in the D7 and D8 regions was pulled down from KSHV-infected HUVEC by an anti-c-Jun antibody, but not an anti-Ets1 antibody or a negative control anti-Tie2 antibody, at 12 hpi. The same DNA fragment was pulled down from KSHV-infected HUVEC by both the anti-Ets1 and anti-c-Jun antibodies, but not by the anti-Tie2 antibody, at 52 hpi. This fragment was not pulled down by any of the antibodies from mock-infected HUVEC (Fig. 5G). Furthermore, DNA of Ang-2 exon 9, outside the Ang-2 promoter region, was not pulled down by any of the antibodies in any of the cells. Taken together, these results confirmed that KSHV transcriptional induction of Ang-2 was dependent on the activation of AP-1 during the early stage of infection and both AP-1 and Ets1 during the late stage.
While Ets1 is known to mediate Ang-2 expression (32, 34), the direct involvement of AP-1 in the upregulation of Ang-2 expression had not been demonstrated before. We therefore tested the effect of AP-1 activation on the Ang-2 promoter in vitro. Overexpression of either c-Jun alone or together with c-Fos activated the Ang-2 promoter by 3- and 3.9-fold, respectively (Fig. 5H), thus further confirming the important role of AP-1 in the transcriptional regulation of Ang-2.
KSHV induction of Ang-2 is mediated by MAPK pathways.
We have previously shown that multiple MAPK pathways mediate KSHV activation of AP-1 (75). Similarly, inhibitors of the ERK, JNK, and p38 pathways reduced KSHV induction and activation of Ets1 (Fig. 6A and B). These results suggested that KSHV induction of Ang-2 could be mediated by MAPK pathways. Indeed, inhibitors of the ERK, JNK, and p38 pathways reduced KSHV induction of both Ang-2 mRNA and protein (Fig. 6C and D). Furthermore, DNs of ERK, JNK, and p38 reduced KSHV activation of the Ang-2 promoter at both 12 and 52 hpi (Fig. 6E and F). Together, these results clearly illustrated that the ERK, JNK, and p38 MAPK pathways regulate Ang-2 expression during KSHV infection by activating AP-1 and Ets1.
FIG. 6.

Transcriptional induction of Ang-2 by KSHV is mediated by multiple MAPK pathways. (A to D) HUVEC were mock or KSHV infected for 52 h with or without inhibitors of ERK, JNK, or p38 MAPK pathways. The cells were examined for Ets1 and β-actin protein expression by Western blotting (A), for Ets1 binding to its consensus element by EMSA (B), and for Ang-2 mRNA by RT-PCR (C). The supernatants were examined for Ang-2 release by Western blotting (D). (E and F) The full-length Ang-2 promoter reporter pEBS1 was cotransfected with the DN of JNK, ERK, or p38 and assayed for reporter activity at 12 hpi. (E) and 52 hpi. (F). In the reporter assays, results represent averages with standard deviations.
DISCUSSION
Angiogenesis is initiated by the destruction of existing blood vessels and is followed by the sprouting and growth of new blood vessels (22). This multistep process is typically regulated in a coordinated fashion by a number of angiogenic factors. Several cytokines, including VEGFs, IL-6, IL-8, and GRO-α, have been implicated in KS angiogenesis (19). In this study, culture medium from de novo KSHV-infected HUVEC alone induced paracrine-dependent angiogenesis (Fig. 3). Of particular interest, we found that KSHV infection of HUVEC induced the expression and release of Ang-2 (Fig. 1) and that Ang-2 was necessary for the KSHV-induced paracrine-dependent angiogenesis (Fig. 3). Furthermore, we detected strong Ang-2 protein expression in KSHV tumors (Fig. 2). These results demonstrate an important role for Ang-2 in promoting KSHV-induced KS angiogenesis. Intriguingly, Ang-2 in the context of mock-infected HUVEC culture supernatants was sufficient to induce angiogenesis (Fig. 3), suggesting that it can exert its proangiogenic effect with or without the coordinated effects of other proangiogenic factors. Coincidentally, a recent study demonstrated that Ang-2 alone induced vascular remodeling and angiogenesis in the absence of VEGF in an Ang-2 transgenic-mouse model (10). Moreover Ang-2 can sensitize ECs to TNF-α and is necessary for the induction of inflammation (23). It remains to be determined whether Ang-2 also promotes angiogenesis by inducing other proangiogenic factors in addition to its own effects on vascular remodeling.
Consistent with our findings, Wang and colleagues in their microarray analysis of KS biopsy specimens identified upregulated Ang-2 gene expression (72). A possible corollary of this observation was that Ang-2 protein levels were significantly higher in the plasma of AIDS patients with KS than in other individuals. Moreover, Ang-2 mRNA expression was also upregulated in response to KSHV infection in either blood or lymphatic ECs (72). Poole and colleagues identified similar upregulated Ang-2 mRNA expression in DMVEC infected with KSHV (60).
Although a number of cytokines and growth factors are known to regulate the expression of Ang-2 (11, 14, 25, 37, 45, 54, 56, 77), the mechanisms and cellular pathways controlling its expression are still not fully understood. We found that KSHV activated AP-1 and Ets1 to synergistically induce Ang-2 expression, with AP-1 functioning early (12 hpi) and both AP-1 and Ets1 acting later (52 hpi) after infection (Fig. 4 and 5). Activated AP-1 and Ets1 can also upregulate VEGFs and their receptors, matrix metalloproteinases, and inflammatory cytokines (15, 61, 74, 80), which could further accelerate tumor angiogenesis and promote metastasis. In agreement with these observations, inhibition of c-Jun with a catalytic DNA molecule resulted in the suppression of vascular permeability, angiogenesis, and inflammation (21).
The ERK MAPK pathway and its upstream effectors, such as phosphatidylinositol 3-kinase and Akt, regulate angiogenesis (12, 30, 48, 66, 69). Importantly, Ang-2 is a direct target of the phosphatidylinositol 3-kinase and Akt pathways (69). We have previously shown that KSHV infection activates the ERK, JNK, and p38 MAPK pathways, and all three mediate KSHV activation of AP-1 (57, 75). In this study, we have shown that all three MAPK pathways mediate KSHV activation of Ets1 (Fig. 6A and B). Consistent with these results, KSHV induction of Ang-2 is mediated by the ERK, JNK, and p38 MAPK pathways (Fig. 6C and D). Therefore, multiple MAPK pathways might promote angiogenesis by inducing the expression of Ang-2 and other angiogenic factors, at least in part via AP-1 and Ets1 (Fig. 7).
FIG. 7.
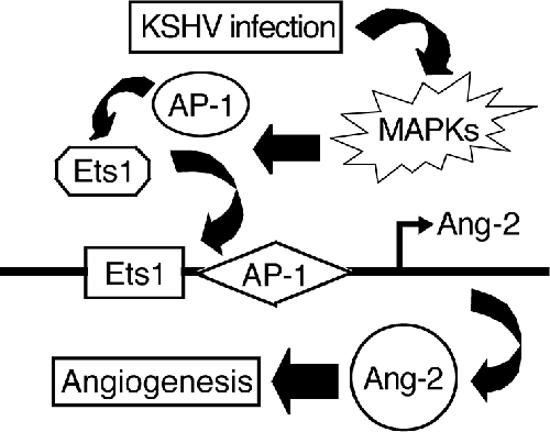
Working model proposing the molecular mechanism by which KSHV induces Ang-2.
It is now accepted that the development of malignant tumors depends not only on the ability of the tumor cells to undergo unlimited proliferation but also on angiogenesis (24). Consistent with this concept, the products of a number of oncogenes not only promote cell growth and survival, but also induce angiogenesis (16, 36, 38, 39, 41). Similarly, inhibition of the p53 tumor suppressor pathway results in the loss of cell cycle checkpoints and facilitates uncontrolled cell growth, as well as enhancing angiogenesis by interfering with the hypoxia pathway and deregulating the expression of either angiogenic factors or antiangiogenic factors (53). Our finding that the expression of Ang-2 is mediated by AP-1 and Ets1 indicates that these two transcription factors might play important roles in early blood vessel remodeling and promotion of angiogenesis during the development of other types of tumor that also contain activated AP-1 and Ets1, such as nasopharyngeal carcinoma (79). Therefore, AP-1, Ets1, and their upstream MAPK pathways could be attractive therapeutic targets for controlling oncogenic angiogenesis. In this context, KSHV infection of HUVEC provides a model for screening angiogenesis inhibitors. Since Ets1 and the AP-1 components c-Fos and c-Jun are the products of oncogenes (4), our findings illustrate the convergence of oncogenesis and angiogenesis pathways in tumor development. Further delineation of the roles of AP-1 and MAPK pathways in blood vessel remodeling and angiogenesis in other conditions, such as inflammation and infection, may reveal commonalties in the pathogenesis of different diseases.
The mechanism by which KSHV induces tumorigenesis is complex. Early-stage KS lesions have features resembling hyperplasia (19). KSHV infection of human primary ECs also does not always lead to cellular transformation (13, 27), even though it can cause chromosome instability (58). It has been proposed that the vast angiogenesis and inflammation presented in the early KS lesions could promote the transition from hyperplasia-like lesions into malignant tumors observed in late-stage KS (19). Our finding that KSHV induces the expression of Ang-2 early during infection suggests that KSHV-induced blood vessel remodeling might precede the clinical manifestation of KS tumor formation. Such early Ang-2-mediated angiogenesis could be important for tumor initiation and growth, not least by augmenting inflammation.
The KSHV genome includes genes that can promote both cell growth and survival and angiogenesis. For example, the products of the KSHV genes for vIRF (ORF-K9), vGPCR, ORF-K1, kaposin (ORF-K12), and LANA possess cellular-transformation potential, either alone or in combination with other cellular oncogenes. The vIRF, vIL-6, vBcl-2 (ORF16), and vIAP (ORF-K7) gene products regulate immune responses and promote cell growth and survival (17, 62). At the same time, a number of KSHV proteins, including vIL-6, vGPCR, vCCL-1, and vCCL-II, directly promote angiogenesis (5, 7, 43, 50, 63, 64). Furthermore, some KSHV proteins mediate the activation of the AP-1 and MAPK pathways. Two KSHV gene products, LANA and vFLIP (ORF-K13), directly activate AP-1 (2, 3). vFLIP and five other KSHV gene products, vGPCR, vPK (ORF36), LAMP (ORF-K15), kaposin B, and ORF49, and the binding of KSHV glycoproteins to cellular receptors activate MAPK pathways (1, 3, 6, 8, 29, 31, 49, 75), all of which could contribute to KSHV-induced angiogenesis. Thus, further delineation of the angiogenic and oncogenic properties of these gene products should help decipher the mechanism(s) of KSHV-induced tumorigenesis.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (CA096512 and DE017333), an American Cancer Society Research Scholar Grant (RSG-04-195), and a Type B Outstanding Abroad Young Scientist Award from the National Science Foundation of China (30328001) to S.-J. Gao and an Association for International Cancer Research Grant (01-242) and a Cancer Research United Kingdom Grant (C7934) to D. J. Blackbourn.
We thank Jiahua Han (Scripps Institute), Lin Mantell (New York University School of Medicine), Melanie Cobb (University of Texas Southwest Medical Center), Charles Vinson (National Institute of Health), B. E. Sawaya (Temple University), and B. W. Ozanne (Cancer Research United Kingdom Beatson Laboratories) for kindly providing us with reagents. We acknowledge Helmut Ostertag for providing 10 of the immunohistochemically analyzed KS paraffin blocks. We thank Sharon Murphy and members of the Gao laboratory for comments on the manuscript.
Footnotes
Published ahead of print on 7 February 2007.
REFERENCES
- 1.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2002. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407-419. [DOI] [PubMed] [Google Scholar]
- 2.An, J., A. K. Lichtenstein, G. Brent, and M. B. Rettig. 2002. The Kaposi's sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood 99:649-654. [DOI] [PubMed] [Google Scholar]
- 3.An, J., Y. Sun, R. Sun, and M. B. Rettig. 2003. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-κB and JNK/AP1 pathways. Oncogene 22:3371-3385. [DOI] [PubMed] [Google Scholar]
- 4.Angel, P., and M. Karin. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072:129-157. [DOI] [PubMed] [Google Scholar]
- 5.Aoki, Y., E. S. Jaffe, Y. Chang, K. Jones, J. Teruya-Feldstein, P. S. Moore, and G. Tosato. 1999. Angiogenesis and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93:4034-4043. [PubMed] [Google Scholar]
- 6.Bais, C., B. Santomasso, O. Coso, L. Arvanitakis, E. G. Raaka, J. S. Gutkind, A. S. Asch, E. Cesarman, M. C. Gershengorn, and E. A. Mesri. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86-89. [DOI] [PubMed] [Google Scholar]
- 7.Boshoff, C., Y. Endo, P. D. Collins, Y. Takeuchi, J. D. Reeves, V. L. Schweickart, M. A. Siani, T. Sasaki, T. J. Williams, P. W. Gray, P. S. Moore, Y. Chang, and R. A. Weiss. 1997. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278:290-294. [DOI] [PubMed] [Google Scholar]
- 8.Brinkmann, M. M., M. Glenn, L. Rainbow, A. Kieser, C. Henke-Gendo, and T. F. Schulz. 2003. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77:9346-9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown, L. F., B. J. Dezube, K. Tognazzi, H. F. Dvorak, and G. D. Yancopoulos. 2000. Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi's sarcoma and cutaneous angiosarcoma. Am. J. Pathol. 156:2179-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bureau, W., P. Van Slyke, J. Jones, R. N. Han, N. L. Ward, D. J. Stewart, and D. J. Dumont. 2006. Chronic systemic delivery of angiopoietin-2 reveals a possible independent angiogenic effect. Am. J. Physiol. Heart Circ. Physiol. 291:H948-H956. [DOI] [PubMed] [Google Scholar]
- 11.Carter, W. B., and M. D. Ward. 2000. HER2 regulatory control of angiopoietin-2 in breast cancer. Surgery 128:153-158. [DOI] [PubMed] [Google Scholar]
- 12.Chen, J., P. R. Somanath, O. Razorenova, W. S. Chen, N. Hay, P. Bornstein, and T. V. Byzova. 2005. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 11:1188-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciufo, D. M., J. S. Cannon, L. J. Poole, F. Y. Wu, P. Murray, R. F. Ambinder, and G. S. Hayward. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen, B., D. Barkan, Y. Levy, I. Goldberg, E. Fridman, J. Kopolovic, and M. Rubinstein. 2001. Leptin induces angiopoietin-2 expression in adipose tissues. J. Biol. Chem. 276:7697-7700. [DOI] [PubMed] [Google Scholar]
- 15.Crowe, D. L., and T. N. Brown. 1999. Transcriptional inhibition of matrix metalloproteinase 9 (MMP-9) activity by a c-fos/estrogen receptor fusion protein is mediated by the proximal AP-1 site of the MMP-9 promoter and correlates with reduced tumor cell invasion. Neoplasia 1:368-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dews, M., A. Homayouni, D. Yu, D. Murphy, C. Sevignani, E. Wentzel, E. E. Furth, W. M. Lee, G. H. Enders, J. T. Mendell, and A. Thomas-Tikhonenko. 2006. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 38:1060-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ensoli, B., and M. Sturzl. 1998. Kaposi's sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 9:63-83. [DOI] [PubMed] [Google Scholar]
- 19.Ensoli, B., M. Sturzl, and P. Monini. 2000. Cytokine-mediated growth promotion of Kaposi's sarcoma and primary effusion lymphoma. Semin. Cancer Biol. 10:367-381. [DOI] [PubMed] [Google Scholar]
- 20.Etoh, T., H. Inoue, S. Tanaka, G. F. Barnard, S. Kitano, and M. Mori. 2001. Angiopoietin-2 is related to tumor angiogenesis in gastric carcinoma: possible in vivo regulation via induction of proteases. Cancer Res. 61:2145-2153. [PubMed] [Google Scholar]
- 21.Fahmy, R. G., A. Waldman, G. Zhang, A. Mitchell, N. Tedla, H. Cai, C. R. Geczy, C. N. Chesterman, M. Perry, and L. M. Khachigian. 2006. Suppression of vascular permeability and inflammation by targeting of the transcription factor c-Jun. Nat. Biotechnol. 24:856-863. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara, N., and R. S. Kerbel. 2005. Angiogenesis as a therapeutic target. Nature 438:967-974. [DOI] [PubMed] [Google Scholar]
- 23.Fiedler, U., Y. Reiss, M. Scharpfenecker, V. Grunow, S. Koidl, G. Thurston, N. W. Gale, M. Witzenrath, S. Rosseau, N. Suttorp, A. Sobke, M. Herrmann, K. T. Preissner, P. Vajkoczy, and H. G. Augustin. 2006. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat. Med. 12:235-239. [DOI] [PubMed] [Google Scholar]
- 24.Folkman, J. 2006. Angiogenesis. Annu. Rev. Med. 57:1-18. [DOI] [PubMed] [Google Scholar]
- 25.Fujiyama, S., H. Matsubara, Y. Nozawa, K. Maruyama, Y. Mori, Y. Tsutsumi, H. Masaki, Y. Uchiyama, Y. Koyama, A. Nose, O. Iba, E. Tateishi, N. Ogata, N. Jyo, S. Higashiyama, and T. Iwasaka. 2001. Angiotensin AT(1) and AT(2) receptors differentially regulate angiopoietin-2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding-epidermal growth factor (EGF)-mediated EGF receptor transactivation. Circ. Res. 88:22-29. [DOI] [PubMed] [Google Scholar]
- 26.Gale, N. W., G. Thurston, S. F. Hackett, R. Renard, Q. Wang, J. McClain, C. Martin, C. Witte, M. H. Witte, D. Jackson, C. Suri, P. A. Campochiaro, S. J. Wiegand, and G. D. Yancopoulos. 2002. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 3:411-423. [DOI] [PubMed] [Google Scholar]
- 27.Gao, S. J., J. H. Deng, and F. C. Zhou. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738-9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao, S. J., L. Kingsley, D. R. Hoover, T. J. Spira, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, P. Parry, Y. Chang, and P. S. Moore. 1996. Seroconversion to antibodies against Kaposi's sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N. Engl. J. Med. 335:233-241. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez, C. M., E. L. Wong, B. S. Bowser, G. K. Hong, S. Kenney, and B. Damania. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 80:3062-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada, K., T. Sasaki, P. A. Koni, M. Natsui, H. Kishimoto, J. Sasaki, N. Yajima, Y. Horie, G. Hasegawa, M. Naito, J. Miyazaki, T. Suda, H. Itoh, K. Nakao, T. W. Mak, T. Nakano, and A. Suzuki. 2005. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 19:2054-2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamza, M. S., R. A. Reyes, Y. Izumiya, R. Wisdom, H. J. Kung, and P. A. Luciw. 2004. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 279:38325-38330. [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa, Y., M. Abe, T. Yamazaki, O. Niizeki, K. Shiiba, I. Sasaki, and Y. Sato. 2004. Transcriptional regulation of human angiopoietin-2 by transcription factor Ets-1. Biochem. Biophys. Res. Commun. 316:52-58. [DOI] [PubMed] [Google Scholar]
- 33.Hawighorst, T., M. Skobe, M. Streit, Y. K. Hong, P. Velasco, L. F. Brown, L. Riccardi, B. Lange-Asschenfeldt, and M. Detmar. 2002. Activation of the tie2 receptor by angiopoietin-1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am. J. Pathol. 160:1381-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegen, A., S. Koidl, K. Weindel, D. Marme, H. G. Augustin, and U. Fiedler. 2004. Expression of angiopoietin-2 in endothelial cells is controlled by positive and negative regulatory promoter elements. Arterioscler. Thromb. Vasc. Biol. 24:1803-1809. [DOI] [PubMed] [Google Scholar]
- 35.Holash, J., P. C. Maisonpierre, D. Compton, P. Boland, C. R. Alexander, D. Zagzag, G. D. Yancopoulos, and S. J. Wiegand. 1999. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994-1998. [DOI] [PubMed] [Google Scholar]
- 36.Kalas, W., J. L. Yu, C. Milsom, J. Rosenfeld, R. Benezra, P. Bornstein, and J. Rak. 2005. Oncogenes and angiogenesis: down-regulation of thrombospondin-1 in normal fibroblasts exposed to factors from cancer cells harboring mutant ras. Cancer Res. 65:8878-8886. [DOI] [PubMed] [Google Scholar]
- 37.Kim, I., J. H. Kim, Y. S. Ryu, M. Liu, and G. Y. Koh. 2000. Tumor necrosis factor-alpha upregulates angiopoietin-2 in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 269:361-365. [DOI] [PubMed] [Google Scholar]
- 38.Knies-Bamforth, U. E., S. B. Fox, R. Poulsom, G. I. Evan, and A. L. Harris. 2004. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 64:6563-6570. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu, M., and E. Ruoslahti. 2005. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 11:1346-1350. [DOI] [PubMed] [Google Scholar]
- 40.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lasorella, A., G. Rothschild, Y. Yokota, R. G. Russell, and A. Iavarone. 2005. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol. Cell. Biol. 25:3563-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lemieux, C., R. Maliba, J. Favier, J. F. Theoret, Y. Merhi, and M. G. Sirois. 2005. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood 105:1523-1530. [DOI] [PubMed] [Google Scholar]
- 43.Liu, C., Y. Okruzhnov, H. Li, and J. Nicholas. 2001. Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 75:10933-10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maisonpierre, P. C., C. Suri, P. F. Jones, S. Bartunkova, S. J. Wiegand, C. Radziejewski, D. Compton, J. McClain, T. H. Aldrich, N. Papadopoulos, T. J. Daly, S. Davis, T. N. Sato, and G. D. Yancopoulos. 1997. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55-60. [DOI] [PubMed] [Google Scholar]
- 45.Mandriota, S. J., and M. S. Pepper. 1998. Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ. Res. 83:852-859. [DOI] [PubMed] [Google Scholar]
- 46.Masood, R., E. Cesarman, D. L. Smith, P. S. Gill, and O. Flore. 2002. Human herpesvirus-8-transformed endothelial cells have functionally activated vascular endothelial growth factor/vascular endothelial growth factor receptor. Am. J. Pathol. 160:23-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masood, R., G. Xia, D. L. Smith, P. Scalia, J. G. Still, A. Tulpule, and P. S. Gill. 2005. Ephrin B2 expression in Kaposi's sarcoma is induced by human herpesvirus type 8: phenotype switch from venous to arterial endothelium. Blood 105:1310-1318. [DOI] [PubMed] [Google Scholar]
- 48.Mavria, G., Y. Vercoulen, M. Yeo, H. Paterson, M. Karasarides, R. Marais, D. Bird, and C. J. Marshall. 2006. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 9:33-44. [DOI] [PubMed] [Google Scholar]
- 49.McCormick, C., and D. Ganem. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739-741. [DOI] [PubMed] [Google Scholar]
- 50.Meads, M. B., and P. G. Medveczky. 2004. Kaposi's sarcoma-associated herpesvirus-encoded viral interleukin-6 is secreted and modified differently than human interleukin-6: evidence for a unique autocrine signaling mechanism. J. Biol. Chem. 279:51793-51803. [DOI] [PubMed] [Google Scholar]
- 51.Mengel, M., M. Werner, and R. von Wasielewski. 1999. Concentration dependent and adverse effects in immunohistochemistry using the tyramine amplification technique. Histochem. J. 31:195-200. [DOI] [PubMed] [Google Scholar]
- 52.Naranatt, P. P., H. H. Krishnan, S. R. Svojanovsky, C. Bloomer, S. Mathur, and B. Chandran. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72-84. [DOI] [PubMed] [Google Scholar]
- 53.North, S., M. Moenner, and A. Bikfalvi. 2005. Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors. Cancer Lett. 218:1-14. [DOI] [PubMed] [Google Scholar]
- 54.Oh, H., H. Takagi, K. Suzuma, A. Otani, M. Matsumura, and Y. Honda. 1999. Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J. Biol. Chem. 274:15732-15739. [DOI] [PubMed] [Google Scholar]
- 55.Orem, J., M. W. Otieno, and S. C. Remick. 2004. AIDS-associated cancer in developing nations. Curr. Opin. Oncol. 16:468-476. [DOI] [PubMed] [Google Scholar]
- 56.Otani, A., H. Takagi, H. Oh, S. Koyama, and Y. Honda. 2001. Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells. Diabetes 50:867-875. [DOI] [PubMed] [Google Scholar]
- 57.Pan, H., J. Xie, F. Ye, and S. J. Gao. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 80:5371-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan, H., F. Zhou, and S. J. Gao. 2004. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64:4064-4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pati, S., M. Cavrois, H. G. Guo, J. S. Foulke, Jr., J. Kim, R. A. Feldman, and M. Reitz. 2001. Activation of NF-κB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi's sarcoma pathogenesis. J. Virol. 75:8660-8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poole, L. J., Y. Yu, P. S. Kim, Q. Z. Zheng, J. Pevsner, and G. S. Hayward. 2002. Altered patterns of cellular gene expression in dermal microvascular endothelial cells infected with Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:3395-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pourtier-Manzanedo, A., C. Vercamer, E. Van Belle, V. Mattot, F. Mouquet, and B. Vandenbunder. 2003. Expression of an Ets-1 dominant-negative mutant perturbs normal and tumor angiogenesis in a mouse ear model. Oncogene 22:1795-1806. [DOI] [PubMed] [Google Scholar]
- 62.Rezaee, S. A., C. Cunningham, A. J. Davison, and D. J. Blackbourn. 2006. Kaposi's sarcoma-associated herpesvirus immune modulation: an overview. J. Gen. Virol. 87:1781-1804. [DOI] [PubMed] [Google Scholar]
- 63.Sodhi, A., S. Montaner, V. Patel, M. Zohar, C. Bais, E. A. Mesri, and J. S. Gutkind. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 60:4873-4880. [PubMed] [Google Scholar]
- 64.Stine, J. T., C. Wood, M. Hill, A. Epp, C. J. Raport, V. L. Schweickart, Y. Endo, T. Sasaki, G. Simmons, C. Boshoff, P. Clapham, Y. Chang, P. Moore, P. W. Gray, and D. Chantry. 2000. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 95:1151-1157. [PubMed] [Google Scholar]
- 65.Sturzl, M., C. Zietz, P. Monini, and B. Ensoli. 2001. Human herpesvirus-8 and Kaposi's sarcoma: relationship with the multistep concept of tumorigenesis. Adv. Cancer Res. 81:125-159. [DOI] [PubMed] [Google Scholar]
- 66.Sun, J. F., T. Phung, I. Shiojima, T. Felske, J. N. Upalakalin, D. Feng, T. Kornaga, T. Dor, A. M. Dvorak, K. Walsh, and L. E. Benjamin. 2005. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc. Natl. Acad. Sci. USA 102:128-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suri, C., P. F. Jones, S. Patan, S. Bartunkova, P. C. Maisonpierre, S. Davis, T. N. Sato, and G. D. Yancopoulos. 1996. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87:1171-1180. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka, S., M. Mori, Y. Sakamoto, M. Makuuchi, K. Sugimachi, and J. R. Wands. 1999. Biologic significance of angiopoietin-2 expression in human hepatocellular carcinoma. J. Clin. Investig. 103:341-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsigkos, S., Z. Zhou, A. Kotanidou, D. Fulton, S. Zakynthinos, C. Roussos, and A. Papapetropoulos. 2006. Regulation of Ang2 release by PTEN/PI3-kinase/Akt in lung microvascular endothelial cells. J. Cell Physiol. 207:506-511. [DOI] [PubMed] [Google Scholar]
- 70.Visconti, R. P., C. D. Richardson, and T. N. Sato. 2002. Orchestration of angiogenesis and arteriovenous contribution by angiopoietins and vascular endothelial growth factor (VEGF). Proc. Natl. Acad. Sci. USA 99:8219-8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.von Wasielewski, R., M. Mengel, S. Gignac, L. Wilkens, M. Werner, and A. Georgii. 1997. Tyramine amplification technique in routine immunohistochemistry. J. Histochem. Cytochem. 45:1455-1459. [DOI] [PubMed] [Google Scholar]
- 72.Wang, H. W., M. W. Trotter, D. Lagos, D. Bourboulia, S. Henderson, T. Makinen, S. Elliman, A. M. Flanagan, K. Alitalo, and C. Boshoff. 2004. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36:687-693. [DOI] [PubMed] [Google Scholar]
- 73.Wang, X. P., Y. J. Zhang, J. H. Deng, H. Y. Pan, F. C. Zhou, and S. J. Gao. 2002. Transcriptional regulation of Kaposi's sarcoma-associated herpesvirus-encoded oncogene viral interferon regulatory factor by a novel transcriptional silencer, Tis. J. Biol. Chem. 277:12023-12031. [DOI] [PubMed] [Google Scholar]
- 74.Westermarck, J., and V. M. Kahari. 1999. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 13:781-792. [PubMed] [Google Scholar]
- 75.Xie, J., H. Pan, S. Yoo, and S. J. Gao. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 79:15027-15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yao, D., T. Taguchi, T. Matsumura, R. Pestell, D. Edelstein, I. Giardino, G. Suske, N. Ahmed, P. J. Thornalley, V. P. Sarthy, H. P. Hammes, and M. Brownlee. 2006. Methylglyoxal modification of mSin3A links glycolysis to angiopoietin-2 transcription. Cell 124:275-286. [DOI] [PubMed] [Google Scholar]
- 77.Ye, F., M. Florian, S. A. Magder, and S. N. Hussain. 2002. Regulation of angiopoietin and Tie-2 receptor expression in non-reproductive tissues by estrogen. Steroids 67:305-310. [DOI] [PubMed] [Google Scholar]
- 78.Yoo, S. M., F. C. Zhou, F. C. Ye, H. Y. Pan, and S. J. Gao. 2005. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology 343:47-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yoshizaki, T. 2002. Promotion of metastasis in nasopharyngeal carcinoma by Epstein-Barr virus latent membrane protein-1. Histol. Histopathol. 17:845-850. [DOI] [PubMed] [Google Scholar]
- 80.Zhang, G., C. R. Dass, E. Sumithran, N. Di Girolamo, L. Q. Sun, and L. M. Khachigian. 2004. Effect of deoxyribozymes targeting c-Jun on solid tumor growth and angiogenesis in rodents. J. Natl. Cancer Inst. 96:683-696. [DOI] [PubMed] [Google Scholar]
- 81.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]