

Abstract
One of the most interesting functions attributed to the adenovirus early region 4 open reading frame 3 (E4 ORF3) protein is its reorganization of promyelocytic leukemia (PML) protein nuclear bodies. These normally punctate structures are reorganized by E4 ORF3 into tracks that eventually surround viral replication centers. PML rearrangement is an evolutionarily conserved function of E4 ORF3, yet its cause and functional relevance remain mysteries. The E4 ORF3 protein coimmunoprecipitates with the PML protein, yet E4 ORF3 still forms tracks in cells that lack PML. The PML protein is a member of a larger protein family termed tripartite motif (TRIM) proteins. TRIM proteins contain a tripartite domain structure in proximity to their N termini that consists of a RING finger domain, followed by one or two B box domains and a C-terminal coiled-coil domain (collectively termed the RBCC domain). The order and spacing of these domains are evolutionarily conserved and thought to mediate protein-protein interactions and other functions. We implemented a proteomic approach to isolate cellular proteins that bind to E4 ORF3. We identified a novel interaction between E4 ORF3 and another TRIM family member, transcriptional intermediary factor 1 alpha (TIF1α). TIF1α functions by recruiting coactivators and/or corepressors to modulate transcription. The interaction between E4 ORF3 and TIF1α was validated by coimmunoprecipitation and binding of recombinant proteins. Indirect immunofluorescence assays demonstrated that TIF1α is reorganized into track structures that contain PML upon E4 ORF3 expression. The RBCC domain of TIF1α is sufficient for E4 ORF3-induced rearrangement, and TIF1α reorganization is conserved across adenovirus serotypes.
Evolutionary pressures hone most viral genomes into ideal models of efficiency. Often, viral polypeptides are found to be multifunctional, targeting different critical cellular pathways (e.g., the adenovirus [Ad] E1A proteins) (2). The Ad early region 4 open reading frame 3 (E4 ORF3) protein is one example of a virally encoded, multifunctional protein (48). However, the mechanisms by which the E4 ORF3 protein accomplishes its known functions have remained elusive.
The most striking consequence of E4 ORF3 expression is a dramatic reorganization of the promyelocytic leukemia (PML) protein nuclear body (also called PML oncogenic domain [POD] and/or ND10). In uninfected cells stained with an antibody against the PML protein, these nuclear substructures appear punctate. Upon expression of the E4 ORF3 protein, the PML nuclear bodies are rearranged into track structures which eventually surround viral replication centers (8, 9). Recently, the E4 ORF3 protein was found to interact with a specific isoform of the PML protein, PMLII (16). It appears, however, that the PML protein is not required for E4 ORF3 track formation since E4 ORF3-containing tracks are observed in PML−/− mouse embryo fibroblasts (16). While the functional consequences of PML track formation during Ad infection have remained elusive, its importance is underscored by evolutionary conservation among different Ad serotypes (43). In addition, disruption of the PML nuclear body is a conserved feature among many viruses by disparate mechanisms (12). The function(s) of the undisrupted, endogenous PML structure also remains enigmatic, although PML nuclear bodies have been linked to many different cellular functions (4).
PML nuclear bodies contain an ever-increasing number of cellular proteins but were named after the promyelocytic leukemia protein, which functions as the nucleator of the structure (18). In patients with acute promyelocytic leukemia, a reciprocal chromosomal translocation fuses the N terminus of PML to the C terminus of retinoic acid receptor alpha (24). The oncogenic fusion protein disrupts the normally punctate appearance of PODs, resulting in a microspeckled pattern (24). A loss of growth control ensues, and in hematopoietic lineages, cellular differentiation is blocked at the promyelocyte stage. Upon treatment of these patients and at the onset of disease remission, the PML nuclear bodies resume their circular appearance (24). These results and others (37) demonstrate the role of PML, and most likely POD, in tumor suppression. Substantial evidence also supports the role of PML in the regulation of diverse cellular processes, including transcription, posttranslational modification, DNA repair, and apoptosis (4).
Of the six proteins produced by adenovirus type 5 (Ad5) E4, two have proven to be essential for the viral life cycle. These are the E4 ORF3 and E4 ORF6 proteins. Described as having redundant functions, the presence of either protein will complement the growth of an otherwise defective mutant virus that lacks the E4 region (6, 17). One function shared by both proteins is the inhibition of the host cell double-strand break repair machinery (48). If uninhibited, this pathway recognizes the linear, double-stranded Ad genome as damaged DNA in the infected host cell. Activation of the host cell nonhomologous end-joining (NHEJ) pathway ensues and results in the end-to-end ligation of Ad genomes (47). These genomic multimers are dead-end molecules, since NHEJ is an imprecise process whereby terminal sequences that contain the viral origins of DNA replication (27) are lost; therefore, viral DNA replication is significantly reduced if this process takes place. Furthermore, multimeric viral genomes are too large to be packaged within the viral capsid (3). Several critical players in the NHEJ pathway have been identified as common targets of E4 ORF3 and E4 ORF6. These include the DNA-protein kinase (PK) catalytic subunit and a multiprotein complex consisting of Mre11, Rad50, and Nbs1 (the MRN complex) (5, 11, 22, 42, 43). While E4 ORF6 directs the proteasome-dependent degradation of components of the MRN complex, E4 ORF3 sequesters MRN proteins into the aforementioned PML tracks (1, 11, 22, 42, 43). In addition, both proteins have been shown to modulate an aggresome response leading to MRN inactivation (22, 42).
The analysis of viral mutants emphasizes the importance of MRN complex inactivation. Growth of the severely deficient mutant virus that lacks the E4 region is rescued by the infection of cell lines that lack specific components of the NHEJ pathway (11). Nevertheless, E4 ORF3 targeting of the MRN complex has been shown to be specific to Ad subgroup C and is not a conserved function among other Ad serotypes (43). In contrast, E4 ORF3 reorganization of PML nuclear bodies is conserved among Ad serotypes (43). The E4 ORF3 and E4 ORF6 proteins also play other roles in the viral life cycle, including cell cycle-independent viral replication, regulation of viral late mRNA splicing, cytoplasmic mRNA accumulation, and late protein translation (14, 30, 31, 38, 40, 41). Mechanisms underlying these functions still remain undiscovered.
The PML protein is a member of a larger protein family called the tripartite motif (TRIM) proteins (also called RBCC domain proteins). Nearly 70 TRIM family members have been identified to date and found to be involved in various processes, including the regulation of proliferation, differentiation, development, and apoptosis (25). Certain TRIM family members are associated with antiviral activities (29). The tripartite motif consists of an N-terminal RING finger, followed by one or two B boxes and a C-terminal coiled-coil domain (Fig. 1B). The order, location, and spacing between the domains are conserved in all family members and are thought to mediate protein-protein interactions and other functions (25, 29). Several of the TRIM proteins have been shown to multimerize, forming both homo- and heteromultimers in vitro (32, 33). The oligomerization domain has been mapped to the coiled-coil region for several of the family members (33). In addition, the RING finger domains of several TRIM proteins have been identified as E3 ubiquitin ligases (25).
FIG. 1.

Ad5 E4 ORF3 protein binds TIF1α. (A) Recombinant E4 ORF3 protein was immobilized on chitin beads and used to isolate cellular binding proteins from uninfected HeLa cell lysates. Shown is a representative silver-stained 10% SDS-polyacrylamide gel. Lane 1, molecular weight standards (size markers are indicated on the left). Lanes 2 to 4, HeLa cell proteins that bound to the intein-CBD tag alone (lane 2), wild-type E4 ORF3 (lane 3), or the N82A mutant E4 ORF3 protein (lane 4). A prominent band present specifically in lane 3 (arrow) was identified by mass spectrometry as TIF1α. (B) Schematic diagrams of PML and TIF1α protein domain organization. R, RING finger domain; B, beta box domain; CC, coiled-coil domain; H, HP1 binding region; N, nuclear hormone receptor interaction domain; P, PHD domain; B, Bromo domain. The 3′ exons used for the expression of different PML isoforms are indicated by an open rectangle.
As a means to further characterize the functions of the E4 ORF3 protein, we implemented a proteomic approach to isolate cellular proteins that bind to Ad5 E4 ORF3. Through these experiments, we identified another TRIM family member as a novel E4 ORF3-interacting partner, namely TRIM24, or the transcriptional intermediary factor 1 alpha (TIF1α) protein. TIF1α is a transcription factor that functions by recruiting either coactivators or corepressors to promoter regions. It was originally identified by a Saccharomyces cerevisiae genetic screen for proteins that would increase the transactivation potential of nuclear hormone receptor transcription (21). TIF1α belongs to a smaller subfamily of proteins that all contain a PHD domain and a Bromo domain at their C termini (36; Fig. 1B). There are four proteins in the TIF1 family. TIF1α is one of the few TRIM proteins shown to heteromultimerize. It is capable of binding to TIF1γ directly, and it has been found in a complex with an isoform of the PML protein, PMLII (33, 52). Additional functions attributed to TIF1α include protein kinase activity and in vitro binding to and phosphorylation of heterochromatin protein 1 (13, 20, 28). Interestingly, TIF1α is also involved in a chromosomal translocation event resulting in an oncogenic fusion protein (21, 52). Upon evaluation of the ORF3-TIF1α interaction, we determined that E4 ORF3 is responsible for reorganizing the TIF1α protein into the PML-containing track structures during Ad infection. E4 ORF3 binds to TIF1α in vitro and in vivo and directs TIF1α reorganization in vivo via the RBCC domain. PML track formation alone is not sufficient for TIF1α reorganization, and most importantly, the reorganization of TIF1α is conserved among other Ad serotypes.
MATERIALS AND METHODS
Cell culture, virus infection, and plasmid transfection.
ATCC HeLa and A549 cells were grown in Dulbecco's minimal essential medium supplemented with 10% calf serum and maintained at 37°C in 5% CO2. All viruses were purified by CsCl equilibrium centrifugation and quantified by optical density at 260 nm (OD260). Unless otherwise noted, virus infections were performed using 200 virus particles/cell for 1 h followed by the addition of fresh medium to infected cells. The following viruses were used: dl309 (wild-type [WT] Ad5 [19]), dl355* (E4 ORF6 mutant [17]), inORF3 (E4 ORF3 mutant [17]), dl355/inORF3 (E4 ORF3 and E4 ORF6 double mutant [17]), and dl355-N82A and dl355-D105A/L106A (E4 ORF6 mutants with the specified point mutations in E4 ORF3 [10]). Experiments to examine E4 ORF3 proteins of different Ad serotypes were performed using infection at 200 particles/cell with wild-type Ad4 or infection at 2,000 particles/cell with Ad E1A-replacement vectors that express hemagglutinin (HA)-tagged wild-type or mutant E4 ORF3 proteins of Ad5 (viruses Ad-CMV-HA-ORF3-WT and Ad-CMV-HA-ORF3-N82A [10, 11]) or HA-tagged wild-type E4 ORF3 proteins from Ad9 or Ad12. All E1-replacement viruses also contained a deletion of E4 open reading frames 1 to 3 (17) to eliminate expression of the natural E4 ORF3 protein.
Transfections were performed according to the manufacturer's instructions, with Fugene 6 reagent (Roche) or Mirus-LT reagent (Mirus Corp.). In all cases, cells were incubated for 24 h after transfection, before subsequent experimentation and/or processing. A 2:1 Fugene 6:DNA ratio was used for the Fugene transfections of HeLa cells, and a 3:1 Mirus-LT:DNA ratio was used for Mirus transfections of A549 cells.
Antibodies.
A mouse monoclonal antibody (MAb) against the T7 epitope (Novagen) was used for immunoprecipitation. Western blots were probed using anti-HA monoclonal antibody 12CA5 (Roche), anti-T7 monoclonal antibody, or anti-TIF1α goat polyclonal antibody (C-18; Santa Cruz Biotechnology). Immunofluorescence was performed using MAbs against TIF1α (MAb 3660; Chemicon), TIF1β (MAb 2023; Affinity BioReagents), and E4 ORF3 (MAb 6A11 [26]) and rabbit polyclonal antibodies against AdDBP, HA (Y-ll; Santa Cruz Biotechnology), and PML (H238; Santa Cruz Biotechnology). Antibodies against E4 ORF3 and DBP were generous gifts from Thomas Dobner, Regensberg Universitat, and Peter van der Vleit, University Medical Center Utrecht, respectively.
Protein expression, purification, and pull-down experiments.
Wild-type and Ad5 N82A mutant E4 ORF3 proteins were cloned and expressed using an IMPACT-CN system (New England Biolabs). Specifically, the pTXB1 vector was used to generate a fusion protein with an intein-chitin binding domain tag attached to the C terminus of Ad5 E4 ORF3. E4 ORF3 proteins were expressed using the BL21(DE3) Rosetta strain of Escherichia coli (Novagen). When cultures reached an OD600 of 0.6 to 0.8, they were cooled to room temperature and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cultures were subsequently incubated for 20 h at 25°C, and cell pellets were frozen prior to E4 ORF3 purification. E. coli cell pellets were lysed by sonication in buffer at a physiological salt concentration {20 mM sodium phosphate [pH 7.5], 140 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine hydrochloride [TCEP]} and immobilized via a 2-h incubation with chitin-agarose beads (New England Biolabs). The beads were then washed six times with the bacterial lysis buffer and once with the target lysate buffer (either F-lysis buffer [50 mM Tris {pH 7.4}, 50 mM NaCl, 10% glycerol, 0.5% Triton X-100] or RIPA buffer [50 mM Tris {pH 8.0}, 150 mM NaCl, 1.0% NP-40, 0.1% sodium dodecyl sulfate {SDS}, 0.5% deoxycholate]) prior to the addition of target lysates. All buffers contained protease inhibitors phenylmethylsulfonyl fluoride (PMSF), benzamidine, pepstatin, leupeptin, and aprotinin. HeLa cell extracts also included NaF and sodium vanadate. Target lysates were generated by the extraction of a HeLa cell pellet in four packed cell volumes of the appropriate lysis buffers. The lysate was incubated for 20 min on ice prior to clarification by centrifugation at 20,000 × g for 30 min. The soluble fraction (target lysate) was transferred to a new tube, and total protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce) and then divided for incubations with beads. E4 ORF3-bound beads were incubated with 10 mg of total HeLa cell protein overnight at 4°C with rotation, followed by five washes in the appropriate target lysate buffer (F-lysis or RIPA). Sample buffer (2× SDS) was then added (1.2% SDS, 150 mM Tris [pH 6.8], 50 mM dithiothreitol), and samples were incubated for 3 min at 95°C. Samples were analyzed using one-dimensional SDS-polyacrylamide gel electrophoresis (PAGE) and stained with mass spectrometry-compatible Silverquest silver stain (Invitrogen). Bands of interest were subsequently excised and frozen at −20°C before shipping to ProTech, Inc. (Norristown, PA) for identification by tandem mass spectrometry.
Recombinant glutathione S-transferase (GST)-TIF1α was expressed in E. coli. Inductions were performed as described above except that 100 μM Zn(SO4) was added to the culture immediately preceding induction with IPTG (33). A 100-ml E. coli cell pellet containing recombinantly expressed GST-TIF1α was lysed by sonication in 10 ml of F-lysis buffer containing protease inhibitors. The lysate was clarified by centrifugation at 20,000 × g, and 4.5 ml of the supernatant was incubated with both wild-type and N82A mutant E4 ORF3 protein-bound beads.
Enhanced yellow fluorescent protein (EYFP)-TIF1α fusion proteins were expressed in mammalian cells using vector pEYFP-N1 (Clontech). This vector was also modified to replace the EYFP gene with a T7 epitope tag and used for T7-TIF1α expression (46).
Indirect immunofluorescence analyses.
HeLa cells were grown on glass coverslips and infected with the viruses described above. At 8 and 18 h postinfection, cells were washed three times with cold phosphate-buffered saline (PBS) and fixed for 5 min with −20°C methanol. Cells were then washed an additional three times with PBS and blocked in PBS containing 10% goat serum for 1 h. Primary and secondary antibodies were diluted in PBS containing 10% goat serum, with primary antibody incubations lasting 1 h and secondary antibody incubations lasting 30 min. Cells were washed three times with cold PBS between antibody incubations and prior to mounting. To prevent cross-reactivity between secondary antibodies, the mouse anti-TIF1α primary and anti-mouse secondary antibodies were added prior to the rat anti-ORF3 antibody, as determined in control experiments. For immunofluorescence experiments with Ad5-infected cells, the primary antibodies used are listed above; the secondary antibodies used were Alexa 546-conjugated goat anti-mouse highly cross-adsorbed immunoglobulin G (IgG) (Molecular Probes) and Alexa 488-conjugated goat anti-rat IgG (Molecular Probes). Wild-type Ad3- and Ad4-infected cells were labeled concurrently with primary antibodies that included rabbit polyclonal anti-DBP and mouse monoclonal anti-TIF1α. Secondary antibodies were added together and included Alexa 546-conjugated goat anti-mouse highly cross-adsorbed IgG (Molecular Probes) and Alexa 350-conjugated goat anti-rabbit IgG (Molecular Probes). Cells infected with viruses expressing HA-tagged E4 ORF3 from other Ad serotypes were labeled concurrently with primary mouse monoclonal anti-TIF1α and rabbit polyclonal anti-HA antibodies. Secondary antibodies were added concurrently and included Alexa 546-conjugated goat anti-mouse highly cross-adsorbed IgG (Molecular Probes) and fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (Zymed). Coverslips were mounted using Immumount (Thermo Shandon). Images were captured with a Zeiss digital deconvolution microscope equipped with an Apotome and Axiovision 4.5 software.
Immunoprecipitation experiments.
HeLa cells were transfected with a vector for the expression of T7-tagged TIF1α, as described above, 24 h prior to infection with Ad E1-replacement E4 ORF3 vectors that express the wild-type E4 ORF3 or the N82A mutant E4 ORF3 protein. Infected cells were incubated for 18 h prior to harvesting and cell lysis. For immunoprecipitation, the method described by Leppard et al. (15, 16) was used, with the addition of 10 mM N-ethyl maleimide to the initial lysis buffer. Briefly, 1.2 × 107 HeLa cells were lysed for 20 min on ice in 0.8 ml of a buffer containing 500 mM NaCl. After a brief sonication (10 s), lysates were clarified by centrifugation at 12,000 × g for 20 min. The supernatant was then diluted 1:1 with a buffer containing a 50 mM NaCl concentration and used for immunoprecipitation. Two micrograms of anti-T7 antibody was then incubated with the lysate for 3 h and 30 min. Immune complexes were captured with 30 μl of protein A (Roche), washed five times with immunoprecipitation buffer, and boiled in 2× sample buffer. Proteins were then separated by SDS-PAGE and transferred to Hybond-P (Amersham). Anti-T7, anti-HA, and anti-TIF1α antibodies were used to probe the Western blots. Immobilon (Millipore) or ECL (Amersham) reagents were used for detection.
RESULTS
Ad5 E4 ORF3 protein binds TIF1α.
A proteomic approach was undertaken to identify novel cellular proteins that bind to Ad5 E4 ORF3. Recombinant Ad5 E4 ORF3 was expressed in E. coli with an intein-chitin binding domain (CBD) tag and immobilized on chitin agarose beads. The immobilized protein was then used to isolate cellular binding proteins from uninfected HeLa cell lysates. In order to narrow the search for relevant targets, the pattern of cellular proteins that bind to wild-type E4 ORF3 was compared, using the intein-CBD tag alone, with that of the nonfunctional point mutant N82A E4 ORF3 (42). HeLa cells were lysed using F-lysis or RIPA buffer in separate experiments, and soluble extracts were incubated with E4 ORF3-containing beads. The beads were washed extensively, and bound proteins were eluted and analyzed by one-dimensional SDS-PAGE (Fig. 1A). Analysis of the binding patterns revealed surprisingly few unique proteins that bound either the wild-type E4 ORF3 protein or the N82A mutant E4 ORF3 protein in comparison to the intein-CBD tag alone (Fig. 1A, lanes 2 to 4). The most prominent protein that bound to wild-type E4 ORF3, but not to that of the N82A mutant or the intein-CBD tag, was specific to F-lysis buffer extracts and had an apparent mobility of ∼160 kDa (Fig. 1A, lane 3, arrow). This band was analyzed by mass spectrometry and identified as the tripartite motif family member TRIM24, or the TIF1α protein.
Ad5 E4 ORF3 coimmunoprecipitates with TIF1α expressed in vivo.
In order to investigate whether E4 ORF3 and TIF1α interact in vivo, coimmunoprecipitation experiments were performed. Since an antibody sufficient for immunoprecipitation of endogenous TIF1α was not available, a transfection/viral infection approach was undertaken. A plasmid for the expression of T7-tagged TIF1α was transfected into HeLa cells 24 h prior to infection with Ad vectors that express HA-tagged wild-type E4 ORF3 or N82A mutant E4 ORF3 (42). Eighteen hours after viral infection, cellular extracts were prepared and T7-TIF1α immunoprecipitated using anti-T7 monoclonal antibody. Immunoprecipitates were resolved by SDS-PAGE, and TIF1α and E4 ORF3 levels were determined by Western blotting using anti-T7 and anti-HA antibodies, respectively (Fig. 2A). These results demonstrated coimmunoprecipitation of wild-type E4 ORF3, but not that of the N82A mutant, with TIF1α (Fig. 2, lanes 5 and 6). Equivalent amounts of the T7-TIF1α protein were immunoprecipitated in both cases (Fig. 2, lanes 1 and 2), and equivalent amounts of wild-type E4 ORF3 and N82A mutant proteins were present in the starting extracts (Fig. 2, lanes 3 and 4). These results demonstrate that E4 ORF3 and TIF1α interact in vivo. An additional Western blot assay performed using anti-TIF1α antiserum demonstrated only a modest overexpression of the T7-tagged protein compared to the level of endogenous TIF1α when untransfected cell extracts were compared to extracts from cells transfected with the T7-TIF1α expression vector (data not shown).
FIG. 2.

E4 ORF3 interacts with TIF1α in vivo and in vitro. (A) The wild-type but not the N82A mutant E4 ORF3 protein coimmunoprecipitates with TIF1α. HeLa cells were transfected with a vector for the expression of T7-tagged TIF1α 24 h prior to infection with Ad vectors that expressed either the wild-type or the N82A HA-tagged mutant E4 ORF3 proteins. Immunoprecipitations were performed using anti-T7 antibody, and Western blots were probed using antibodies against T7 (left panel) and HA (right panel). Lanes 1 and 2, immunoprecipitated T7-TIF1α, indicated by an arrow. Lanes 3 and 4, wild-type and N82A mutant proteins present in the soluble extract prior to immunoprecipitation. Lanes 5 and 6, wild-type and N82A mutant proteins present in the T7 immunoprecipitate. The E4 ORF3 protein is indicated by an arrow. Antibody heavy and light chains are evident in lanes 5 and 6. (B) E4 ORF3 and TIF1α form a direct interaction. Recombinant wild-type and N82A mutant E4 ORF3 proteins were expressed in E. coli, coupled to chitin beads and incubated with an E. coli lysate containing recombinant GST-TIF1α. The beads were washed and bound GST-TIF1α was examined by Western blotting. Lane 1, input soluble GST-TIF1α. Lanes 2 and 3, GST-TIF1α present in the flowthrough of the wild-type and the N82A mutant E4 ORF3 incubations. Lanes 4 and 5, GST-TIF1α bound to the wild-type and the N82A mutant E4 ORF3 beads. Molecular size markers are shown, and GST-TIF1α is indicated by an arrow.
Ad5 E4 ORF3 forms a direct interaction with TIF1α in vitro.
The ability of E4 ORF3 to form a direct interaction with TIF1α was explored through recombinant protein pull-down experiments. Experiments were performed identically to those used for the initial proteomic pull-down experiment (Fig. 1); however, E. coli lysates containing recombinant GST-TIF1α were substituted for the HeLa cell extract. Wild-type E4 ORF3 fused to the intein-CBD tag was immobilized on chitin-agarose beads and incubated with E. coli lysate containing GST-TIFα, and the beads were precipitated and washed extensively. Again, the N82A mutant E4 ORF3 was used as a negative control. Pull-down of GST-TIF1α by E4 ORF3 was visualized by SDS-PAGE and by Western blotting for TIF1α (Fig. 2B) or by Coomassie blue staining (data not shown). E4 ORF3 protein amounts were normalized by Coomassie blue staining prior to TIF1α Western blotting analysis (data not shown). The wild-type E4 ORF3 protein, but not the N82A mutant protein, bound to the recombinant GST-TIF1α protein (Fig. 2B, lanes 4 and 5, arrow). Similar TIF1α protein amounts were present in the flowthrough for both the WT and the N82A mutant incubations (Fig. 2B, lanes 2 and 3), indicating an excess of TIF1α in both incubations. Since the two proteins bind in the absence of any other eukaryotic proteins, these data demonstrate that Ad5 E4 ORF3 is capable of forming a direct interaction with TIF1α. Since both proteins were expressed in E. coli, it is highly unlikely that any posttranslational modifications of either protein are required for the interaction to occur.
Endogenous TIF1α colocalizes with Ad5 E4 ORF3 and is redistributed into PML-containing tracks during Ad infection.
During Ad infection, E4 ORF3 expression reorganizes PML nuclear bodies into track structures that eventually surround viral replication centers. In order to observe whether E4 ORF3 alters the localization of endogenous TIF1α in vivo, indirect immunofluorescence was performed (Fig. 3). In mock-infected HeLa cells, TIF1α staining appeared granular and generally uniform throughout the nucleus, with some underlying foci (Fig. 3A). This is consistent with previous reports (35, 52). Eighteen hours after infection with viruses that express the wild-type E4 ORF3 (dl309 and dl355), the TIF1α protein was reorganized into track-like structures (Fig. 3B and E). Significant colocalization of TIF1α and E4 ORF3 was evident in cells infected with these viruses (Fig. 3C, D, F, and G). However, TIF1α reorganization was not complete, and the TIF1α tracks appeared less delineated than those formed with E4 ORF3 (Fig. 3B and E versus C and F). Furthermore, the reorganization was not apparent at 8 h postinfection (data not shown), distinguishing TIF1α reorganization from PML rearrangement, where PML track formation induced by E4 ORF3 was complete by 6 h postinfection (10, 11). Despite these differences, TIF1α did colocalize with PML in E4 ORF3-induced tracks (Fig. 3N to P). The reorganization of TIF1α during Ad infection was E4 ORF3 dependent, as infection with a virus that is capable of replicating to near wild-type levels but lacks the E4 ORF3 protein (inORF3) did not redistribute the TIF1α protein (Fig. 3H). As expected, infection of cells with a mutant virus that expresses nonfunctional E4 ORF3 (i.e., dl355-N82A) also did not cause TIF1α reorganization (Fig. 3K). TIF1α reorganization was specific, since no effect of E4 ORF3 expression was observed when TIF1β localization was evaluated (Fig. 4A to D). Finally, transfection of a plasmid that expresses E4 ORF3 in the absence of viral infection was sufficient to bring about this TIF1α reorganization (Fig. 4E to G). We conclude that the Ad5 E4 ORF3 protein is both necessary and sufficient to induce TIF1α rearrangement at late times after Ad infection into PML-containing track structures.
FIG. 3.

Wild-type E4 ORF3 protein is necessary for the reorganization of TIF1α into PML-containing tracks. The localization of endogenous TIF1α, with and without Ad infection, was examined by indirect immunofluorescence. (A) TIF1α localization (Alexa-546) in uninfected HeLa cells. (B to P) HeLa cells were infected with the following viruses: B to D, wild-type Ad5 dl309; E to G, mutant virus dl355; H to J, mutant virus inORF3; K to M, mutant virus dl355-N82A; N to P, mutant virus dl355. TIF1α localization (Alexa-546) was examined as shown in panels A, B, E, H, K, and N. E4 ORF3 localization (Alexa-488) was examined as shown in panels C, F, and L. Ad DBP localization (Alexa-350) was examined as shown in panel I. PML localization (Alexa-488) was examined as shown in panel O. Merge, panels D, G, J, M, and P.
FIG. 4.
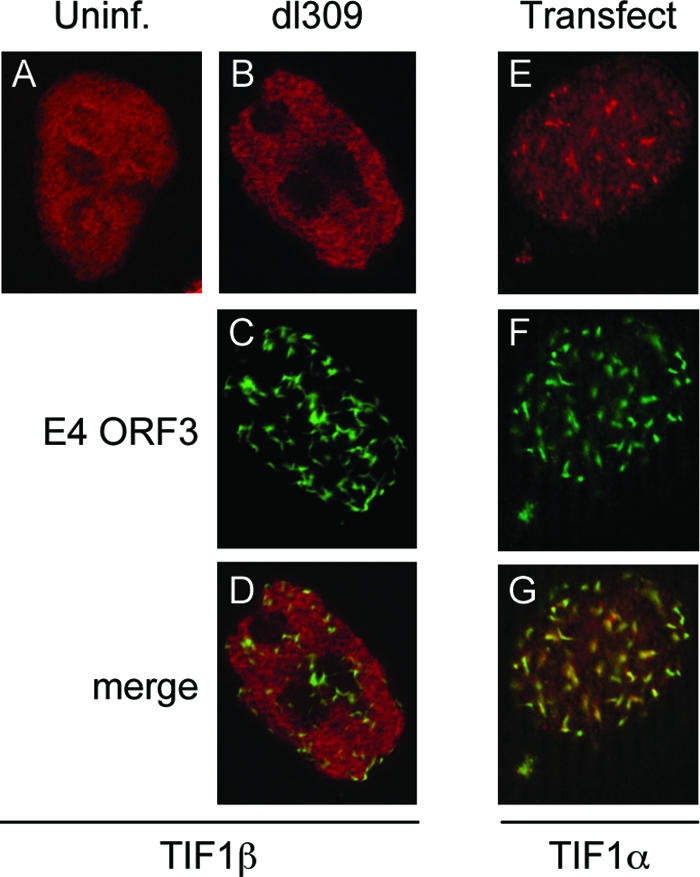
E4 ORF3 is sufficient to reorganize TIF1α but does not reorganize TIF1β. (A) Endogenous TIF1β localization was examined in uninfected HeLa cells (Alexa-546). (B to D) HeLa cells were infected with mutant virus dl355, and TIF1β localization (Alexa-546) and E4 ORF3 localization (Alexa-488) were examined as shown in panels B and C, respectively. (D) Merge. (E to G) HeLa cells were transfected with a vector for the expression of wild-type HA-tagged E4 ORF3. After 24 h, endogenous TIF1α localization (Alexa-546) and E4 ORF3 localization (Alexa-488) were examined as shown in panels E and F, respectively. (G) Merge.
PML track formation induced by E4 ORF3 is not sufficient for TIF1α reorganization.
Upon infection with the E4 ORF3 protein from a double point mutant, dl355-D105A/L106A, the PML nuclear bodies formed an elongated track structure throughout the nucleus (10). Although this mutant E4 ORF3 protein reorganized PML, the dl355-D105A/L106A mutant virus was severely defective for virus growth and viral DNA replication. This defect was attributed to the inability of this double mutant's E4 ORF3 protein to reorganize components of the MRN complex (11), thus genetically separating the functions of E4 ORF3 in PML from those in MRN rearrangement. We analyzed whether the dl355-D105A/L106A mutant virus was able to alter TIF1α nuclear distribution (Fig. 5). Upon analysis by immunofluorescence, this mutant virus was found to be unable to reorganize the TIF1α protein (Fig. 5A), although the mutant E4 ORF3 protein clearly formed track structures in infected cell nuclei (Fig. 5B) that contained PML (10). Prior results demonstrated that the D105A/L106A mutant E4 ORF3 protein accumulated to levels similar to those of the wild-type protein (10, 11). These data suggest that PML rearrangement and TIF1α reorganization are genetically separable and that PML track formation alone is not sufficient to induce changes in TIF1α localization.
FIG. 5.

TIF1α reorganization by E4 ORF3 is conserved across Ad serotypes. HeLa cells were infected with the dl355-D105A/L106A mutant virus (A to C), the wild-type Ad4 virus (D to F), and Ad vectors expressing HA-tagged E4 ORF3 from Ad9 (G to I) or Ad12 (J to L). Endogenous TIF1α localization (Alexa-546) was examined as shown in panels A, D, G, and J. Ad4-infected cells were identified by staining for Ad DBP (Alexa-350) (E). E4 ORF3 localization (Alexa-488) was examined as shown in panels B, H, and K. (C, F, I, and L) Merge.
TIF1α reorganization by E4 ORF3 is evolutionarily conserved.
We analyzed whether TIF1α rearrangement is an evolutionarily conserved function of E4 ORF3. Cells were infected with wild-type Ad of different serotypes as well as with Ad E1A-replacement vectors that express HA-tagged E4 ORF3 proteins of different Ad serotypes. TIF1α reorganization was analyzed by immunofluorescence (Fig. 5). Wild-type Ad4 reorganized TIF1α in a manner that was indistinguishable from that of Ad5 (Fig. 5D). Since the anti-Ad5 E4 ORF3 monoclonal antibody did not recognize the Ad4 or the Ad3 E4 ORF3 protein (data not shown), virus-infected cells were identified using a polyclonal antibody that detected Ad DBP from different Ad serotypes (Fig. 5E). Ad9 and Ad12 E4 ORF3 proteins also resulted in similar reorganizations of the TIF1α proteins (Fig. 5G and J). With Ad9 and Ad12, the E4 ORF3 protein was visualized using an anti-HA antibody (Fig. 5H and K), and significant colocalization of TIF1α by these serotypes' E4 ORF3 proteins was evident (Fig. 5I and L). These data suggest that TIF1α reorganization brought about by E4 ORF3 is an evolutionarily conserved feature among the E4 ORF3 proteins of Ad subgroups A, C, D, and E.
E4 ORF3 protein targets the N-terminal RBCC domain of TIF1α.
As a means of identifying the interaction domains required for the reorganization of TIF1α by E4 ORF3, a transfection/virus infection approach was used. TIF1α was expressed in A549 cells as an EYFP fusion protein. The localization of full-length TIF1α and two deletion mutants was examined upon E4 ORF3 expression. The first deletion mutant (EYFP-TIF1α-RBCC) expressed only the N-terminal RBCC domain of TIF1α fused to EYFP, while the second mutant lacked only the RBCC domain of TIF1α (EYFP-TIF1α-ΔRBCC). A549 cells were transfected with expression vectors for these proteins 24 h prior to infection with the viral mutant dl355 that expresses the E4 ORF3 protein, and EYFP-TIF1α localization was examined by immunofluorescence (Fig. 6). In uninfected cells, the full-length EYFP-TIF1α protein localized to the nucleus and displayed a uniformly granular pattern (Fig. 6A), reminiscent of the endogenous staining pattern (Fig. 3). The TIF1α-RBCC domain alone and the TIF1α-ΔRBCC mutant exhibited diffuse localization throughout the cell, with increased nuclear staining, in the absence of Ad infection (Fig. 6E and I). Upon infection with dl355, both the full-length TIF1α and the TIF1α-RBCC domain were reorganized and colocalized with E4 ORF3 (Fig. 6B to D and F to H). Localization of the TIF1α-ΔRBCC mutant proteins was not altered by dl355 infection, despite the formation of E4 ORF3-containing track structures (Fig. 6J to L). These data suggest that E4 ORF3 interacts with the N-terminal RBCC domain of TIF1α conserved in TRIM family members and that this domain is sufficient for E4 ORF3 reorganization of TIF1α.
FIG. 6.

The N-terminal RBCC domain of TIF1α is sufficient for reorganization by E4 ORF3. A549 cells were transfected with vectors for the expression of EYFP-TIF1α wild-type and mutant proteins with EYFP fused to full-length TIF1α (A to D), EYFP fused to the TIF1α RBCC domain (E to H), and EYFP fused to TIF1α-ΔRBCC (I to L). EYFP-TIF1α localization was examined in uninfected cells (A, E, and I) and in cells infected with mutant virus dl355 (B, F, and J). E4 ORF3 localization was examined as shown in panels C, G, and K. (D, H, and L) Merge.
DISCUSSION
We have identified a novel, evolutionarily conserved protein-protein interaction between the Ad E4 ORF3 protein and the cellular transcriptional regulator TIF1α. While these results do not identify the functional relevance underlying the interaction, its evolutionary conservation among E4 ORF3 proteins of different Ad serotypes strongly implicates its importance. An analogous situation underlies PML track formation by Ad E4 ORF3, first described over 10 years ago (8, 9). While the reorganization of PML into tracks is readily apparent upon E4 ORF3 expression, functional data defining the rearrangement have remained elusive. Recently, an interaction between E4 ORF3 and the PMLII isoform was identified (16). This observation may relate to the current study since TIF1α was previously shown to coimmunoprecipitate with PMLII (52). This suggests that the E4 ORF3-TIF1α interaction may be indirect via PMLII binding. However, we believe this is unlikely based on two lines of evidence. First, the interaction of wild-type E4 ORF3 with TIF1α was reproduced when both proteins were produced recombinantly (Fig. 2). This result demonstrates that the E4 ORF3-TIF1α interaction is direct. Second, the D105A/L106A mutant E4 ORF3 protein uncouples PML track formation from TIF1α reorganization (Fig. 5). Therefore, PML track formation alone is not sufficient for TIF1α reorganization. These results also suggest that the interaction of E4 ORF3 with TIF1α does not require posttranslational modifications of either protein. Phosphorylation and sumoylation of TIF1α have been demonstrated (13, 39), but these modifications would not be expected to occur in E. coli. In fact, the ubiquitination pathway is absent in E. coli, and therefore, ubiquitin-like protein modification pathways are also absent (25).
Several TRIM family members that display antiviral activities have been identified (29). These include TRIM1, TRIM5α, TRIM19 (PML), TRIM22, and TRIM30. Also called retroviral restriction factor, TRIM5α confers resistance to human immunodeficiency virus (HIV) infection upon its expression. This ability has been linked to the RING finger domain of the protein (44). These and other observations have led to a hypothesis that TRIM5α targets the HIV capsid and other viral components for degradation. The TRIM1 protein is thought to play a similar role in viral inhibition (50). In addition, the antiviral role that the interferon-inducible PML protein plays has been well established (34). It is possible that TIF1α is involved in a cellular antiviral response. The time course of TIF1α reorganization during Ad infection, however, suggests that this may not be the case since TIF1α reorganization by E4 ORF3 was apparent only at later times of infection. In addition, TIF1α levels are unaffected by viral infection (M. Yondola and P. Hearing, unpublished data), and it does not appear to be an interferon-inducible gene (52; M. Yondola and P. Hearing, unpublished data). If TIF1α were part of an antiviral response, it might be expected to be augmented by one of these mechanisms, although we do not rule out an antiviral role for the TIF1α protein. It is possible that the reorganization of TIF1α late during Ad infection may reflect a late antiviral function of this protein by analogy to the regulation of the HIV capsid protein by other TRIM family members.
The E4 ORF3 protein was shown to activate a glucocorticoid-responsive promoter (49), which may reflect regulation of the transcriptional activity of TIF1α by E4 ORF3. Downstream glucocorticoid signaling targets were also activated by the E4 ORF3 protein (49). TIF1α has been shown to increase the transactivation potential of nuclear hormone receptors in a ligand-dependent manner (21, 52). The glucocorticoid promoter is TIF1α responsive, like promoters that are regulated by the retinoic acid receptor and estrogen receptor (45). Although limited, these results suggest that glucocorticoid signaling and/or signaling by other nuclear hormone receptors may play a beneficial role for Ad. TIF1α reorganization by E4 ORF3 may facilitate this process.
Some of the more enigmatic functions of E4 ORF3 also could be TIF1α related. The E4 ORF3 protein plays undefined roles in viral DNA replication, cell cycle-independent viral replication, late mRNA splicing and accumulation, and late gene expression (7, 14, 23, 30, 31, 38, 40, 41). Of these possibilities, the transcriptional nature of the TIF1α protein suggests a possible role in the activation of viral late gene expression and/or cellular gene expression. The regulation of viral late gene expression does not appear to be influenced by E4 ORF3 expression since late gene products accumulate to approximately the same levels with or without E4 ORF3 (6, 17). However, the functional redundancy of the different major late promoter transcription factors (51) suggests that future experiments should address this possibility more closely. Alternatively, sequestration of TIF1α by E4 ORF3 may inhibit the transcriptional activity of TIF1α to down-regulate host gene expression. In addition, TIF1α functions as a tumor suppressor (52), which may provide a link between TIF1α regulation by E4 ORF3 and Ad cell cycle-independent viral replication (14, 40, 41).
As we explore the functional consequences of this interaction, it is important to note that TIF1α reorganization marks a novel function of the E4 ORF3 protein. Similar to PML in its evolutionary conservation (43), TIF1α reorganization by E4 ORF3 can be uncoupled from PML track formation, using the dl355-D105A/L106A virus mutant (Fig. 5). With this mutant, TIF1α localization remains unaltered in a situation analogous to its deficiency in MRN reorganization (11). Distinct from MRN rearrangement (43), TIF1α is reorganized by E4 ORF3 proteins of different Ad serotypes (Fig. 5). Thus, E4 ORF3-induced reorganization of PML, MRN, and TIF1α are three genetically separable functions of this Ad regulatory protein. Assuming that evolutionary pressures would eliminate superfluous interactions, it will be interesting to explore the underlying functional relevance of E4 ORF3-TIF1α interaction.
Acknowledgments
We thank several colleagues for the generous gifts of antibodies, including Thomas Dobner for antibody against E4 ORF3 and Peter van der Vleit for antibody against DBP. We acknowledge the excellent technical assistance of Mary Anderson. We thank Chris Gordon for advice on microscopy and members of our laboratory for informed discussions.
This work was supported by NIH grant CA028146 to P.H. M.A.Y. was supported by NIH training grant CA009176.
Footnotes
Published ahead of print on 7 February 2007.
REFERENCES
- 1.Araujo, F. D., T. H. Stracker, C. T. Carson, D. V. Lee, and M. D. Weitzman. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 79:11382-11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berk, A. J. 2005. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24:7673-7685. [DOI] [PubMed] [Google Scholar]
- 3.Bett, A. J., L. Prevec, and F. L. Graham. 1993. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 67:5911-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borden, K. L. 2002. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol. Cell. Biol. 22:5259-5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer, J., K. Rohleder, and G. Ketner. 1999. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263:307-312. [DOI] [PubMed] [Google Scholar]
- 6.Bridge, E., and G. Ketner. 1989. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 63:631-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bridge, E., S. Medghalchi, S. Ubol, M. Leesong, and G. Ketner. 1993. Adenovirus early region 4 and viral DNA synthesis. Virology 193:794-801. [DOI] [PubMed] [Google Scholar]
- 8.Carvalho, T., J. S. Seeler, K. Ohman, P. Jordan, U. Pettersson, G. Akusjarvi, M. Carmo-Fonseca, and A. Dejean. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131:45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doucas, V., A. M. Ishov, A. Romo, H. Juguilon, M. D. Weitzman, R. M. Evans, and G. G. Maul. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10:196-207. [DOI] [PubMed] [Google Scholar]
- 10.Evans, J. D., and P. Hearing. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77:5295-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79:6207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 8:365-374. [DOI] [PubMed] [Google Scholar]
- 13.Fraser, R. A., D. J. Heard, S. Adam, A. C. Lavigne, B. Le Douarin, L. Tora, R. Losson, C. Rochette-Egly, and P. Chambon. 1998. The putative cofactor TIF1alpha is a protein kinase that is hyperphosphorylated upon interaction with liganded nuclear receptors. J. Biol. Chem. 273:16199-16204. [DOI] [PubMed] [Google Scholar]
- 14.Goodrum, F. D., and D. A. Ornelles. 1999. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J. Virol. 73:7474-7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guccione, E., K. J. Lethbridge, N. Killick, K. N. Leppard, and L. Banks. 2004. HPV E6 proteins interact with specific PML isoforms and allow distinctions to be made between different POD structures. Oncogene 23:4662-4672. [DOI] [PubMed] [Google Scholar]
- 16.Hoppe, A., S. J. Beech, J. Dimmock, and K. N. Leppard. 2006. Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J. Virol. 80:3042-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, M. M., and P. Hearing. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 63:2605-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishov, A. M., A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss III, and G. G. Maul. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones, N., and T. Shenk. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683-689. [DOI] [PubMed] [Google Scholar]
- 20.Le Douarin, B., A. L. Nielsen, J. M. Garnier, H. Ichinose, F. Jeanmougin, R. Losson, and P. Chambon. 1996. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 15:6701-6715. [PMC free article] [PubMed] [Google Scholar]
- 21.Le Douarin, B., C. Zechel, J. M. Garnier, Y. Lutz, L. Tora, P. Pierrat, D. Heery, H. Gronemeyer, P. Chambon, and R. Losson. 1995. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 14:2020-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu, Y., A. Shevchenko, and A. J. Berk. 2005. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 79:14004-14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medghalchi, S., R. Padmanabhan, and G. Ketner. 1997. Early region 4 modulates adenovirus DNA replication by two genetically separable mechanisms. Virology 236:8-17. [DOI] [PubMed] [Google Scholar]
- 24.Melnick, A., and J. D. Licht. 1999. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93:3167-3215. [PubMed] [Google Scholar]
- 25.Meroni, G., and G. Diez-Roux. 2005. TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. Bioessays 27:1147-1157. [DOI] [PubMed] [Google Scholar]
- 26.Nevels, M., B. Tauber, E. Kremmer, T. Spruss, H. Wolf, and T. Dobner. 1999. Transforming potential of the adenovirus type 5 E4orf3 protein. J. Virol. 73:1591-1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicolas, A. L., P. L. Munz, E. Falck-Pedersen, and C. S. Young. 2000. Creation and repair of specific DNA double-strand breaks in vivo following infection with adenovirus vectors expressing Saccharomyces cerevisiae HO endonuclease. Virology 266:211-224. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen, A. L., J. A. Ortiz, J. You, M. Oulad-Abdelghani, R. Khechumian, A. Gansmuller, P. Chambon, and R. Losson. 1999. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 18:6385-6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nisole, S., J. P. Stoye, and A. Saib. 2005. TRIM family proteins: retroviral restriction and antiviral defense. Nat. Rev. Microbiol. 3:799-808. [DOI] [PubMed] [Google Scholar]
- 30.Nordqvist, K., K. Öhman, and G. Akusjärvi. 1994. Human adenovirus encodes two proteins which have opposite effects on accumulation of alternatively spliced mRNAs. Mol. Cell. Biol. 14:437-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohman, K., K. Nordqvist, and G. Akusjarvi. 1993. Two adenovirus proteins with redundant activities in virus growth facilitates tripartite leader mRNA accumulation. Virology 194:50-58. [DOI] [PubMed] [Google Scholar]
- 32.Peng, H., G. E. Begg, D. C. Schultz, J. R. Friedman, D. E. Jensen, D. W. Speicher, and F. J. Rauscher III. 2000. Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 295:1139-1162. [DOI] [PubMed] [Google Scholar]
- 33.Peng, H., I. Feldman, and F. J. Rauscher III. 2002. Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors: a potential mechanism for regulating the switch between coactivation and corepression. J. Mol. Biol. 320:629-644. [DOI] [PubMed] [Google Scholar]
- 34.Regad, T., and M. K. Chelbi-Alix. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274-7286. [DOI] [PubMed] [Google Scholar]
- 35.Remboutsika, E., Y. Lutz, A. Gansmuller, J. L. Vonesch, R. Losson, and P. Chambon. 1999. The putative nuclear receptor mediator TIF1alpha is tightly associated with euchromatin. J. Cell Sci. 112:1671-1683. [DOI] [PubMed] [Google Scholar]
- 36.Reymond, A., G. Meroni, A. Fantozzi, G. Merla, S. Cairo, L. Luzi, D. Riganelli, E. Zanaria, S. Messali, S. Cainarca, A. Guffanti, S. Minucci, P. G. Pelicci, and A. Ballabio. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20:2140-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salomoni, P., and P. P. Pandolfi. 2002. The role of PML in tumor suppression. Cell 108:165-170. [DOI] [PubMed] [Google Scholar]
- 38.Sandler, A. B., and G. Ketner. 1989. Adenovirus early region 4 is essential for normal stability of late nuclear RNAs. J. Virol. 63:624-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seeler, J. S., A. Marchio, R. Losson, J. M. Desterro, R. T. Hay, P. Chambon, and A. Dejean. 2001. Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: role of SUMO modification. Mol. Cell. Biol. 21:3314-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shepard, R. N., and D. A. Ornelles. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 78:9924-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shepard, R. N., and D. A. Ornelles. 2003. E4orf3 is necessary for enhanced S-phase replication of cell cycle-restricted subgroup C adenoviruses. J. Virol. 77:8593-8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348-352. [DOI] [PubMed] [Google Scholar]
- 43.Stracker, T. H., D. V. Lee, C. T. Carson, F. D. Araujo, D. A. Ornelles, and M. D. Weitzman. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 79:6664-6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stremlau, M., C. M. Owens, M. J. Perron, M. Kiessling, P. Autissier, and J. Sodroski. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848-853. [DOI] [PubMed] [Google Scholar]
- 45.Teyssier, C., C. Y. Ou, K. Khetchoumian, R. Losson, and M. R. Stallcup. 2006. Transcriptional intermediary factor 1alpha mediates physical interaction and functional synergy between the coactivator-associated arginine methyltransferase 1 and glucocorticoid receptor-interacting protein 1 nuclear receptor coactivators. Mol. Endocrinol. 20:1276-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ueki, N., and M. J. Hayman. 2003. Signal-dependent N-CoR requirement for repression by the Ski oncoprotein. J. Biol. Chem. 278:24858-24864. [DOI] [PubMed] [Google Scholar]
- 47.Weiden, M. D., and H. S. Ginsberg. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 91:153-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weitzman, M. D., and D. A. Ornelles. 2005. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 24:7686-7696. [DOI] [PubMed] [Google Scholar]
- 49.Wienzek, S., and M. Dobbelstein. 2001. Viral and cellular factors that target the promyelocytic leukemia oncogenic domains strongly activate a glucocorticoid-responsive promoter. J. Virol. 75:5391-5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yap, M. W., S. Nisole, C. Lynch, and J. P. Stoye. 2004. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. USA 101:10786-10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young, C. S. 2003. The structure and function of the adenovirus major late promoter. Curr. Top. Microbiol. Immunol. 272:213-249. [DOI] [PubMed] [Google Scholar]
- 52.Zhong, S., L. Delva, C. Rachez, C. Cenciarelli, D. Gandini, H. Zhang, S. Kalantry, L. P. Freedman, and P. P. Pandolfi. 1999. A RA-dependent, tumour-growth suppressive transcription complex is the target of the PML-RARalpha and T18 oncoproteins. Nat. Genet. 23:287-295. [DOI] [PubMed] [Google Scholar]