

Abstract
Maedi-visna virus (MVV) is a lentivirus of sheep causing chronic inflammatory disease of the lungs (maedi) and the nervous system (visna). We have previously shown that a duplicated sequence in the long terminal repeat (LTR) of MVV is a determinant of cell tropism. Here, we demonstrate that deletion of a CAAAT sequence from either one of the repeats resulted in poor virus growth in sheep choroid plexus cells. A duplication in the LTR encompassing the CAAAT sequence was found in four neurological field cases that were sequenced, but no duplication was present in the LTRs from seven maedi cases; one maedi isolate was mixed. These results indicate that the duplication in the LTR is associated with neurovirulence.
Maedi-visna virus (MVV) is a lentivirus of sheep, mainly affecting the lungs (maedi) and the central nervous system (CNS) (visna) (44). The primary target cells of MVV infection are cells of the monocyte/macrophage lineage, and virus replication is restricted until differentiation of the monocytes to macrophages (17, 21). However, various cell types are permissive for infection in vitro and in experimental infection by certain MVV strains (19). The identity of the cellular receptor(s) for MVV has not been elucidated, but it appears that it is a common molecule that is expressed on many cell types or that the viruses have a choice of many receptors (8, 20, 29). Cell tropism of MVV is therefore probably mainly determined by virus and/or host factors that act after virus entry. We have shown that a repeat sequence in the long terminal repeat (LTR) of MVV extends the cell tropism of the virus from being strictly macrophagetropic to being able to grow in a variety of cell types (1).
The LTR of MVV is divided into U3, R, and U5 regions, like other retroviral LTRs. The U3 region contains the promoter and enhancer regions of the virus, and unlike human immunodeficiency virus (HIV), no transactivation response element has been identified in the R region. The Tat protein of MVV has in most studies been shown to activate transcription 2- to 8-fold (compared to 100-fold activation by HIV type 1 [HIV-1] Tat) and has been suggested to be analogous to the Vpr protein in HIV-1 (6, 16, 56). The LTR has been associated with tissue tropism in a number of retroviruses (11, 14, 32, 35, 38, 39, 49, 55).
Epidemiological and experimental data suggest that there are strain differences in organ tropism of MVV. During the maedi-visna epizootic in Iceland from 1933 to 1965, maedi, the pulmonary affection, was most prevalent. However, in some flocks, visna was the main cause of disease and death (34). The neurovirulent MVV strains K1514 and K1772 are descendants of virus that was isolated from the brains of sheep from one of these flocks (45). These viruses contain a repeat sequence within the LTR that determines the cell tropism of MVV (1). Neurological disease is common in lentiviral infections; a large proportion of those infected with HIV develop neurological complications (see reference 54 for a review). A number of studies have shown that HIV strains entering the brain are predominantly macrophagetropic (22). However, studies of neurovirulent variants of simian immunodeficiency virus (SIV) have shown that macrophage tropism is not sufficient for neurological disease (30), and several studies have implicated the LTRs in neurotropism of HIV and SIV (2, 13, 14, 52).
In this study we determined a sequence within the repeat sequence of the MVV LTR which was necessary for growth in cell types other than macrophages. Furthermore, we sequenced the LTRs from field cases of maedi and visna and found a duplication in the visna cases but not in most of the maedi cases.
MATERIALS AND METHODS
Cells.
Sheep choroid plexus (SCP) cells were established as described previously (37, 46). Monocyte-derived macrophages were isolated from peripheral blood samples from healthy sheep as previously described (48). The cells were maintained in Dulbecco's modified Eagle medium (Gibco) supplemented with 2 mM glutamine, 100 IU of penicillin per ml, 100 IU of streptomycin per ml, and 10% lamb serum.
Transfections.
Fetal ovine synovial cells and SCP cells were transfected using Lipofectamine 2000 as specified by the manufacturer (Invitrogen).
Construction of recombinant viruses.
The molecular MVV clone KV1772 is contained in two plasmids, as described previously (48). The deletions were generated by PCR-mediated site-directed mutagenesis using the oligonucleotide primers listed in Table 1. The deletions were introduced only into the 3′ U3 region, since the U3 region in both the 5′ and 3′ LTRs should be derived from the 3′ LTR of the transfected DNA after one round of replication (43). All constructs and both 5′ and 3′ LTRs of the progeny of transfected cells were confirmed by sequencing. Sheep blood-derived macrophages were used for preparation of virus stocks.
TABLE 1.
Oligonucleotide primers used for construction of LTR mutants
| Oligonucleotide primer | Oligonucleotide primer sequence |
|---|---|
| V-8614 | 5′-AGAAACGGGGATGGTACAAAT |
| V-8917-AP1 | 5′-GTGTCATCCTTCTGGTAACAGTGAC |
| V-8917-2AP1 | 5′-TACATTTGCTTCTGGTAACAGTGAC |
| V-9012-CAAAT | 5′-CTTGCGGTTACCTGTGTCATCC |
| V-9018-AML | 5′-GCAGAACTACATTTGCTGTGTC |
| V-9156 | 5′-GAGCTTTCAGGCAGGCAGGAG |
RT assay.
Viral particles from 0.5 ml of cell-free supernatants from infected cells were pelleted at 14,000 rpm for 1 h in a microcentrifuge. Reverse transcriptase (RT) activity was determined as described previously (3).
Amplification of DNA from formalin-fixed, paraffin-embedded samples.
DNA was extracted by the method of Smith et al. (50). A thin section of paraffin-embedded tissue was trimmed of excess paraffin and put into 500 μl of lysis buffer (10 mM Tris, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.45% Tween 20, and 0.45% Nonidet P-40) containing bovine serum albumin (100 μg/ml) and proteinase K (100 μg/ml). The reaction mixture was incubated at 55°C overnight, boiled for 10 min, and then centrifuged at 14,000 rpm for 10 min in a microcentrifuge. Six microliters of the supernatant were used in a 40-μl PCR mixture. Nested PCR was performed by using Pfu polymerase (Stratagene) for the first PCR round of 35 cycles with primers V-8856 (forward, 5′-GAGAACAAATGCCTACCCTGG-3′) and V-9124 (reverse, 5′-CTTTCCTTCGAGACTCTCCA-3′) and an annealing temperature of 55°C. For the second round of nested PCR, 3 μl of the first-round PCR was used in a 40-μl reaction mixture using Taq polymerase (New England Biolabs) and primers V-8897 (forward, 5′-GTAGAGTTATAGGAAGGCCG-3′) and V-9045 (reverse, 5′-GCTGCTACATGACTTAGCAC-3′). The reaction conditions were the same as for the first round. The PCR products were cloned using a TOPO TA cloning kit (Invitrogen) and sequenced.
Generation of LTR/reporter gene constructs and enzyme assays.
A SacI site was introduced into the multiple cloning site of the reporter plasmid pCAT-Basic (Promega) for cloning HindIII8643-SacI9128 fragments containing the 3′ LTR of the virus. The luciferase plasmids were constructed by replacing a XhoI-HindIII fragment containing the HIV-1 LTR in pBlue3′LTR-luc, which is a pBluescript KS(+) derivative (28). The 5′ LTR from nucleotides 8852-280 was cloned into this plasmid (nucleotide numbering is that of KV1772 [4]). pBudCE4.1/lacZ/CAT (Invitrogen) was used as an internal control for transfection efficiencies.
Chloramphenicol acetyltransferase (CAT) was assayed using the phase extraction method as described by Seed and Sheen (42). Luciferase was assayed using the Enhanced Luciferase Assay kit (Pharmingen) as specified by the manufacturer. β-Galactosidase was quantified using chlorophenol red β-galactopyranoside (CPRG) as a substrate (47).
RESULTS
Identification of sequences within the repeats in the MVV LTR U3 region that enhance replication in SCP cells.
In previous studies, we demonstrated that a duplication in the LTR determines the cell tropism of MVV (1). The region in the LTR that needs to be duplicated for replication in SCP cells contains a number of known transcription factor binding sites, including degenerate AP-1 sites, a CAAATG sequence which constitutes a consensus E-box (CANNTG) (15) and an AML/PEA-2 site, which has been implicated in the replication of equine infectious anemia virus in fibroblasts (31, 35) and has also been suggested to be important in controlling MVV replication (51).
We introduced deletions of these binding sites in one copy of the repeats in the molecular clone VA4. The molecular clones VA4 and VA3 are identical except for a 54-bp duplication in the LTR of VA4. The sequence that is duplicated overlaps the 43-bp repeat in the molecular clone KV1772 by 34 bp (1) (Fig. 1; see Fig. 4). Fetal ovine synovial cells were transfected with the plasmid constructs bearing deletions, and macrophages were used for preparation of virus stocks, since they are permissive for virus with and without a repeat (1). Replication of the chimeric viruses was tested in SCP cells and macrophages. The cells were infected with equal amounts of virus as determined by RT activity. A multiplicity of infection (MOI) of 0.5 was used. The MOI was estimated by using a titrated stock of KV1772 MVV and determining the correlation between 50% tissue culture infective dose and RT activity. Virus replication was monitored by taking samples daily and measuring RT activity. Deletion of the AP-1 sites or the AML/PEA-2 site had no effect on replication of the virus in SCP cells. However, deletion of the CAAAT sequence from either copy of the duplication reduced replication markedly in SCP cells but had no effect on replication in macrophages (Fig. 2). The replication experiments were repeated three times with the same result. The identity of the virus strains was confirmed by PCR and sequencing at the end of each experiment. No revertants were detected.
FIG. 1.

Portion of the U3 region of the LTR of the MVV recombinant clones VA3 and VA4 comprising the duplications studied in this work. The repeated sequence is marked with vertical lines. The potential transcription factor binding sites where the sequence is identical to the consensus sequence are indicated by a solid line under the sequence; dotted lines below the sequence indicate sites that deviate from the consensus sequence by one nucleotide. Nucleotide deletions are indicated by dashes. The ability of each virus strain to replicate (+) or not (−) in SCP cells is indicated on the right.
FIG. 4.

Sequences of the regions in the LTRs comprising the duplications in the molecular clones KV1772, VA3, and VA4 and in field cases of visna and maedi. Duplicated sequences are shown only once, but are shown with a thick black line above the sequence. Identical nucleotides (dots) and deletions (dashes) are indicated. The CAAAT sequence is underlined.
FIG. 2.
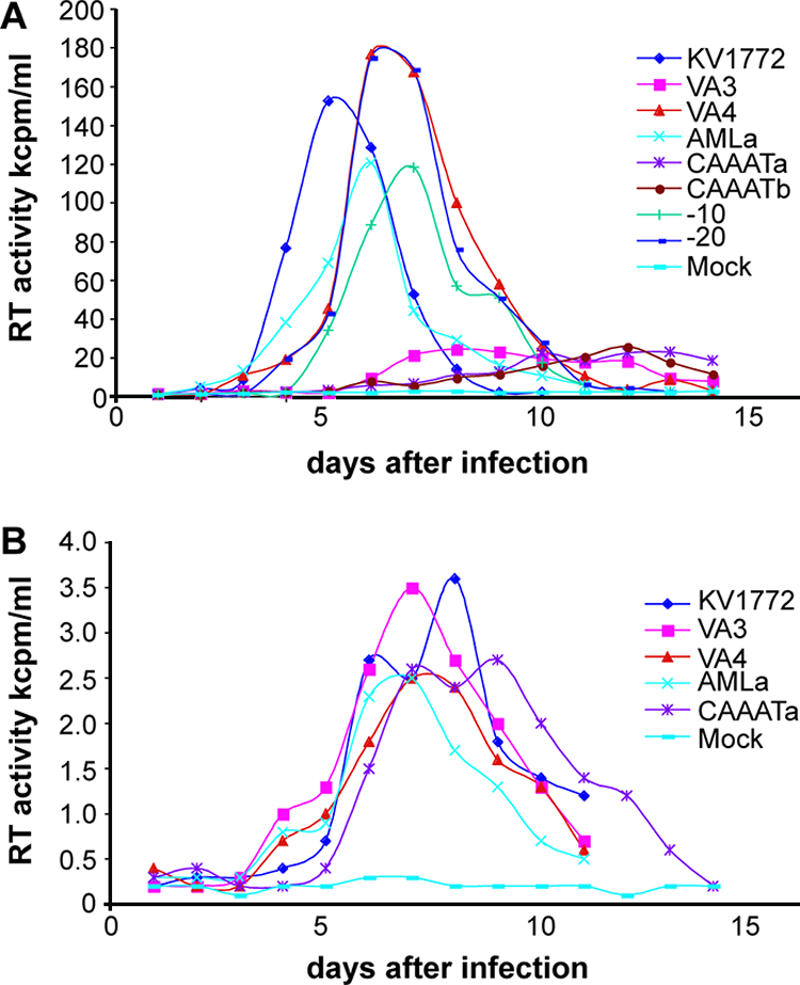
Growth curves of the various LTR deletion variants in SCP cells (A) and macrophages (B) as measured by RT assays. Virus strains with one or two API sites (−10 and −20 respectively), the AML site (AMLa), and CAAAT (CAAATa) deleted from the first copy of the repeat in the LTR and CAAAT deleted from the second copy of the repeat (CAAATb) are shown. kcpm, counts per minute (in thousands).
Transcriptional activity of the LTRs.
To determine whether the cell tropism is controlled at the transcriptional level, the promoter activities of the LTRs with and without a repeat were tested in transient transfections with reporter plasmids. First, LTRs with the 54-bp repeat of VA4 and without the repeat (VA3) were cloned into the pCAT-Basic plasmid and were transiently transfected into SCP cells. Transactivation by MVV infection was also tested by infecting the cells with MVV (MOI of 1) for 48 h before transfection. No difference could be detected between the LTRs with and without a repeat, and prior infection resulted in three- to fourfold transactivation (Table 2). These results are in accordance with the results of other studies (24, 25, 41). The loss of replication activity in SCP cells that we see in the viruses without a repeat in the LTR can therefore not be detected in the context of transcription of a reporter gene in transient transfection.
TABLE 2.
Effect of the repeat sequence in the LTR on CAT activity
| Strain | Relative CAT activity in cellsa:
|
Fold transactivation | |
|---|---|---|---|
| Uninfected | Infected | ||
| VA3 | 1,184 | 4,433 | 3.7 |
| VA4 | 1,442 | 5,196 | 3.6 |
CAT activity is expressed as counts per minute.
We then went on to determine the importance of the CAAAT sequence and the AML site by deleting these sites from the LTR without a repeat using a luciferase reporter plasmid. In MVV, no transactivation responsive element has been identified in the R sequence comparable to the transactivation response element in HIV-1. However, sequences 3′ of the promoter have been reported to contribute to transactivation (10, 25). We therefore included downstream sequences in this new construct as explained in Materials and Methods. SCP cells were transfected and, again, there was no detectable difference between the LTRs with and without a repeat. However, there was significantly less transcription if the CAAAT sequence was deleted from the LTR without a repeat, and deletion of the AML site also resulted in less transcription. These sequences are well conserved in the LTRs of MVV. Deletion of the TATA box abolished transcription almost completely (Fig. 3).
FIG. 3.

Relative luciferase activity of the LTRs with and without a repeat (VA4 and VA3, respectively) and with deletions of the CAAAT sequence and the AML site from VA3 in SCP cells. pBudCE4.1/lacZ/CAT was cotransfected for normalization of transfection efficiency. The experiments were repeated three times, and the mean values plus standard deviations (error bars) are shown in comparison to VA3 values.
Importance of extended cell tropism in vivo.
The visna virus strain KV1772 is a descendant of virus that was originally isolated from the brain of a visna-affected sheep. To investigate the possibility that the repeat sequence in the LTR, and thus extended cell tropism, is associated with neurotropism, PCR products from the LTRs of formalin-fixed, paraffin-embedded samples of four field cases of visna and eight cases of maedi were sequenced. Three of the visna samples (strains 8, 9, and 10) were from the herd that was the source of virus in our transmission experiments (strains K1514 and K1772). The 43-bp duplication that is present in these tissue culture-adapted strains was also found in the original field samples (Fig. 4). The fourth visna sample was from a sheep that had both maedi and visna and came from a different farm several years later. This MVV isolate had a 34-bp repeat in the LTR that overlapped the repeats in the other visna virus samples by 14 nucleotides encompassing the CAAAT sequence. The maedi samples were taken from three farms over a period of 4 years. All of these samples had LTRs without a repeat, but one was mixed (Fig. 4). Thus, a repeat sequence was significantly more frequent in the visna strains than in the maedi strains (P = 0.005, Fisher's exact test). However, since three of the visna samples were from the same herd, and may have been related, there may have been only two independent visna strains. Even so, the association of visna with the repeat sequence is still significant (P = 0.045).
DISCUSSION
Deletion of a CAAAT sequence from either copy of a duplication in the LTR caused deficient virus replication in SCP cells but had no effect on replication in macrophages. This sequence constitutes an E-box and could be expected to influence transcription of the viral genome in SCP cells. However, when LTR sequences with or without a duplication of the CAAAT sequence were put in front of a reporter gene and SCP cells were transfected with the DNA, no difference was detected. These results are in accordance with a number of studies that have shown no correlation of the presence of repeats in the LTR of MVV and transcriptional activity (10, 24, 25, 41). In a comprehensive study to localize transcriptional control elements in the LTR of MVV using CAT reporter plasmids in various cell types, Hess et al. (25) found that the AP-4 and AP-1 sites proximal to the TATA box were necessary for promoter activity both in SCP cells and macrophages. Deleting one copy of the 43-bp repeat of MVV strain K1514 had no effect on transcriptional activity. However, deletion of both repeats abolished the activity of the promoter in SCP cells almost completely, whereas it had little effect in macrophages, which suggests that a different set of transcription factors control the expression of MVV in the different cell types. Our finding that both the CAAAT sequence and the AML site, when present in one copy, contribute to the transcription efficiency in SCP cells further supports the notion that transcription is synergistically controlled by a repertoire of transcription factors and that the repeated sequence creates a redundancy of transcription factor binding sites (25). The loss of virus replication that we detected in this study may be controlled at the level of chromatin that cannot be detected by transient transfection (12, 33). Chromatin remodeling has been shown to modulate gene expression, and in fact, there is increasing evidence that the lentiviruses are regulated at the level of chromosomally integrated virus (9, 12, 26, 27, 53).
Eight maedi samples contained LTRs without a duplication, and this seems to be most common in virus isolated from other breeds of sheep as well, where maedi is most prevalent (5, 10, 40). A repeat sequence encompassing the CAAAT sequence was found in all brain-derived visna samples, however, implicating a repeat of the CAAAT motif in neurotropism. Three of the four visna strains in this study originated in the herd that was the source of virus used in the transmission experiments that the neurovirulent viruses K1514 and K1772 are derived from. These viruses replicate to high titers in SCP cells. The fourth strain, 2219, had a different duplication, but it also encompassed the CAAAT sequence. Unfortunately, this strain was not available for examining the replication in SCP cells. However, this strain was reported to grow to much higher titers in SCP cells than the maedi strains (23). The route of access to the CNS is most likely by the bloodstream, but the mechanism by which these viruses cross the blood-brain barrier has not been elucidated. A study of the distribution of viral antigens in the CNS in an experimental infection using the neurovirulent MVV strain K1772 revealed a variety of cell types productively infected. These included lymphocytes, plasma cells, macrophages, endothelial cells, pericytes, fibroblasts, and choroidal epithelial cells (19). In contrast to these findings, others have reported that, although other strains of MVV can enter a variety of cell types, productive infection is strictly confined to macrophages (7, 18). Together, these results suggest that the invasion of the brain by MVV is in most cases probably dependent on activated T cells or monocytes entering the brain and recruiting macrophages that may be infected, in accordance with the Trojan horse hypothesis (36). However, if the virus strain can infect the cells which comprise the blood-brain barrier, i.e., endothelial cells or cells of the choroid plexus, it may have easier access to the brain and, hence, be more neuroinvasive.
Acknowledgments
We thank Steinunn Árnadóttir for help with formalin-fixed, paraffin-embedded tissue samples.
This work was supported by grants from the Icelandic Research Fund, the University of Iceland Research Fund, and the Icelandic Research Fund for Graduate Students.
Footnotes
Published ahead of print on 7 February 2007.
REFERENCES
- 1.Agnarsdottir, G., H. Thorsteinsdottir, T. Oskarsson, S. Matthiasdottir, B. St. Haflidadottir, O. S. Andresson, and V. Andresdottir. 2000. The long terminal repeat is a determinant of cell tropism of maedi-visna virus. J. Gen. Virol. 81:1901-1905. [DOI] [PubMed] [Google Scholar]
- 2.Ait-Khaled, M., J. E. McLaughlin, M. A. Johnson, and V. C. Emery. 1995. Distinct HIV-1 long terminal repeat quasispecies present in nervous tissues compared to that in lung, blood and lymphoid tissues of an AIDS patient. AIDS 9:675-683. [DOI] [PubMed] [Google Scholar]
- 3.Andresdottir, V., X. Tang, G. Agnarsdottir, O. S. Andresson, G. Georgsson, R. Skraban, S. Torsteinsdottir, B. Rafnar, E. Benediktsdottir, S. Matthiasdottir, S. Arnadottir, S. Hognadottir, P. A. Palsson, and G. Petursson. 1998. Biological and genetic differences between lung- and brain-derived isolates of maedi-visna virus. Virus Genes 16:281-293. [DOI] [PubMed] [Google Scholar]
- 4.Andresson, O. S., J. E. Elser, G. J. Tobin, J. D. Greenwood, M. A. Gonda, G. Georgsson, V. Andresdottir, E. Benediktsdottir, H. M. Carlsdottir, and E. O. Mantyla. 1993. Nucleotide sequence and biological properties of a pathogenic proviral molecular clone of neurovirulent visna virus. Virology 193:89-105. [DOI] [PubMed] [Google Scholar]
- 5.Angelopoulou, K., G. D. Brellou, T. Greenland, and I. Vlemmas. 2006. A novel deletion in the LTR region of a Greek small ruminant lentivirus may be associated with low pathogenicity. Virus Res. 118:178-184. [DOI] [PubMed] [Google Scholar]
- 6.Barros, S. C., V. Andresdottir, and M. Fevereiro. 2005. Cellular specificity and replication rate of Maedi Visna virus in vitro can be controlled by LTR sequences. Arch. Virol. 150:201-213. [DOI] [PubMed] [Google Scholar]
- 7.Brodie, S. J., L. D. Pearson, M. C. Zink, H. M. Bickle, B. C. Anderson, K. A. Marcom, and J. C. DeMartini. 1995. Ovine lentivirus expression and disease. Virus replication, but not entry, is restricted to macrophages of specific tissues. Am. J. Pathol. 146:250-263. [PMC free article] [PubMed] [Google Scholar]
- 8.Bruett, L., and J. E. Clements. 2001. Functional murine leukemia virus vectors pseudotyped with the visna virus envelope show expanded visna virus cell tropism. J. Virol. 75:11464-11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bukrinsky, M. 2006. SNFing HIV transcription. Retrovirology 3:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell, B. J., and R. J. Avery. 1996. Sequence analysis and transcriptional activity of the LTR of OLV-CU1, a North American ovine lentivirus. J. Gen. Virol. 77:2999-3004. [DOI] [PubMed] [Google Scholar]
- 11.Celander, D., and W. A. Haseltine. 1984. Tissue-specific transcription preference as a determinant of cell tropism and leukaemogenic potential of murine retroviruses. Nature 312:159-162. [DOI] [PubMed] [Google Scholar]
- 12.Chen, J., T. Malcolm, M. C. Estable, R. G. Roeder, and I. Sadowski. 2005. TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. J. Virol. 79:4396-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corboy, J. R., J. M. Buzy, M. C. Zink, and J. E. Clements. 1992. Expression directed from HIV long terminal repeats in the central nervous system of transgenic mice. Science 258:1804-1808. [DOI] [PubMed] [Google Scholar]
- 14.Corboy, J. R., and P. J. Garl. 1997. HIV-1 LTR DNA sequence variation in brain-derived isolates. J. Neurovirol. 3:331-341. [DOI] [PubMed] [Google Scholar]
- 15.Fisher, F., and C. R. Goding. 1992. Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. EMBO J. 11:4103-4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gdovin, S. L., and J. E. Clements. 1992. Molecular mechanisms of visna virus Tat: identification of the targets for transcriptional activation and evidence for a post-transcriptional effect. Virology 188:438-450. [DOI] [PubMed] [Google Scholar]
- 17.Gendelman, H. E., O. Narayan, S. Kennedy-Stoskopf, P. G. Kennedy, Z. Ghotbi, J. E. Clements, J. Stanley, and G. Pezeshkpour. 1986. Tropism of sheep lentiviruses for monocytes: susceptibility to infection and virus gene expression increase during maturation of monocytes to macrophages. J. Virol. 58:67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gendelman, H. E., O. Narayan, S. Molineaux, J. E. Clements, and Z. Ghotbi. 1985. Slow, persistent replication of lentiviruses: role of tissue macrophages and macrophage precursors in bone marrow. Proc. Natl. Acad. Sci. USA 82:7086-7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgsson, G., D. J. Houwers, P. A. Palsson, and G. Petursson. 1989. Expression of viral antigens in the central nervous system of visna-infected sheep: an immunohistochemical study on experimental visna induced by virus strains of increased neurovirulence. Acta Neuropathol. 77:299-306. [DOI] [PubMed] [Google Scholar]
- 20.Gilden, D. H., M. Devlin, and Z. Wroblewska. 1981. The use of vesicular stomatitis (visna virus) pseudotypes to demonstrate visna virus receptors in cells from different species. Arch. Virol. 67:181-185. [DOI] [PubMed] [Google Scholar]
- 21.Gorrell, M. D., M. R. Brandon, D. Sheffer, R. J. Adams, and O. Narayan. 1992. Ovine lentivirus is macrophagetropic and does not replicate productively in T lymphocytes. J. Virol. 66:2679-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorry, P. R., G. Bristol, J. A. Zack, K. Ritola, R. Swanstrom, C. J. Birch, J. E. Bell, N. Bannert, K. Crawford, H. Wang, D. Schols, E. De Clercq, K. Kunstman, S. M. Wolinsky, and D. Gabuzda. 2001. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J. Virol. 75:10073-10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gudnadottir, M., and P. A. Palsson. 1967. Transmission of maedi by inoculation of a virus grown in tissue culture from maedi-affected lungs. J. Infect. Dis. 117:1-6. [DOI] [PubMed] [Google Scholar]
- 24.Hess, J. L., J. E. Clements, and O. Narayan. 1985. cis- and trans-acting transcriptional regulation of visna virus. Science 229:482-485. [DOI] [PubMed] [Google Scholar]
- 25.Hess, J. L., J. A. Small, and J. E. Clements. 1989. Sequences in the visna virus long terminal repeat that control transcriptional activity and respond to viral trans-activation: involvement of AP-1 sites in basal activity and trans-activation. J. Virol. 63:3001-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiebenthal-Millow, K., and F. Kirchhoff. 2002. The most frequent naturally occurring length polymorphism in the HIV-1 LTR has little effect on proviral transcription and viral replication. Virology 292:169-175. [DOI] [PubMed] [Google Scholar]
- 27.Imai, K., and T. Okamoto. 2006. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 281:12495-12505. [DOI] [PubMed] [Google Scholar]
- 28.Jeeninga, R. E., M. Hoogenkamp, M. Armand-Ugon, M. de Baar, K. Verhoef, and B. Berkhout. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 74:3740-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyall, J. W., N. Solanky, and L. S. Tiley. 2000. Restricted species tropism of maedi-visna virus strain EV-1 is not due to limited receptor distribution. J. Gen. Virol. 81:2919-2927. [DOI] [PubMed] [Google Scholar]
- 30.Mankowski, J. L., M. T. Flaherty, J. P. Spelman, D. A. Hauer, P. J. Didier, A. M. Amedee, M. Murphey-Corb, L. M. Kirstein, A. Munoz, J. E. Clements, and M. C. Zink. 1997. Pathogenesis of simian immunodeficiency virus encephalitis: viral determinants of neurovirulence. J. Virol. 71:6055-6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maury, W., S. Bradley, B. Wright, and R. Hines. 2000. Cell specificity of the transcription-factor repertoire used by a lentivirus: motifs important for expression of equine infectious anemia virus in nonmonocytic cells. Virology 267:267-278. [DOI] [PubMed] [Google Scholar]
- 32.Maury, W., R. J. Thompson, Q. Jones, S. Bradley, T. Denke, P. Baccam, M. Smazik, and J. L. Oaks. 2005. Evolution of the equine infectious anemia virus long terminal repeat during the alteration of cell tropism. J. Virol. 79:5653-5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada, M., and K. T. Jeang. 2002. Differential requirements for activation of integrated and transiently transfected human T-cell leukemia virus type 1 long terminal repeat. J. Virol. 76:12564-12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palsson, P. A. 1976. Maedi and visna in sheep. Front. Biol. 44:17-43. [PubMed] [Google Scholar]
- 35.Payne, S. L., K. La Celle, X. F. Pei, X. M. Qi, H. Shao, W. K. Steagall, S. Perry, and F. Fuller. 1999. Long terminal repeat sequences of equine infectious anaemia virus are a major determinant of cell tropism. J. Gen. Virol. 80:755-759. [DOI] [PubMed] [Google Scholar]
- 36.Peluso, R., A. Haase, L. Stowring, M. Edwards, and P. Ventura. 1985. A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology 147:231-236. [DOI] [PubMed] [Google Scholar]
- 37.Petursson, G., N. Nathanson, G. Georgsson, H. Panitch, and P. A. Palsson. 1976. Pathogenesis of visna. I. Sequential virologic, serologic, and pathologic studies. Lab. Investig. 35:402-412. [PubMed] [Google Scholar]
- 38.Rosen, C. A., W. A. Haseltine, J. Lenz, R. Ruprecht, and M. W. Cloyd. 1985. Tissue selectivity of murine leukemia virus infection is determined by long terminal repeat sequences. J. Virol. 55:862-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen, C. A., J. G. Sodroski, R. Kettman, A. Burny, and W. A. Haseltine. 1985. Trans activation of the bovine leukemia virus long terminal repeat in BLV-infected cells. Science 227:320-322. [DOI] [PubMed] [Google Scholar]
- 40.Sargan, D. R., I. D. Bennet, C. Cousens, D. J. Roy, B. A. Blacklaws, R. G. Dalziel, N. J. Watt, and I. McConnell. 1991. Nucleotide sequence of EV1, a British isolate of maedi-visna virus. J. Gen. Virol. 72:1893-1903. [DOI] [PubMed] [Google Scholar]
- 41.Sargan, D. R., K. A. Sutton, I. D. Bennet, I. McConnell, and G. D. Harkiss. 1995. Sequence and repeat structure variants in the long terminal repeat of maedi-visna virus EV1. Virology 208:343-348. [DOI] [PubMed] [Google Scholar]
- 42.Seed, B., and J. Y. Sheen. 1988. A simple phase-extraction assay for chloramphenicol acyltransferase activity. Gene 67:271-277. [DOI] [PubMed] [Google Scholar]
- 43.Shimotohno, K., and H. M. Temin. 1982. Spontaneous variation and synthesis in the U3 region of the long terminal repeat of an avian retrovirus. J. Virol. 41:163-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sigurdsson, B. 1954. Observations on three slow infections of sheep. Br. Vet. J. 110:255-270. [Google Scholar]
- 45.Sigurdsson, B., and P. A. Palsson. 1958. Visna of sheep. A slow demyelinating infection. Br. J. Exp. Pathol. 39:519-528. [PMC free article] [PubMed] [Google Scholar]
- 46.Sigurdsson, B., H. Thormar, and P. A. Palsson. 1960. Cultivation of visna virus in tissue culture. Arch. Gesamte Virusforsch. 10:368-380. [DOI] [PubMed] [Google Scholar]
- 47.Simon, J. A., and J. T. Lis. 1987. A germline transformation analysis reveals flexibility in the organization of heat shock consensus elements. Nucleic Acids Res. 15:2971-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skraban, R., S. Matthiasdottir, S. Torsteinsdottir, G. Agnarsdottir, B. Gudmundsson, G. Georgsson, R. H. Meloen, O. S. Andresson, K. A. Staskus, H. Thormar, and V. Andresdottir. 1999. Naturally occurring mutations within 39 amino acids in the envelope glycoprotein of maedi-visna virus alter the neutralization phenotype. J. Virol. 73:8064-8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Small, J. A., C. Bieberich, Z. Ghotbi, J. Hess, G. A. Scangos, and J. E. Clements. 1989. The visna virus long terminal repeat directs expression of a reporter gene in activated macrophages, lymphocytes, and the central nervous systems of transgenic mice. J. Virol. 63:1891-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith, S. A., D. F. Easton, D. G. Evans, and B. A. Ponder. 1992. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat. Genet. 2:128-131. [DOI] [PubMed] [Google Scholar]
- 51.Sutton, K. A., C. T. Lin, G. D. Harkiss, I. McConnell, and D. R. Sargan. 1997. Regulation of the long terminal repeat in visna virus by a transcription factor related to the AML/PEBP2/CBF superfamily. Virology 229:240-250. [DOI] [PubMed] [Google Scholar]
- 52.Thompson, K. A., S. J. Kent, M. E. Gahan, D. F. Purcell, C. A. McLean, S. Preiss, C. J. Dale, and S. L. Wesselingh. 2003. Decreased neurotropism of nef long terminal repeat (nef/LTR)-deleted simian immunodeficiency virus. J. Neurovirol. 9:442-451. [DOI] [PubMed] [Google Scholar]
- 53.Van Lint, C., S. Emiliani, M. Ott, and E. Verdin. 1996. Transcriptional activation and chromatin remodeling of the human immunodeficiency virus type 1 promoter in response to histone acetylation. J. Virol. 15:1112-1120. [PMC free article] [PubMed] [Google Scholar]
- 54.van Marle, G., and C. Power. 2005. Human immunodeficiency virus type 1 genetic diversity in the nervous system: evolutionary epiphenomenon or disease determinant? J. Neurovirol. 11:107-128. [DOI] [PubMed] [Google Scholar]
- 55.van Opijnen, T., R. E. Jeeninga, M. C. Boerlijst, G. P. Pollakis, V. Zetterberg, M. Salminen, and B. Berkhout. 2004. Human immunodeficiency virus type 1 subtypes have a distinct long terminal repeat that determines the replication rate in a host-cell-specific manner. J. Virol. 78:3675-3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villet, S., B. A. Bouzar, T. Morin, G. Verdier, C. Legras, and Y. Chebloune. 2003. Maedi-visna virus and caprine arthritis encephalitis virus genomes encode a Vpr-like but no Tat protein. J. Virol. 77:9632-9638. [DOI] [PMC free article] [PubMed] [Google Scholar]