

Abstract
Nef, an accessory protein of human and simian immunodeficiency viruses, is a critical determinant of pathogenesis that promotes the progression from infection to AIDS. The pathogenic effects of Nef are in large part dependent on its ability to downregulate the macrophage and T-cell coreceptor, CD4. It has been proposed that Nef induces downregulation by linking the cytosolic tail of CD4 to components of the host-cell protein trafficking machinery. To identify these components, we developed a novel Nef-CD4 downregulation system in Drosophila melanogaster S2 cells. We found that human immunodeficiency virus type 1 (HIV-1) Nef downregulates human CD4 in S2 cells and that this process is subject to the same sequence requirements as in human cells. An RNA interference screen targeting protein trafficking genes in S2 cells revealed a requirement for clathrin and the clathrin-associated, plasma membrane-localized AP2 complex in the downregulation of CD4. The requirement for AP2 was confirmed in the human cell line HeLa. We also used a yeast three-hybrid system and glutathione S-transferase pull-down analyses to demonstrate a robust, direct interaction between HIV-1 Nef and AP2. This interaction requires a dileucine motif in Nef that is also essential for downregulation of CD4. Together, these results support a model in which HIV-1 Nef downregulates CD4 by promoting its accelerated endocytosis by a clathrin/AP2 pathway.
CD4 is a type I transmembrane glycoprotein that is expressed on a subset of T lymphocytes and cells of the macrophage/monocyte lineage. Expression of CD4 on these cells is critical to the development and function of the immune system (9). In addition, CD4 acts as the primary coreceptor for human immunodeficiency virus type 1 (HIV-1) and HIV-2 and simian immunodeficiency virus (SIV), which are the etiological agents of AIDS. Binding of virions to CD4 is the initial step in the entry of these viruses into the target cells. Over time, infection leads to the depletion of CD4+ T lymphocytes and the ensuing collapse of the immune system that is characteristic of AIDS. Strikingly, the progression from infection to full-blown AIDS is critically dependent on the viral accessory protein, Nef (33, 45, 50, 72).
Nef is a 27- to 35-kDa myristoylated protein (Fig. 1) that interferes with various signaling and trafficking pathways in the host cells, leading to increased viral spread and pathogenesis (reviewed in references 25 and 66). One of the most extensively studied effects of Nef on host cells is the downregulation of CD4 (24, 32). Although it may seem counterintuitive that immunodeficiency viruses benefit from downregulating their own coreceptor, this effect strongly correlates with enhanced viral pathogenesis (26, 77). This is probably because CD4 downregulation prevents superinfection (53) and promotes virion release (69), leading to a controlled and productive infection.
FIG. 1.
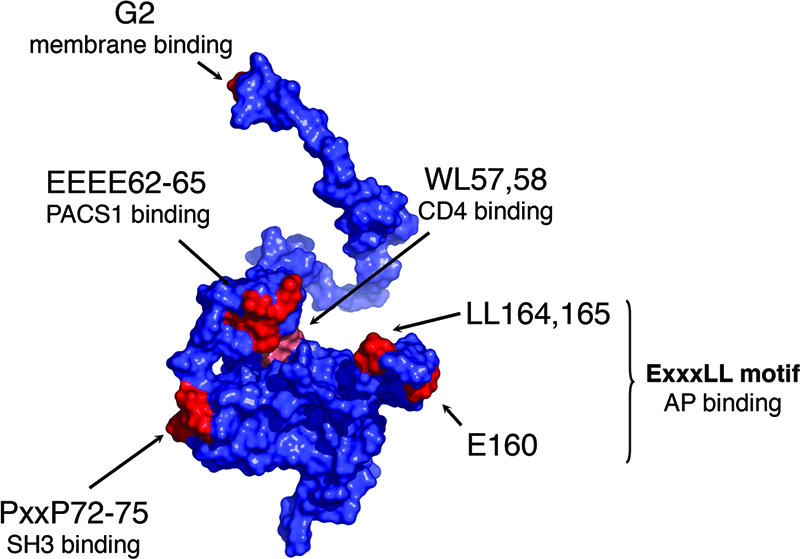
(A) Surface representation of the three-dimensional structure of HIV-1 Nef. The representation is a composite assembled from two reported nuclear magnetic resonance structures (accession codes 1QA5 and 2NEF) with an additional loop region (residues 159 to 173) modeled and drawn by using PyMOL (20). Residues that are relevant to the present study are indicated in red. Residues are numbered according to the NL4-3 Nef sequence.
The cellular pathways and molecular machinery that Nef uses to downregulate CD4 have still not been fully elucidated, despite extensive study. An important and unresolved issue is the pathway by which CD4 is depleted from the cell surface. According to one model, Nef facilitates the endocytic removal of CD4 from the plasma membrane (2, 11, 64). An alternative model posits that Nef interferes with the transport of newly synthesized CD4 from the trans-Golgi network (TGN) to the cell surface (48, 68). In both cases, Nef is also believed to redirect CD4 from endosomes or the TGN to lysosomes, where the receptor is degraded (2, 64). Previous reports have provided evidence for both enhanced endocytosis (64) and intracellular retention (68) of CD4 upon Nef expression, but it is not known which of these processes is most important for CD4 downregulation.
The molecular mechanism used by Nef to modulate CD4 expression is also uncertain. Nef has been proposed to act as a physical link between the cytosolic tail of CD4 and protein sorting devices in the host cells (4, 11, 18, 19, 23, 28, 30, 34, 39, 41, 46, 58, 70). Indeed, a site comprising tryptophan and leucine residues (indicated as WL57,58 in Fig. 1) on a folded region of Nef interacts with the cytosolic tail of CD4 (30). In addition, a dileucine-containing sequence (LL164,165 in Fig. 1) within a flexible loop of Nef has been shown to interact with the clathrin-associated, heterotetrameric adaptor protein (AP) complexes AP1, AP2, and AP3 (11, 19, 28, 41, 46, 58). AP2 is involved in internalization from the plasma membrane, whereas AP1 and AP3 participate in sorting events at the TGN and/or endosomes, making these complexes logical candidates to mediate CD4 downregulation by Nef. Both the WL57,58 (28, 49) and LL164,165 (11, 18, 27, 41) sites on Nef are essential for CD4 downregulation, thus supporting the functional significance of these interactions and the role of Nef as a linker. However, different studies have produced conflicting results regarding which specific AP complexes and AP subunits interact with Nef (11, 19, 27, 39, 41, 46).
To determine which AP complexes are functionally required for Nef-induced CD4 downregulation in vivo and to identify other host cell factors that might be involved in this process, we undertook an RNA interference (RNAi) approach. This approach was based on the coexpression of HIV-1 Nef and human CD4 in Drosophila melanogaster S2 cells, which are highly amenable to RNAi treatment (1, 21, 57, 62, 78). We found that Nef was capable of downregulating human CD4 in S2 cells with the same amino acid sequence requirements as in human T lymphocytes, demonstrating that the protein trafficking machinery exploited by Nef is conserved between Drosophila and humans. RNAi-mediated depletion of 68 different components of the trafficking machinery revealed that clathrin and AP2 were required for the Nef-induced downregulation of CD4 in S2 cells; in contrast, the depletion of AP1, AP3, or other clathrin adaptors had no effect. RNAi experiments with a human cell line confirmed the requirement for AP2. We also used a yeast three-hybrid (Y3H) system to demonstrate a robust interaction between HIV-1 Nef and the αC-σ2 hemicomplex of AP2. Moreover, glutathione S-transferase (GST) pull-down analyses showed that HIV-1 Nef binds directly to recombinant AP2. These interactions are specifically dependent on the Nef dileucine (LL164,165) motif, which was previously characterized as essential for CD4 downregulation. Since AP2 is exclusively localized to plasma membrane clathrin-coated pits and plasma membrane-derived clathrin-coated vesicles, these results support a model in which HIV-1 Nef downregulates CD4 by inducing its AP2-mediated capture into clathrin-coated pits, leading to enhanced CD4 internalization from the cell surface.
MATERIALS AND METHODS
Cells, antibodies, and other reagents.
Drosophila Schneider 2 (S2) cells were provided by Mary Lilly (Cell Biology and Metabolism Branch, NICHD, NIH, Bethesda, MD), CD4+ Jurkat T cells (JM) were obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD), and HeLa cells were purchased from the American Type Culture Collection (Manassas, VA). Mouse monoclonal antibody to human CD4 was purchased from Caltag (Burlingame, CA). Mouse monoclonal antibody to human CD71 (transferrin receptor [TfR]) was purchased from Sigma (St. Louis, MO). Allophycocyanin-conjugated and phycoerythrin (PE)-conjugated secondary antibodies to mouse immunoglobulin G (IgG) were purchased from Jackson Immunoresearch (West Grove, PA). AlexaFluor 594-conjugated secondary antibody to mouse IgG was purchased from Invitrogen (Carlsbad, CA). Horseradish peroxidase-conjugated secondary antibodies were purchased from GE Healthcare (Piscataway, NJ). HIV-1 Nef antiserum was obtained from the NIH AIDS Research and Reference Reagent Program and was originally deposited by Ronald Swanstrom (76). Blasticidin and copper (II) sulfate (CuSO4) were obtained from Invitrogen and Sigma, respectively.
Cell culture and transfections.
S2 cells were grown in Schneider's Drosophila medium (Invitrogen) and passaged every 2 to 3 days; JM cells were grown in RPMI 1640 medium (Invitrogen) and passaged every 2 to 3 days; HeLa cells were grown in Dulbecco's modified Eagle medium (Invitrogen) and passaged every 3 to 4 days. In each case, the medium was supplemented with 100 U of penicillin/ml, 0.1 mg of streptomycin/ml, 2 mM l-glutamine, and 10% (vol/vol) fetal bovine serum (FBS). Stable S2 clones were generated by cotransfection of linearized pAc.CD4, pMt.NL4-3 Nef, and pCo-BLAST (Invitrogen) in a 4:4:1 ratio; these clones were cultured without antibiotics for 1 week, after which colonies were selected and maintained in growth medium containing 25 μg of blasticidin/ml. All transfections of S2 and JM cells were performed by using an Amaxa Biosystems Nucleofector II (Amaxa, Gaithersburg, MD) with the recommended reagents. One day after transient transfection, the expression of Nef was induced in S2 cells by treatment with 0.5 mM CuSO4 for 24 h. HeLa cells were transiently transfected with the indicated plasmids by using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer.
Recombinant DNA constructs.
Human CD4 cDNA was amplified by PCR and inserted into the EcoRI and SalI sites of pAc5.1/V5-His (Invitrogen), an expression vector that contains the constitutive Drosophila 5C actin promoter, to generate the Drosophila expression vector pAc.CD4. The same method was used to subclone the CD4-dileucine mutant (LL413,414AA) cDNA, generating pAc.CD4 LL413,414AA. Wild-type HIV-1 Nef (NL4-3 variant) was subcloned into the EcoRI-XhoI sites of pMt/V5-His (Invitrogen) downstream of the Drosophila metallothionein promoter that is responsive to Cu2+ to generate pMt.NL4-3 Nef. The NL4-3 Nef coding sequence was also subcloned into the EcoRI-SalI sites of pIRES2-eGFP (Clontech, Mountain View, CA), a plasmid containing an internal ribosome entry site (IRES) for the coexpression of enhanced green fluorescence protein (eGFP) and a gene of interest from the same bicistronic transcript, producing pNL4-3 Nef.IRES.GFP. The HIV-1 Nef alleles NA7 (kindly provided by Jacek Skowronski, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY), DH12-3, and 248 and the SIVmac239 Nef allele were also subcloned into pMt/V5-His and pIRES2-eGFP by using the same restriction sites. The following mutants were also generated in pMt.NL4-3 Nef and pNL4-3 Nef.IRES.GFP using site-directed mutagenesis: G2A, WL57,58AA, EEEE62-65AAAA, PP72,75AA, and LL164,165AA. Drosophila cDNAs for the AP subunits μ1 (CG9388), μ2 (CG7057), and μ3 (CG3035), as well as Drosophila GGA (CG3002) were obtained from the Drosophila Genomics Resource Center (Bloomington, IN). Drosophila clathrin light-chain cDNA (dCLC; CG6948) was kindly provided by Henry Chang (Purdue University, West Lafayette, IN). Each Drosophila cDNA was amplified by PCR without a stop codon and inserted into the EcoRI and ApaI sites of pAc5.1/V5-His such that they are expressed with C-terminal V5 epitope tags. All constructs were verified by nucleotide sequence analysis.
Generation of templates for the production of dsRNAs.
The following oligonucleotides were used to make templates for the double-stranded RNAs (dsRNAs) that were not commercially available: CD4, 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTGACATCGTGGTGCTAGCTTTCCAG-3′ and 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTTGGATTCCAGCAGGACCTG-3′; dCLC, 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTTTACGGGTGGATCTGCATCAG--3′ and 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTAACCAGTCGTCCAGCTCCTTC-3′; dTsg101, 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTTAGCAACTTTCCACCGTATCCCAC-3′ and 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTGCATATCAATATGTTCGCGCTC-3′; GFP, 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTATGGTGAGCAAGGGCGAGGAG-3′ and 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTGCTCGTCCATGCCGAGAG-3′; and NL4-3 Nef, 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTGGGTGGGAGCAGTATCTCGAG-3′ and 5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGTGCAGCTCTCGGGCCACG-3′.
These primers include T7 RNA polymerase initiation sites on both ends of the gene fragment and were designed according to the method of Foley and O'Farrell (22). dsDNA fragments of the corresponding genes with flanking T7 sites were generated by using standard PCR techniques using the primer pairs and a cDNA template. The dTsg101 (erupted) cDNA was kindly provided by Ken Moberg (Emory University, Atlanta, GA).
RNAi.
The Expression Arrest Drosophila RNAi library, a collection of dsDNAs representing most open reading frames in the Drosophila genome, with flanking T7 RNA polymerase initiation sites was purchased from Open Biosystems (Huntsville, AL) and used as templates for the production of dsRNAs. We selected 68 genes that have been implicated in trafficking processes in animals and yeasts for use in the present study. In vitro transcription reactions were carried out with the MEGAscript T7 kit (Ambion, Austin, TX) at 37°C for 16 h, after which the RNA was purified by agarose gel electrophoresis and quantified spectrophotometrically. S2 cells were incubated for 1 h in serum-free growth medium containing 30 ng of dsRNA per ml. This was followed by the addition of FBS to a final concentration of 10% (vol/vol) and incubation for 3 days, with passage of the cells as necessary. On the fourth day, CuSO4 was added to a final concentration of 0.5 mM, and the cells were incubated for another 24 h before fluorescence-activated cell sorting (FACS) analysis. HeLa cells were transfected with small interfering RNA (siRNA) duplexes (Dharmacon, Lafayette, CO) as described previously (61). Briefly, HeLa cells received two rounds of siRNA transfections for a total of 6 days. On the fifth day, cells were transiently transfected with DNA plasmids using Lipofectamine 2000 (Invitrogen). On day 6, the siRNA-treated cells were prepared for FACS and immunoblot analysis.
RNA duplexes (siRNAs) used for RNAi in human cells.
The following sequences were used to generate RNAs specifically targeting human trafficking genes and were ordered from Dharmacon as 21-nucelotide duplexes with 3′ dTdT overhangs: CHC, 5′-AACCUGCGGUCUGGAGUCAAC-3′; μ1, 5′-AAGGCAUCAAGUAUCGGAAGA-3′; μ2, 5′-AAGUGGAUGCCUUUCGGGUCA-3′; and μ3, 5′-AAGGAGAACAGUUCUUGCGGC-3′ (54, 61). For control experiments, we used the siCONTROL nontargeting siRNA 1 (Dharmacon).
FACS analysis.
S2, JM, and HeLa cells were stained with the appropriate antibodies and prepared for FACS analysis as previously described (41). For JM and HeLa cells transfected with pIRES.GFP constructs, GFP fluorescence was used to identify the transfected cells. In each case, the amount of cell-associated fluorescence was measured with a FACSCalibur flow cytometer and analyzed by using CellQuest software (Becton Dickinson, Franklin Lakes, NJ).
Immunofluorescence microscopy.
S2 cells were grown on glass coverslips coated with poly-l-lysine (Sigma). One day after the addition of CuSO4, the cells were fixed for 15 min with 3.75% (wt/vol) paraformaldehyde in phosphate-buffered saline (PBS), permeabilized for 10 min with 0.1% (wt/vol) Triton X-100 in PBS, and incubated for 1 h in blocking buffer (PBS plus 4% FBS). This was followed by incubation with blocking buffer plus monoclonal antibody to CD4 (1:100 dilution). After several washes with PBS, cells were incubated with AlexaFluor 594 secondary antibody, washed again, and mounted on slides. All steps were performed at room temperature. Cells were imaged on a Zeiss LSM510 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY) with a 63X Plan Apochromat 1.4 NA objective lens, using the 543- nm line of a HeNe laser. Emission was collected over the range of 560 to 660 nm with the appropriate filter sets.
Immunoblotting.
Cells were lysed for 20 min at 4°C by using 1% (vol/vol) Triton X-100 in 300 mM NaCl-50 mM Tris-HCl-5 mM EDTA (pH 7.4) supplemented with a protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). The cells were centrifuged at 13,000 rpm for 3 min to remove insoluble material, and NuPAGE LDS sample buffer (Invitrogen) was added to the supernatants. After boiling, samples were separated by electrophoresis on NuPAGE Bis-Tris gels (Invitrogen). The gels were then transferred to nitrocellulose membranes. Membranes were blocked in PBS-T (PBS plus 0.01% [vol/vol] Tween 20) plus 5% milk (wt/vol) for 1 h. Primary antibodies were added in the same milk mixture for 1 h or overnight. After three washes with PBS-T, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h and washed again, and proteins were detected by using ECL Plus (GE Healthcare).
Yeast three-hybrid assays.
Y3H assays were performed as previously described (41). Wild-type or mutant NL4-3 Nef was expressed as a GAL4BD fusion protein in the pBridge vector (Clontech) along with either rat σ1A or σ2. The large AP subunits (either mouse γ1 or rat αC) were expressed as GAL4AD fusion proteins in the pGADT7 vector (Clontech) as described previously (41). Resequencing of the original rat αC cDNA insert (41) revealed the presence of a missense mutation that changed Ala-131 to Thr. This was reverted back to Ala by site-directed mutagenesis and verified again by sequencing. The Saccharomyces cerevisiae strain HF7c was transformed with pairs of pBridge and pGADT7 vectors. Transformants were selected on plates lacking Leu, Trp, and Met. Colony growth was assayed 3 to 5 days after the transformants were transferred to plates lacking Leu, Trp, Met, and His.
Recombinant protein expression and purification and pull-down experiments.
The NL4-3 Nef coding sequence was subcloned into the BamHI and EcoRI site of the pHis-Parallel2 vector (75) and overexpressed as an N-terminal hexahistidine-tagged fusion protein in Escherichia coli BL21(DE3). Nef LL164,165AA mutant was generated by using the QuikChange site-directed mutagenesis kit (Stratagene). Bacteria were induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and grown at 18°C overnight. Harvested cells were lysed in Tris-buffered saline (TBS; 50 mM Tris-HCl [pH 8.0], 500 mM NaCl), and the supernatant was applied to a HisTrap column (GE Healthcare). Protein was concentrated and applied to a Superdex 200 column (GE Healthcare) in TDN buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 5 mM dithiothreitol). The four subunits of AP2 (residues 1 to 621 from rat αC adaptin, residues 1 to 591 from rat β2 adaptin, residues 1 to 141 from rat μ2 adaptin, and residues 1 to 143 from rat σ2 adaptin; hereafter referred to as AP2core) were coexpressed in E. coli Rosetta2 (DE3) cells (Novagen, San Diego, CA) from the pST39 vector (35). Expression was under the control of the T7 promoter, with each gene having its own ribosome-binding site. The β2 “trunk” was produced as an N-terminal, hexahistidine fusion and the αC “trunk” was produced as a C-terminal GST fusion. For AP2, bacteria were grown at 37°C to an optical density at 600 nm of 0.8, the temperature was lowered to 18°C, and expression was induced by the addition of 0.2 mM IPTG. Protein expression was allowed to continue for 12 h, the cells were harvested and lysed in TBS, and insoluble material was removed by centrifugation. The complex was bound to a HisTrap column, extensively washed with TBS, and eluted with TBS containing 0.25 M imidazole. The AP2core complex was then bound to glutathione-Sepharose (GE Healthcare), extensively washed, and eluted with TBS containing 10 mM glutathione. AP2core was finally purified by gel filtration on a Superdex 200 (GE Healthcare) column in TDN buffer. Recombinant GST was produced from BL21(DE3) cells and purified using glutathione-Sepharose affinity column followed by a Superdex S200 (GE Healthcare) gel filtration chromatography in TDN. Pull-down experiments were described previously (47). Briefly, GST-AP2core and GST-ɛ-ear domain of AP4 were immobilized onto glutathione-Sepharose. Nef (wild-type) or Nef LL164,165AA (100 mM final concentration) was incubated with the immobilized GST fusion proteins for 30 min on ice. The beads were subjected to a 1,000 × g centrifugation step, the supernatant was removed, and the beads were washed with TBS supplemented with 0.1% (wt/vol) Triton X-100. This step was performed two additional times, and the beads were resuspended in LDS sample buffer, boiled, and resolved on a 4 to 12% NuPAGE gel (Invitrogen).
RESULTS
Downregulation of human CD4 by HIV-1 Nef in Drosophila S2 cells.
To identify host cell factors that are required for the downregulation of CD4 by Nef, we performed an RNAi screen using the Drosophila phagocytic cell line, S2. This system was chosen because the efficiency of RNAi and transient-transfection approaches 100%, RNAi treatment is performed by the simple addition of dsRNAs to the culture medium, and an RNAi library targeting most of the genome is commercially available at a reasonable price. This approach was predicated on the assumption that HIV-1 Nef would be able to downregulate human CD4 in these Drosophila cells. To determine whether this was the case, S2 cells were transiently cotransfected with a plasmid driving constitutive expression of human CD4 (pAc.CD4) and another plasmid driving Cu2+-inducible expression of the NL4-3 variant of HIV-1 Nef (pMt.NL4-3 Nef). Immunoblot analysis revealed that both CD4 and Nef were expressed in the S2 cells; as expected, Nef was apparent only in cells incubated with Cu2+ (Fig. 2A). Importantly, in the absence of Nef expression, CD4 was detected on the cell surface by fluorescence-activated cell sorting (FACS) (Fig. 2B) and immunofluorescence microscopy (Fig. 2C). Induction of Nef expression by the addition of Cu2+ led to an ∼3-fold reduction in the surface level of CD4 (Fig. 2B) and its redistribution to intracellular vesicles (Fig. 2C). These effects of Nef expression on CD4 distribution were similar to those previously demonstrated in human cells (2, 24, 68).
FIG. 2.
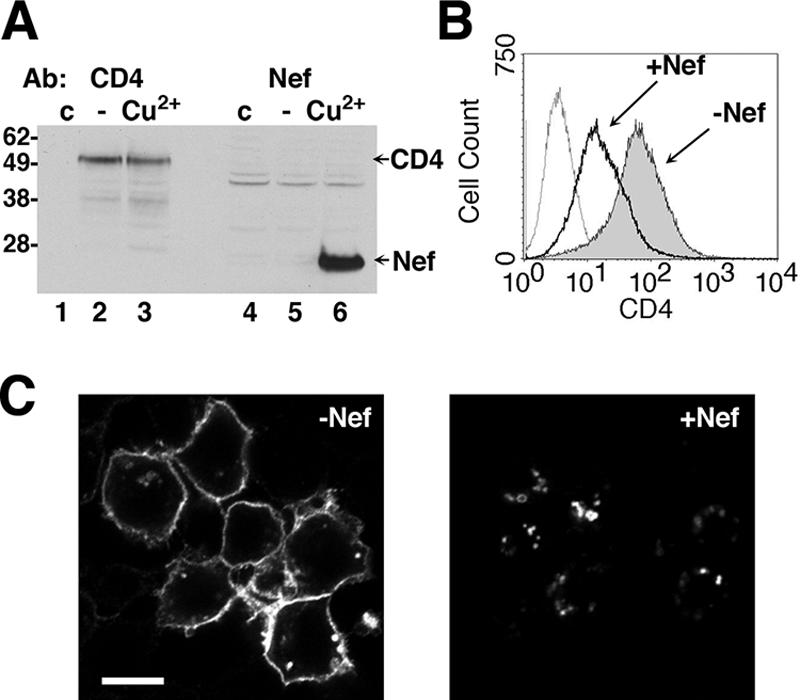
Downregulation of human CD4 by HIV-1 Nef in Drosophila S2 cells. (A) Immunoblot analysis of S2 cells 48 h after transient transfection with pAc.CD4 and pMt.Nef and 24 h after induction of Nef expression with CuSO4. Lysates from an untransfected control (lanes 1 and 4) and doubly transfected cells either without (lanes 2 and 5) or with (lanes 3 and 6) induction were analyzed by immunoblotting with antibodies to CD4 and Nef. The positions of molecular mass markers (in kilodaltons) are shown on the left. (B) S2 cells were transiently transfected with pAc.CD4 and pMt.Nef, incubated without (shaded gray) or with (bold line) CuSO4, and stained with a mouse monoclonal antibody to human CD4 and PE-conjugated anti-mouse IgG. Untransfected cells were also stained as a negative control (dotted line). Cells were analyzed by FACS. The x axis represents CD4 fluorescence on a logarithmic scale, and the y axis represents the number of cells on a linear scale. (C) Immunofluorescence microscopy of CD4 in S2 cells. The cells were transiently transfected with pAc.CD4 and pMt. Nef, incubated for 24 h, and either left untreated (i.e., −Nef) or treated with CuSO4 (i.e., +Nef) for another 24 h, fixed, permeabilized, and stained with mouse monoclonal antibody to human CD4 and AlexaFluor 594-conjugated donkey antibody to mouse IgG. Stained cells were examined by confocal microscopy. Bar, 5 μm.
Determinants of Nef-induced CD4 downregulation in S2 cells.
Next, we tested the ability of various Nef alleles to downregulate CD4 in Drosophila S2 and human CD4+ JM cells. Despite high sequence variability among different HIV-1 and SIV clades, the ability to downregulate CD4 in human cells is a highly conserved feature of Nef proteins (5, 37, 41, 51). We observed that the HIV-1 Nef variants NA7, DH12-3, and 248, as well as the SIV Nef variant SIVmac239, reduced CD4 surface levels to a similar extent in both S2 and JM cells (Fig. 3).
FIG. 3.

Comparison of the downregulation of CD4 by Nef from various HIV-1 and SIV variants in Drosophila S2 and human JM CD4+ T cells. (A) FACS histograms of Drosophila S2 cells that were doubly transfected with pAc.CD4 and pMt vectors encoding a variety of HIV-1 (NL4-3, NA7, DH12-3, and 248) and SIV Nef (mac239) alleles, incubated without (shaded gray) or with CuSO4 (bold line), and stained with a mouse monoclonal antibody to human CD4 and PE-conjugated anti-mouse IgG. Uninduced control cells were also stained with an isotype antibody control and PE anti-mouse IgG (dotted line). (B) Bar graph depicting levels of cell surface CD4 in Drosophila S2 cells cotransfected with pAc.CD4 and pMt.Nef (various HIV-1 and SIV alleles) (dark gray) and in human JM CD4+ cells transfected with pNef.IRES.GFP (various HIV-1 and SIV alleles) (light gray). Induction of Nef expression and FACS analysis were performed as described in Materials and Methods. For S2 cells the control represents uninduced cells transfected with NL4-3 Nef. For JM cells, the control represents cells transfected with empty vector, pIRES2-eGFP. In order to compare between experiments, CD4 surface levels were represented as a percentage of the control condition. Values are the mean relative CD4 surface level percentage ± the standard error of the mean (SEM) from three independent experiments.
Further evidence for the suitability of the S2 cell system was obtained by pairwise comparisons of the activity of various Nef mutants in S2 and JM T cells (see reference 66 for a recent review of functional sites on Nef). S2 cells were cotransfected with pAc.CD4 and pMt.NL4-3 Nef wild-type and mutant plasmids (see Materials and Methods). CD4+ JM cells were transfected with pNL4-3 Nef.IRES.GFP plasmids. CD4 surface levels were measured by FACS analysis. For both S2 and JM cells (Fig. 4), mutation of the myristoylation site (G2), the CD4-binding site (WL57,58), and the dileucine sequence (LL164,165) of Nef (see Fig. 1) abrogated its ability to downregulate CD4. On the other hand, mutation of the acidic cluster (EEEE62-65) and the polyproline motif (P72,75) (see Fig. 1) that are required for major histocompatibility complex class I (MHC-I) downregulation (49, 60, 66) had no effect in the ability of Nef to downregulate CD4 in both S2 and JM cells (Fig. 4). Similar observations were made by immunofluorescence microscopy (Fig. 5). We also measured Nef expression by immunoblotting lysates from the transiently transfected S2 cells. We observed variable expression of these proteins in the total cell population. Nevertheless, consistently lower amounts of expression of the EEEE62-65AAAA and PP72,75AA mutants were sufficient to drive levels of downregulation that were comparable to those elicited by higher amounts of wild-type Nef, whereas similar levels of expression of the G2A and LL164,165AA null mutants with respect to wild type had no effect on CD4 (data not shown). This suggests that the expression levels of all of the constructs were saturating. Another hallmark of CD4 downregulation in mammalian systems is the requirement of a dileucine motif (LL413,414) on the CD4 cytosolic tail (2, 3). Mutation of this motif to di-alanine in pAc.CD4 also prevented CD4 downregulation by pMt.NL4-3 Nef in S2 cells (Fig. 4A).
FIG. 4.

Determinants of Nef-induced CD4 downregulation in Drosophila S2 and human JM CD4+ T cells. (A) FACS histograms of Drosophila S2 cells cotransfected with pAc.CD4 (wild-type or LL413,414AA mutant) and pMt.Nef (wild-type or various mutants). FACS analysis of uninduced (shaded gray), induced (bold line), and background fluorescence (dotted line) was performed as described in the legend to Fig. 2 and in Materials and Methods. (B) Bar graph depicting levels of cell surface CD4 in Drosophila S2 cells cotransfected with pAc.CD4 and pMt.Nef (wild type or various mutants) (dark gray) and in human CD4+ JM cells transfected with pIRES.Nef.GFP (wild type or mutants) (light gray). In this case the control represents relative CD4 surface levels in the presence of the empty vector pIRES2-eGFP. Induction of Nef expression and FACS analysis were performed as described in Materials and Methods. CD4 surface levels were calculated as described in the legend to Fig. 3. Values are the mean ± the SEM from three independent experiments.
FIG. 5.

Immunofluorescence microscopy of CD4 in S2 cells. Cells were transiently transfected with pAc.CD4 and pMt.NL4-3 Nef (wild type or the indicated mutant), incubated for 24 h, and left untreated (C, E, G, I, and K) or treated with CuSO4 (D, F, H, J, and L) for another 24 h, fixed, permeabilized, and stained with mouse monoclonal antibody to human CD4 and AlexaFluor 594-conjugated donkey antibody to mouse IgG. Untransfected cells treated in a similar fashion (A and B) were also stained as a control for antibody specificity. Stained cells were examined by confocal microscopy. Bar, 10 μm.
Taken together, these data provided strong evidence that the downregulation of CD4 in S2 and JM cells by HIV-1 Nef occurs by the same mechanism and likely involves homologous host-cell trafficking proteins. S2 cells are therefore a suitable system for an RNAi screen aimed at identifying host-cell proteins involved in Nef-induced CD4 downregulation.
RNAi screen in S2 cells reveals a requirement for clathrin and AP2 in the Nef-induced downregulation of CD4.
We used the vectors described above to generate a stable cell line expressing CD4 constitutively and Nef in a Cu2+-inducible manner. Several clones were assayed by FACS for CD4 surface levels before and after the induction of Nef expression with Cu2+. Of these clones, the cell line D6 exhibited the most uniform surface expression of CD4 and consistent, inducible downregulation by Nef (data not shown; see also Fig. 6 and Table 1). The ability of RNAi to reduce protein expression levels in the D6 cells was tested by the addition of dsRNAs targeted against CD4 and Nef (Fig. 6A). Compared to a negative control targeting GFP, the dsRNA directed to CD4 knocked down surface levels of this protein by ∼85% (see Table 1), as measured by FACS (Fig. 6A). In addition, treatment of D6 cells with dsRNA to Nef completely abolished the Cu2+-induced downregulation of CD4 (Fig. 6A), demonstrating that Nef expression had been effectively eliminated. The nearly complete elimination of surface CD4 expression and Nef function by the control dsRNAs was taken as a general indicator for the effectiveness of RNAi treatment in this system. Moreover, several V5-epitope-tagged Drosophila proteins were targeted for RNAi knockdown in transiently transfected S2 cells and subjected to immunoblot analysis (Fig. 6C). We observed that, upon treatment with gene-specific dsRNAs, there was a marked reduction in the expression of all of these proteins relative to treatment with a nonspecific dsRNA (Fig. 6C). We did not measure endogenous protein levels in the cells from the RNAi screening due to the lack of available antibodies for most targets.
FIG. 6.

Effect of selected RNAi treatments on Nef-induced CD4 downregulation in stably transfected S2 cells. (A) CD4 expression profiles of S2 cells stably expressing CD4 and Nef, after treatment with dsRNAs targeting GFP (nontargeting control), CD4 (no CD4 expression control), and Nef (no downregulation control). (B) CD4 expression profiles of S2 cells stably expressing CD4 and Nef, after treatment with dsRNAs targeting the clathrin subunits CHC and CLC; α-COP; AP complex subunits μ1, μ2, and μ3; and GGA. In each case, a portion of the S2 cells treated with dsRNAs were induced with CuSO4 for 24 h, whereas the remainder were left untreated. CD4 surface expression was measured by FACS as described in the legend to Fig. 2 and in Materials and Methods, with the exception that an isotype control antibody was used to stain the cells as a negative antibody control. (C) Immunoblot analysis of lysates from S2 cells transiently transfected with V5-epitope-tagged Drosophila genes (μ1, μ2, μ3, GGA, and CLC). After transfection, each group of cells was seeded into two culture wells and received dsRNA targeting either GFP (negative control; lanes 1, 3, 5, 7, and 9) or the specific transgene (lanes 2, 4, 6, 8, and 10). Lysates were subjected to SDS-PAGE and probed with anti-V5 monoclonal antibody. The positions of molecular mass markers (in kilodaltons) are shown on the left.
TABLE 1.
Raw data from the Nef-CD4 S2 RNAi screena
| Control target gene or human orthologue | CG no. | No. of trials | Mean surface rfu ± SEMb
|
Fold downregulationc | |
|---|---|---|---|---|---|
| CD4 | CD4 plus Nef | ||||
| Control target gene | |||||
| GFP | 10 | 54.84 ± 3.71 | 26.33 ± 1.92 | 2.08 | |
| CD4 | 7 | 8.78 ± 1.10 | 5.28 ± 0.69 | 1.66 | |
| NL4-3 Nef | 5 | 55.62 ± 4.65 | 55.10 ± 2.21 | 1.01 | |
| Human ortholog | |||||
| Intersectin | 1099 | 4 | 65.48 ± 4.31 | 29.08 ± 5.75 | 2.25 |
| Auxilin/GAK | 1107 | 4 | 63.12 ± 2.62 | 26.79 ± 3.43 | 2.36 |
| Ykt6 | 1515 | 2 | 93.20 ± 11.87 | 34.36 ± 0.99 | 2.71 |
| CVAK104 | 1951 | 3 | 59.11 ± 3.86 | 24.04 ± 4.21 | 2.46 |
| CALM/AP180 | 2520 | 5 | 70.98 ± 4.04 | 25.16 ± 7.55 | 2.82 |
| PI4K II α | 2929 | 4 | 58.37 ± 2.41 | 20.50 ± 2.35 | 2.85 |
| GGA | 3002 | 5 | 60.08 ± 1.87 | 26.63 ± 4.97 | 2.26 |
| AP3 μ3 | 3035 | 3 | 61.05 ± 3.06 | 26.13 ± 3.72 | 2.34 |
| β-TrCP | 3412 | 3 | 67.80 ± 7.28 | 28.08 ± 3.51 | 2.41 |
| TOM1 | 3529 | 3 | 62.18 ± 2.05 | 25.27 ± 3.11 | 2.46 |
| Cortactin | 3637 | 4 | 53.17 ± 3.23 | 28.70 ± 4.60 | 1.85 |
| Numb | 3779 | 3 | 64.39 ± 6.52 | 20.16 ± 2.91 | 3.19 |
| Syx5d | 4214 | 2 | 67.95 ± 1.70 | 44.69 ± 5.08 | 1.52 |
| AP2 α | 4260 | 4 | 70.24 ± 7.95 | 25.54 ± 1.87 | 2.75 |
| EEA1 | 5020 | 2 | 68.32 ± 5.97 | 23.82 ± 6.76 | 2.87 |
| Sec6 | 5341 | 5 | 73.75 ± 1.85 | 30.37 ± 2.38 | 2.43 |
| PI3K Vps34 | 5373 | 3 | 69.02 ± 7.64 | 22.89 ± 3.10 | 3.02 |
| Rab7 | 5915 | 4 | 70.40 ± 3.17 | 27.12 ± 2.88 | 2.60 |
| EHD1 | 6148 | 4 | 69.59 ± 4.17 | 30.41 ± 3.19 | 2.29 |
| Snx3 | 6359 | 3 | 55.46 ± 7.95 | 19.89 ± 5.95 | 2.79 |
| Cog8 | 6488 | 3 | 68.54 ± 3.01 | 27.75 ± 5.99 | 2.47 |
| STAM1 | 6521 | 3 | 55.36 ± 4.37 | 23.19 ± 3.84 | 2.39 |
| Synaptojanin | 6562 | 4 | 67.02 ± 1.75 | 33.52 ± 4.24 | 2.00 |
| Clathrin light chain | 6948 | 4 | 87.90 ± 6.10 | 53.82 ± 8.48 | 1.63 |
| Cbl | 7037 | 3 | 65.00 ± 14.46 | 22.07 ± 1.70 | 2.95 |
| AP2 μ2 | 7057 | 4 | 77.53 ± 9.14 | 52.43 ± 7.01 | 1.48 |
| GMX-33 | 7085 | 3 | 81.10 ± 8.33 | 34.91 ± 9.28 | 2.32 |
| Vps39 | 7146 | 3 | 59.18 ± 3.56 | 26.62 ± 2.68 | 2.22 |
| Arp3 | 7558 | 3 | 57.90 ± 3.55 | 33.12 ± 6.05 | 1.75 |
| BIG | 7578 | 4 | 79.55 ± 8.02 | 37.45 ± 1.50 | 2.12 |
| α-COP | 7961 | 4 | 130.12 ± 17.20 | 90.48 ± 8.72 | 1.44 |
| Arf6 | 8156 | 4 | 63.82 ± 6.28 | 28.46 ± 8.68 | 2.24 |
| Snx6 | 8282 | 3 | 48.77 ± 3.55 | 14.90 ± 1.15 | 3.27 |
| Rab8 | 8287 | 3 | 60.87 ± 5.32 | 25.21 ± 2.40 | 2.41 |
| Arf1 | 8385 | 3 | 73.78 ± 4.43 | 29.51 ± 3.35 | 2.50 |
| GBF1 | 8487 | 2 | 60.42 ± 1.67 | 36.60 ± 7.87 | 1.65 |
| Rabenosyn-5 | 8506 | 3 | 59.38 ± 3.86 | 26.10 ± 3.29 | 2.27 |
| Epsin | 8532 | 3 | 63.78 ± 4.24 | 26.71 ± 0.51 | 2.39 |
| Amphiphysin | 8604 | 3 | 63.82 ± 5.42 | 30.13 ± 6.63 | 2.12 |
| Hsc70 | 8937 | 2 | 76.43 ± 2.45 | 22.97 ± 1.32 | 3.33 |
| Clathrin heavy chain | 9012 | 3 | 93.28 ± 6.17 | 92.80 ± 5.57 | 1.01 |
| AP1 γ | 9113 | 2 | 75.58 ± 4.60 | 28.47 ± 5.87 | 2.65 |
| Rabex-5 | 9139 | 4 | 67.11 ± 6.37 | 24.80 ± 3.98 | 2.71 |
| AP1 μ1 | 9388 | 4 | 67.39 ± 2.40 | 29.52 ± 5.60 | 2.28 |
| Rab35 | 9575 | 3 | 76.33 ± 3.97 | 38.16 ± 4.57 | 2.00 |
| Dab | 9695 | 3 | 68.73 ± 7.44 | 27.75 ± 4.37 | 2.48 |
| TSG101 | 9712 | 4 | 109.87 ± 9.74 | 34.28 ± 4.87 | 3.20 |
| Arp2 | 9901 | 3 | 63.93 ± 6.14 | 28.82 ± 2.65 | 2.22 |
| Rab9 | 9994 | 3 | 61.49 ± 1.81 | 23.45 ± 2.31 | 2.62 |
| Vps8 | 10144 | 3 | 68.46 ± 1.17 | 27.74 ± 2.42 | 2.47 |
| Pak1/2 | 10295 | 3 | 64.77 ± 6.90 | 24.87 ± 2.17 | 2.60 |
| AAK1 | 10637 | 3 | 61.26 ± 1.45 | 22.45 ± 3.77 | 2.73 |
| HIP1 | 10971 | 2 | 65.53 ± 6.55 | 26.60 ± 0.76 | 2.46 |
| AP3 δ | 10986 | 3 | 69.80 ± 2.31 | 24.06 ± 1.34 | 2.90 |
| ARH | 11804 | 2 | 72.08 ± 7.39 | 26.55 ± 3.64 | 2.72 |
| Lyst | 11814 | 3 | 66.18 ± 6.19 | 26.98 ± 3.42 | 2.45 |
| Stonin | 12473 | 2 | 72.11 ± 12.54 | 16.87 ± 0.14 | 4.27 |
| ALIX | 12876 | 3 | 68.57 ± 5.86 | 32.56 ± 3.79 | 2.11 |
| Endophilin | 14296 | 2 | 78.68 ± 3.29 | 27.76 ± 8.17 | 2.83 |
| USP2 | 14619 | 3 | 63.62 ± 4.15 | 26.98 ± 6.20 | 2.36 |
| Vps25 | 14750 | 2 | 80.96 ± 20.57 | 19.81 ± 0.82 | 4.09 |
| Vps26 | 14804 | 3 | 68.03 ± 11.48 | 30.67 ± 4.35 | 2.22 |
| Eps15 | 16932 | 2 | 82.77 ± 16.30 | 21.80 ± 0.07 | 3.80 |
| Rab10 | 17060 | 4 | 69.12 ± 4.94 | 32.97 ± 7.53 | 2.10 |
| Rab21 | 17515 | 2 | 74.66 ± 15.15 | 21.97 ± 3.29 | 3.40 |
| Vps41e | 18028 | 3 | 70.99 ± 10.22 | 46.38 ± 10.27 | 1.53 |
| Dynamin | 18102 | 4 | 69.33 ± 7.19 | 26.81 ± 4.27 | 2.59 |
| Rabip4 | 31064 | 5 | 66.84 ± 3.07 | 29.22 ± 6.91 | 2.29 |
Drosophila RNAi targets are listed with the CG number in the second column, with the corresponding human ortholog(s) listed in the first column. Data represent the average CD4 surface level before and after the induction of Nef expression in relative fluorescence units (rfu).
The standard errors of the mean for RNAi experiments with n = 2 are not considered statistically significant but were used to judge the likelihood of reproducibility of subsequent experiments.
By definition, a fold downregulation of 1.00 is equal to no effect, i.e., the CD4 levels are not affected by Nef induction. RNAi against Nef provides a positive control and approaches total inhibition of downregulation (1.01-fold), as does RNAi targeting CHC.
This treatment resulted in mildly toxic effects and was therefore not considered as a positive hit for the RNAi screen.
The standard error of the mean for Vps41 is large compared to the mean values for CD4 levels ± Nef and was not considered reproducible.
A total of 68 components of the protein trafficking machinery were screened for their contribution to the Nef-induced downregulation of CD4 in D6 S2 cells (Table 1). The list of targets included clathrin and various clathrin-associated proteins, non-clathrin coat proteins, components of the multivesicular body (MVB) and endosomal recycling machineries, actin-associated proteins, phosphoinositide metabolism enzymes, components of the ubiquitin-modification machinery, and miscellaneous others. For each protein tested in the screen, D6 cells were treated with a specific dsRNA, incubated for 3 days, and then split into two parallel cultures. One culture was left uninduced, while the other was induced by the addition of Cu2+ for 24 h. The CD4 surface levels of both uninduced and induced cells were measured by FACS. The entire screen was conducted in duplicate, and most targets were tested additional times for confirmation.
The results of the RNAi screen are shown in Fig. 6B (histograms for selected targets) and Fig. 7 (scatter plot of all of the data), and summarized in Table 1. Most RNAi treatments, including the nontargeting negative control, fall along a single regression line with a slope of 0.45 on the scatter plot (Fig. 7), indicating that these dsRNAs had no effect on the ability of Nef to downregulate CD4. This slope corresponds to an average downregulation of 2.2-fold for the entire data set, roughly equivalent to the observed value of ∼2-fold in untreated cells. Because in most cases we do not know if these RNAi treatments caused effective elimination of the target proteins, we cannot rule out an involvement of all of the targets that tested negative in this screen. In addition, some targets that produced small effects were difficult to replicate beyond the initial screen due to mild toxicity (see Table 1). A few dsRNAs, however, produced reliable outliers, evidence that they interfered with the ability of Nef to modulate CD4 expression (Fig. 6B and 7 and Table 1). These included dsRNAs targeting the clathrin heavy chain (CHC), the clathrin light chain (CLC), the μ2 subunit of AP2, and α-COP (Fig. 6B and 7). We also observed a mild inhibition in cells treated with a dsRNA targeting Vps41, but the relatively large variation between experimental replicates precluded the assignment of this target as a true hit.
FIG. 7.

Results of the RNAi screen of 68 components of the protein trafficking machinery for their involvement in Nef-induced CD4 downregulation in S2 cells. Mean ± the SEM (n = 2 to 10) CD4 levels of cells treated with dsRNAs targeting 68 candidate and control genes (see Table 1), represented on an x-y plot. Each datum point represents surface CD4 levels, as measured by FACS, for cells treated with a particular dsRNA. The position on the x axis indicates the amount of CD4 on the cell surface without induction, while the position on the y axis indicates the amount of CD4 on the cell surface upon induction of Nef expression, in relative fluorescence units (rfu). According to this rubric, datum points that have the same amount of CD4 expression without or with induction of Nef expression indicate dsRNAs that completely inhibited the ability of Nef to downregulate CD4. A least-squares fit regression line (solid black line) for the entire data set with a 95% confidence interval (broken lines) and the line y = x (gray line) indicating the position of no downregulation have been added to the plot.
The CHC dsRNA displayed the strongest inhibition of Nef function and was nearly as effective as the dsRNA that targeted Nef itself (Fig. 6B and 7). A dsRNA targeting CLC had a weaker, but reproducible effect on Nef function (Fig. 6B and 7). These findings are consistent with models of Nef-induced CD4 downregulation that invoke a role of clathrin-dependent trafficking intermediaries. Nevertheless, a requirement for clathrin in the Nef-induced downregulation of CD4 had not been directly demonstrated prior to this study. A dsRNA targeting the μ2 subunit of AP2 reduced Nef activity by roughly half in this system (Fig. 6B and 7). dsRNAs targeting the γ and μ1 subunits of AP1 and the δ and μ3 subunits of AP3, either alone (Fig. 6B and Table 1) or in combinations (data not shown), had no effect on CD4 downregulation. Because AP1 and AP3 have been proposed to act as mediators of CD4 downregulation (11, 19, 41, 68), we directly tested the efficacy of the μ1 and μ3 dsRNAs on protein expression. Immunoblot analysis clearly indicated a strong reduction of protein levels (Fig. 6C), supporting the conclusion that neither AP1 nor AP3 are required for Nef-mediated CD4 downregulation in S2 cells. Drosophila does not have orthologs of the subunits of a fourth, non-clathrin-associated AP complex found in human cells, AP4 (7), so this complex can be definitively ruled out as a required mediator of Nef effects on CD4.
The effect of Nef on CD4 was also inhibited by treatment of the D6 cells with dsRNA against α-COP (Fig. 6B and 7). This effect was somewhat unusual in that higher levels of CD4 were found on the cell surface regardless of Nef induction. α-COP is a subunit of the heteroheptameric COP-I complex that is primarily involved in endoplasmic reticulum and Golgi transport processes (for a review, see reference 44), although a role for COP-I in endosomal traffic has also been proposed (31, 80). In this regard, Nef has been previously shown to interact with the β-COP subunit of COP-I (4), but the functional significance of this interaction remains a matter of debate (40, 59).
AP2 requirement for Nef-induced CD4 downregulation in human cells.
We next assessed the requirement for clathrin and the different AP complexes in the human cell line HeLa. RNAi treatment reduced the expression of μ1A, μ2, and μ3A compared to treatment with nonspecific RNAi (Fig. 8A). RNAi against μ2 increased surface levels of TfR and RNAi against μ3A increased surface levels of LAMP1 as determined by FACS (Fig. 8B), indicators of impaired AP2 and AP3 function, respectively (38, 55). We then measured surface levels of CD4 in the RNAi-treated HeLa cells upon expression of wild-type NL4-3 Nef or the Nef-null mutant LL164,165AA. As shown in Fig. 8C, nontargeting RNAi-treated cells had robust downregulation, whereas cells depleted of μ2 showed decreased downregulation. It should be noted that, in both S2 and HeLa cells, depletion of μ2 was only partially effective in blocking the effects of Nef on CD4. This may represent the residual effect of low levels of AP2 in the RNAi-treated cells or the activity of a partially redundant pathway. We are unable distinguish between these two possibilities at present. In agreement with a previous study (67), we did not observe a requirement for either μ1A or μ3A (Fig. 8C). We attempted to perform these experiments in cells depleted of CHC but were unable to obtain reproducible results because these cells proved refractory to transient transfection after RNAi treatment.
FIG. 8.

Analysis of AP2 knockdown in human cells. HeLa cells were treated with either nontargeting or specific RNAi oligonucleotide duplexes targeting μ1A, μ2, or μ3A as described in Materials and Methods. After 6 days of treatment, the cells were transfected with expression vectors for CD4 and either pNL4-3 Nef.IRES.GFP (wild-type) or the inactive LL164,165AA mutant. (A) Immunoblotting of RNAi-treated HeLa cell lysates using rabbit antisera to human μ1A, μ2, or μ3A. Two nonspecific bands on the μ2 blot are indicated by asterisks. The specific band migrates at ∼50 kDa and is drastically reduced in the RNAi-treated cells. (B) Surface TfR or LAMP1 were measured by FACS as a functional indicator of impaired AP2 or AP3 function, respectively. The μ2 RNAi-treated cells exhibit a strong increase in TfR, a finding consistent with a decrease in AP2-dependent endocytosis. The increase in surface LAMP1 in the μ3 RNAi-treated cells is indicative of improper AP3-dependent intracellular sorting. (C) Dependence of Nef-mediated CD4 downregulation on AP2. HeLa cells transfected as described above and processed for FACS in order to detect CD4 surface levels; in each case GFP fluorescence was used as a marker for cells that had been transfected with the Nef expression vector. Histograms for cells transfected with NefLL164,165AA (shaded gray), wild-type Nef (bold line), and an isotype control (dotted line) are presented. Nontargeting, μ1A, and μ3A RNAi-treated cells exhibit a decrease in surface CD4 in the presence of Nef. In the μ2 RNAi-treated cells this decrease is less marked.
Physical interaction of Nef with AP2 demonstrated in a yeast thee-hybrid system.
The RNAi screen pointed to AP2 as a likely mediator of Nef effects on CD4. However, there was little evidence in the literature for a direct interaction of Nef with AP2. We had previously reported interactions of Nef with the γ-σ1 and δ-σ3 hemicomplexes of AP1 and AP3, respectively, using a Y3H system (41). However, we had failed to detect interactions of Nef with the analogous αC-σ2 hemicomplex (41). We recently became aware that the αC subunit clone that was used for the Y3H analyses had a single missense mutation that resulted in a substitution of alanine to threonine at codon 131. This mutation placed a hydrophilic side chain within the hydrophobic core of the α subunit “trunk” domain (see reference 16), with potentially deleterious effects on protein structure. We reverted this mutation to code for alanine and repeated the Y3H analyses. We then observed that Nef was able to interact with the wild-type αC-σ2 hemicomplex (Fig. 9). To ascertain the functional relevance of this interaction, we tested the sequence requirements by mutating residues that are contained within the dileucine motif of Nef. This motif has the sequence ENTSLL (residues 160 to 165) and fits the canonical motif [DE]XXXL[LI] that binds to AP complexes (8). We found that mutation of either L164 or LL164,165 abrogated interaction with αC-σ2 (Fig. 9). Mutation of E160 exhibited a partial loss of binding activity, whereas mutation of either T162 or S163 to alanine had no effect (see Fig. 9; the loss of binding activity of the E160A mutant is more apparent in the presence of 3-aminotriazole). Mutation of the WL57,58, EEEE62-65, or PxxP72-75 functional sites on Nef (Fig. 1), which are not required for CD4 downregulation (Fig. 4 and 5) (49, 60, 66, 71), also had no effect on the interaction of Nef with αC-σ2 (Fig. 9). These results indicated that Nef binding to the αC-σ2 hemicomplex is specifically dependent on the Nef ENTSLL motif in a manner that parallels the requirements for Nef downregulation of CD4 in cells (Fig. 4) (11, 27, 41). It is worth noting that the interaction of Nef with αC-σ2 reported here is much stronger than that previously shown with μ2 (19; see also reference 41).
FIG. 9.

Yeast three-hybrid analysis of Nef-AP2 interactions. GAL4BD-Nef and the σ1 or σ2 subunits of AP1 and AP2, respectively, were expressed from pBridge; γ1 and αC were expressed as fusions with GAL4AD from pGADT7. Yeast strains coexpressing Nef (wild-type NL4-3 variant or selected mutants) with either γ1-σ1 or αC-σ2 hemicomplexes were inoculated on agar plates made with medium with (+) or without (−) histidine (His) and in the absence or presence of 3 mM 3-aminotriazole (3AT). Growth indicates the occurrence of interactions.
Direct interaction of Nef and AP2 in vitro.
We next decided to test whether recombinant forms of Nef and AP2 interact in vitro. Honing et al. (36) previously showed that dileucine motifs from the phosphorylated tail of CD4, LIMP-II, and tyrosinase bound in vitro to a recombinant “core” AP2 complex comprising the N-terminal “trunk” domains of α and β2, plus the full-length μ2 and σ2 subunits. In addition, these authors showed that the dileucine-binding site was not contained within the C-terminal domain of μ2 (36). We therefore produced a similar AP2 core construct (denoted AP2core), with an additional deletion of the μ2 C-terminal domain. The αC trunk domain was expressed as a GST fusion protein in order to perform a pull-down assay. Using this assay, we observed that wild-type Nef, but not the LL164,165AA Nef mutant, bound to GST-AP2core, as determined by Coomassie blue staining (Fig. 10A) and immunoblot analysis (Fig. 10B). In contrast, neither Nef construct interacted with GST-ɛ fusion protein used as a negative control (Fig. 10). These observations thus demonstrated a direct and specific interaction of Nef, through its dileucine motif, with the fully assembled AP2core complex.
FIG. 10.

Direct interaction of Nef and AP2 detected in vitro. Purified recombinant proteins were prepared as described in Materials and Methods, subjected to a GST pull-down assay and SDS-PAGE, and stained with Coomassie blue (A) and immunoblotted with Nef antibody (B). Recombinant proteins were run individually in lane 1 (Nef LL164,165AA), lane 2 (wild-type NL4-3 Nef), lane 3 (GST-AP2core), and lane 4 (GST-ɛ-ear). Recombinant Nef proteins were incubated with GST-ɛ-ear (lanes 5 and 6) (control) or with GST-AP2core (lanes 7 and 8). Wild-type Nef is visible as a 27-kDa band in lane 8 in both panels A and B. This experiment is representative of three experiments with similar results. Molecular mass markers are visible on the left side of the Coomassie blue-stained gel. The masses in kilodaltons are indicated.
DISCUSSION
Recent studies have begun to exploit the ease and efficiency of RNAi screens in Drosophila S2 cells to identify host cell factors that are required for infection by human pathogens such as bacteria and fungi (1, 21, 57, 62, 78). We have now used this model system to investigate the molecular machinery involved in an aspect of viral pathogenesis: the mechanism by which primate immunodeficiency viruses downregulate their own coreceptor, CD4. An RNAi screen of 68 components of the protein trafficking machinery in S2 cells showed that CD4 downregulation by the HIV-1 Nef protein requires clathrin and the heterotetrameric AP2 complex, which are components of protein coats involved in rapid endocytosis from the plasma membrane in the host cells. The requirement for AP2 was confirmed in human HeLa cells. In contrast, other heterotetrameric (i.e., AP1 and AP3) and monomeric (i.e., GGA, Epsin, Dab, ARH, and Stonin) clathrin adaptors appear to be dispensable for CD4 downregulation (see Table 1). In addition, we used a Y3H system to demonstrate a specific interaction of Nef with a combination of the αC and σ2 subunits of AP2. Finally, a GST pull-down assay showed a direct and specific interaction of Nef with the heterotetrameric AP2core complex in vitro. Both the function and the interaction of Nef in these assays exhibited the same dependence on a dileucine sequence in Nef that was previously identified as being critical for CD4 downregulation (11, 27) and enhancement of viral infectivity (18). Our observations thus support a model in which Nef links the cytosolic tail of CD4 to clathrin-AP2 coats at the plasma membrane, leading to its endocytic removal from the cell surface.
Role of clathrin and AP2 in Nef-induced CD4 downregulation.
As the major devices for protein sorting at different stages of the endocytic and secretory pathways, clathrin-AP coats were long suspected to play roles in CD4 downregulation by Nef (11, 18, 19, 23, 28, 39, 41, 46, 58). This hypothesis was affirmed by the discovery that downregulation is strictly dependent on a Nef dileucine sequence (11, 27) that fits the [DE]XXXL[LI] consensus motif for signals that mediate clathrin-dependent sorting events and interaction with clathrin-associated AP complexes (8). Much work has been done to elucidate the exact role of clathrin and the various AP complexes in the downregulation of CD4. However, the evidence has thus far been largely indirect, and different studies have produced conflicting results.
Nef was shown localize to clathrin-AP2-coated pits at the plasma membrane and to promote the recruitment of CD4 to such pits (12, 23, 28). This is consistent with the finding that Nef accelerates CD4 internalization from the cell surface (2, 64). These observations led to the testing for an involvement of AP2 in downregulation. Expression of an AP2-μ2 subunit construct rendered incapable of binding YXXØ-type endocytic signals by mutation of aspartate-176 to alanine (55) was found to block the HIV-1 Nef-dependent redistribution of CD4 to endosomes in HeLa cells (6). This finding is puzzling, however, because neither HIV-1 Nef nor CD4 have YXXØ-type signals; instead, downregulation depends on dileucine-containing sequences in both Nef and CD4 (2, 11, 15, 23, 27). Since YXXØ and dileucine signals most likely have different binding sites on AP2 (36, 41, 52, 56, 63), it is unclear how such a mutant could have a “dominant-negative” effect on CD4 downregulation by Nef. Subsequent experiments showed that depletion of μ2 by RNAi caused only a slight inhibition of Nef-mediated CD4 downregulation in HeLa cells and in T cells (42, 68). More complete inhibition required overexpression of a dominant-negative mutant of Eps15, a regulator of endocytosis, in conjunction with RNAi-mediated μ2 depletion in T cells (42). Attempts to demonstrate a clear physical connection between Nef and AP2 have similarly yielded conflicting results. Yeast two-hybrid (Y2H) assays have been used to detect a very weak interaction of HIV-1 Nef with μ2 (19), whereas an interaction with the AP2 β2 subunit was observed by using a chemical cross-linking approach (27). Thus, the role of AP2 in downregulation remained unclear from all of this work. Our findings that RNAi-mediated depletion of either clathrin or AP2 results in profound inhibition of CD4 downregulation by Nef and that HIV-1 Nef interacts robustly and specifically with AP2 αC-σ2 in Y3H assays and with the AP2 core complex in pull-down assays (all demonstrated here for the first time) now provide strong support to the AP2-dependent, endocytic model of Nef action on CD4.
Recognition of dileucine signals by AP2.
Upon correction of a point mutation in the original AP2-αC subunit construct used in Y3H assays (41), we were able to detect a specific and robust interaction between the αC-σ2 hemicomplex and Nef. This interaction requires the coexpression of both AP2 subunits and is not observed with single subunits (data not shown). This probably reflects the need of hemicomplex assembly for proper folding and stability of the constituent subunits. In addition, the Y3H interaction is strictly dependent on the LL164,165 dileucine sequence and partially dependent on the upstream E160 residue of Nef. It is not dependent, however, on neighboring residues or other functional motifs. These requirements exactly match those defined for CD4 downregulation (11, 14, 27) (Fig. 4 and 5), indicating that the interactions are likely to be functionally relevant. These interactions are analogous to those of Nef with the γ-σ1 subunits of AP1 and the δ-σ3 subunits of AP3 (41), suggesting that these three complexes bind to Nef in a similar manner. Moreover, like the corresponding AP1 and AP3 hemicomplexes, the AP2 αC-σ2 hemicomplex would be expected to bind other [DE]XXXL[LI]-type dileucine signals involved in internalization from the cell surface. The easy detection of these interactions with the Y3H now opens the way for further analyses of the mechanism of recognition of dileucine signals.
Do AP1 and AP3 participate in Nef-induced CD4 downregulation?
HIV-1 Nef was found to localize to the area of the Golgi complex (39, 48), in addition to the plasma membrane, and to induce retention of CD4 in the Golgi region (10, 48). These observations supported a model in which intracellular retention of newly synthesized or recycling CD4 contributes to downregulation of the receptor (48, 68). The area of the Golgi complex houses the TGN and a subset of endosomes, both of which have clathrin coats containing AP1 and AP3. Indeed, a variety of biochemical analyses showed interactions of Nef with AP1 and AP3 that were much stronger than those with AP2. Specifically, HIV-1 Nef was found to interact with AP1 and AP3 from cell extracts by GST pull-down assays (11, 39, 41), with the μ1A subunit of AP1 and the μ3A subunit of AP3 by Y2H assays (19, 46), and with the γ-σ1 and δ-σ3 hemicomplexes of AP1 and AP3, respectively, by Y3H assays (41). These interactions were all dependent on the dileucine sequence of Nef, indicating that they may be functionally relevant for CD4 downregulation. However, RNAi-mediated depletion of AP1 and AP3 subunits has been reported to have no effect on Nef-induced CD4 downregulation in human T cells and astrocytes (67), a finding that we have now replicated in S2 and HeLa cells. Therefore, it is unclear what roles, if any, Nef interactions with AP1 and AP3 play in CD4 downregulation. It is possible that these complexes participate in the postendocytic routing of internalized CD4 but, clearly, this potential role has no impact on the ability of Nef to decrease surface CD4 levels.
Postendocytic fate of internalized CD4.
The downregulation of CD4 by Nef involves not only its removal from the cell surface but also its targeting to lysosomes for degradation (59, 64, 73). Thus, it is likely that Nef also functions to prevent the recycling of internalized CD4 to the plasma membrane and/or to promote its delivery to lysosomes, perhaps following the MVB pathway. However, the depletion of various components of endosomal recycling (e.g., EHD1, Rabenosyn-5, Arf6, Rab35, and Rabip4; Table 1) or MVB pathways (e.g., TSG101, STAM1, ALIX, and Vps25; Table 1) had no effect on the ability of Nef to decrease surface levels of CD4. This observation does not necessarily imply a lack of Nef involvement in these processes because of the following caveats: (i) the RNAi treatment may not have caused sufficient depletion of the target proteins to elicit an effect, (ii) the right target proteins may not have been picked for depletion, and (iii) inhibition of recycling or lysosomal delivery may affect the intracellular distribution of the internalized protein (as may also be the case for AP1 and AP3 depletion), without preventing the reduction in surface CD4 levels. An assessment of the possible role on these intracellular sorting events will require an expansion of the set of machinery components targeted for depletion and assays that do not rely on measurement of cell surface levels (i.e., FACS) but on the localization of downregulated CD4 (e.g., fluorescence microscopy).
Distinct mechanisms for CD4 and MHC-I downregulation.
Although CD4 is the cell surface protein that is most effectively downregulated by Nef, other cell surface proteins also undergo various degrees of Nef-induced downregulation. Among the latter are certain MHC-I haplotypes (29, 49, 67, 74, 79), the downregulation of which is thought to allow HIV-1 to evade immune surveillance (13, 17). Strikingly, Nef-induced MHC-I downregulation appears to occur by a mechanism that is quite distinct from that of CD4 downregulation. Indeed, downregulation of MHC-I by Nef primarily involves the misrouting of newly synthesized molecules from the TGN to the lysosomes (43) and requires AP1 but not AP3 (67). This process is independent of the Nef dileucine sequence (65) and instead depends on an acidic cluster (EEEE62-65) and a polyproline motif (PXXP72,75) (Fig. 1) (49, 60, 66). In addition, Nef promotes the association of MHC-I and AP1 with sequence requirements that are identical to those required for downregulation (49, 67, 81). Thus, Nef is a multifunctional “connector” molecule capable of using distinct interfaces to link the cytosolic tails of different transmembrane proteins to specific AP complexes. These alternative modes of interaction endow Nef with the ability to interfere with protein trafficking at different stages of the secretory and endocytic pathways.
Acknowledgments
We thank Peter Yang for help with Y3H assays, Bridgette Beach for constructing the AP2core expression vector, Xiaolin Zhu and Nora Tsai for technical assistance, and Katy Janvier for critical reading of the manuscript.
This study was supported by the Intramural Programs of NICHD (J.S.B.) and NIDDK (J.H.H.) and by the NIH Intramural AIDS Targeted Antiviral Program (J.S.B. and J.H.H.). R.C. was supported by the NIH-Cambridge and Gates-Cambridge Graduate Scholarships. W.J.S. is the recipient of NICHD Career Transition Award K22 HD54602.
Footnotes
Published ahead of print on 31 January 2007.
REFERENCES
- 1.Agaisse, H., L. S. Burrack, J. A. Philips, E. J. Rubin, N. Perrimon, and D. E. Higgins. 2005. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 309:1248-1251. [DOI] [PubMed] [Google Scholar]
- 2.Aiken, C., J. Konner, N. R. Landau, M. E. Lenburg, and D. Trono. 1994. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76:853-864. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, S. J., M. Lenburg, N. R. Landau, and J. V. Garcia. 1994. The cytoplasmic domain of CD4 is sufficient for its down-regulation from the cell surface by human immunodeficiency virus type 1 Nef. J. Virol. 68:3092-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benichou, S., M. Bomsel, M. Bodeus, H. Durand, M. Doute, F. Letourneur, J. Camonis, and R. Benarous. 1994. Physical interaction of the HIV-1 Nef protein with beta-COP, a component of non-clathrin-coated vesicles essential for membrane traffic. J. Biol. Chem. 269:30073-30076. [PubMed] [Google Scholar]
- 5.Benson, R. E., A. Sanfridson, J. S. Ottinger, C. Doyle, and B. R. Cullen. 1993. Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J. Exp. Med. 177:1561-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blagoveshchenskaya, A. D., L. Thomas, S. F. Feliciangeli, C. H. Hung, and G. Thomas. 2002. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell 111:853-866. [DOI] [PubMed] [Google Scholar]
- 7.Boehm, M., and J. S. Bonifacino. 2001. Adaptins: the final recount. Mol. Biol. Cell 12:2907-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonifacino, J. S., and L. M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395-447. [DOI] [PubMed] [Google Scholar]
- 9.Bowers, K., C. Pitcher, and M. Marsh. 1997. CD4: a coreceptor in the immune response and HIV infection. Int. J. Biochem. Cell. Biol. 29:871-875. [DOI] [PubMed] [Google Scholar]
- 10.Brady, H. J., D. J. Pennington, C. G. Miles, and E. A. Dzierzak. 1993. CD4 cell surface downregulation in HIV-1 Nef transgenic mice is a consequence of intracellular sequestration. EMBO J. 12:4923-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bresnahan, P. A., W. Yonemoto, S. Ferrell, D. Williams-Herman, R. Geleziunas, and W. C. Greene. 1998. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr. Biol. 8:1235-1238. [DOI] [PubMed] [Google Scholar]
- 12.Burtey, A., J. Z. Rappoport, J. Bouchet, S. Basmaciogullari, J. Guatelli, S. M. Simon, S. Benichou, and A. Benmerah. 2007. Dynamic Interaction of HIV-1 Nef with the clathrin-mediated endocytic pathway at the plasma membrane. Traffic 8:61-76. [DOI] [PubMed] [Google Scholar]
- 13.Cohen, G. B., R. T. Gandhi, D. M. Davis, O. Mandelboim, B. K. Chen, J. L. Strominger, and D. Baltimore. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661-671. [DOI] [PubMed] [Google Scholar]
- 14.Coleman, S. H., R. Madrid, N. Van Damme, R. S. Mitchell, J. Bouchet, C. Servant, S. Pillai, S. Benichou, and J. C. Guatelli. 2006. Modulation of cellular protein trafficking by human immunodeficiency virus type 1 Nef: role of the acidic residue in the ExxxLL motif. J. Virol. 80:1837-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman, S. H., N. Van Damme, J. R. Day, C. M. Noviello, D. Hitchin, R. Madrid, S. Benichou, and J. C. Guatelli. 2005. Leucine-specific, functional interactions between human immunodeficiency virus type 1 Nef and adaptor protein complexes. J. Virol. 79:2066-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins, B. M., A. J. McCoy, H. M. Kent, P. R. Evans, and D. J. Owen. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell 109:523-535. [DOI] [PubMed] [Google Scholar]
- 17.Collins, K. L., B. K. Chen, S. A. Kalams, B. D. Walker, and D. Baltimore. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397-401. [DOI] [PubMed] [Google Scholar]
- 18.Craig, H. M., M. W. Pandori, and J. C. Guatelli. 1998. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc. Natl. Acad. Sci. USA 95:11229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craig, H. M., T. R. Reddy, N. L. Riggs, P. P. Dao, and J. C. Guatelli. 2000. Interactions of HIV-1 Nef with the mu subunits of adaptor protein complexes 1, 2, and 3: role of the dileucine-based sorting motif. Virology 271:9-17. [DOI] [PubMed] [Google Scholar]
- 20.DeLano, W. L. 2002. The PyMOL user's manual. DeLano Scientific, San Carlos, CA.
- 21.Elwell, C., and J. N. Engel. 2005. Drosophila melanogaster S2 cells: a model system to study Chlamydia interaction with host cells. Cell Microbiol. 7:725-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foley, E., and P. H. O'Farrell. 2004. Functional dissection of an innate immune response by a genome-wide RNAi screen. PLoS Biol. 2:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foti, M., A. Mangasarian, V. Piguet, D. P. Lew, K. H. Krause, D. Trono, and J. L. Carpentier. 1997. Nef-mediated clathrin-coated pit formation. J. Cell Biol. 139:37-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia, J. V., and A. D. Miller. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by Nef. Nature 350:508-511. [DOI] [PubMed] [Google Scholar]
- 25.Geyer, M., O. T. Fackler, and B. M. Peterlin. 2001. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2:580-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glushakova, S., J. Munch, S. Carl, T. C. Greenough, J. L. Sullivan, L. Margolis, and F. Kirchhoff. 2001. CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4+ T-cell depletion in human lymphoid tissue ex vivo. J. Virol. 75:10113-10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenberg, M., L. DeTulleo, I. Rapoport, J. Skowronski, and T. Kirchhausen. 1998. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 8:1239-1242. [DOI] [PubMed] [Google Scholar]
- 28.Greenberg, M. E., S. Bronson, M. Lock, M. Neumann, G. N. Pavlakis, and J. Skowronski. 1997. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. EMBO J. 16:6964-6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenberg, M. E., A. J. Iafrate, and J. Skowronski. 1998. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grzesiek, S., S. J. Stahl, P. T. Wingfield, and A. Bax. 1996. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 35:10256-10261. [DOI] [PubMed] [Google Scholar]
- 31.Gu, F., and J. Gruenberg. 1999. Biogenesis of transport intermediates in the endocytic pathway. FEBS Lett. 452:61-66. [DOI] [PubMed] [Google Scholar]
- 32.Guy, B., M. P. Kieny, Y. Riviere, C. Le Peuch, K. Dott, M. Girard, L. Montagnier, and J. P. Lecocq. 1987. HIV F/3′ ORF encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 330:266-269. [DOI] [PubMed] [Google Scholar]
- 33.Hanna, Z., D. G. Kay, N. Rebai, A. Guimond, S. Jothy, and P. Jolicoeur. 1998. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 95:163-175. [DOI] [PubMed] [Google Scholar]
- 34.Harris, M. P., and J. C. Neil. 1994. Myristoylation-dependent binding of HIV-1 Nef to CD4. J. Mol. Biol. 241:136-142. [DOI] [PubMed] [Google Scholar]
- 35.Hierro, A., J. Kim, and J. H. Hurley. 2005. Polycistronic expression and purification of the ESCRT-II endosomal trafficking complex. Methods Enzymol. 403:322-332. [DOI] [PubMed] [Google Scholar]
- 36.Honing, S., D. Ricotta, M. Krauss, K. Spate, B. Spolaore, A. Motley, M. Robinson, C. Robinson, V. Haucke, and D. J. Owen. 2005. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol. Cell 18:519-531. [DOI] [PubMed] [Google Scholar]
- 37.Hua, J., W. Blair, R. Truant, and B. R. Cullen. 1997. Identification of regions in HIV-1 Nef required for efficient downregulation of cell surface CD4. Virology 231:231-238. [DOI] [PubMed] [Google Scholar]
- 38.Janvier, K., and J. S. Bonifacino. 2005. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16:4231-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janvier, K., H. Craig, D. Hitchin, R. Madrid, N. Sol-Foulon, L. Renault, J. Cherfils, D. Cassel, S. Benichou, and J. Guatelli. 2003. HIV-1 Nef stabilizes the association of adaptor protein complexes with membranes. J. Biol. Chem. 278:8725-8732. [DOI] [PubMed] [Google Scholar]
- 40.Janvier, K., H. Craig, S. Le Gall, R. Benarous, J. Guatelli, O. Schwartz, and S. Benichou. 2001. Nef-induced CD4 downregulation: a diacidic sequence in human immunodeficiency virus type 1 Nef does not function as a protein sorting motif through direct binding to beta-COP. J. Virol. 75:3971-3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janvier, K., Y. Kato, M. Boehm, J. R. Rose, J. A. Martina, B. Y. Kim, S. Venkatesan, and J. S. Bonifacino. 2003. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 gamma-sigma1 and AP-3 delta-sigma3 hemicomplexes. J. Cell Biol. 163:1281-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin, Y. J., C. Y. Cai, X. Zhang, H. T. Zhang, J. A. Hirst, and S. J. Burakoff. 2005. HIV Nef-mediated CD4 down-regulation is adaptor protein complex 2 dependent. J. Immunol. 175:3157-3164. [DOI] [PubMed] [Google Scholar]
- 43.Kasper, M. R., J. F. Roeth, M. Williams, T. M. Filzen, R. I. Fleis, and K. L. Collins. 2005. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J. Biol. Chem. 280:12840-12848. [DOI] [PubMed] [Google Scholar]
- 44.Kirchhausen, T. 2000. Three ways to make a vesicle. Nat. Rev. Mol. Cell. Biol. 1:187-198. [DOI] [PubMed] [Google Scholar]
- 45.Kirchhoff, F., T. C. Greenough, D. B. Brettler, J. L. Sullivan, and R. C. Desrosiers. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332:228-232. [DOI] [PubMed] [Google Scholar]
- 46.Le Gall, S., L. Erdtmann, S. Benichou, C. Berlioz-Torrent, L. Liu, R. Benarous, J. M. Heard, and O. Schwartz. 1998. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8:483-495. [DOI] [PubMed] [Google Scholar]
- 47.Lefrancois, S., K. Janvier, M. Boehm, C. E. Ooi, and J. S. Bonifacino. 2004. An ear-core interaction regulates the recruitment of the AP-3 complex to membranes. Dev. Cell 7:619-625. [DOI] [PubMed] [Google Scholar]
- 48.Mangasarian, A., M. Foti, C. Aiken, D. Chin, J. L. Carpentier, and D. Trono. 1997. The HIV-1 Nef protein acts as a connector with sorting pathways in the Golgi and at the plasma membrane. Immunity 6:67-77. [DOI] [PubMed] [Google Scholar]
- 49.Mangasarian, A., V. Piguet, J. K. Wang, Y. L. Chen, and D. Trono. 1999. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 73:1964-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariani, R., F. Kirchhoff, T. C. Greenough, J. L. Sullivan, R. C. Desrosiers, and J. Skowronski. 1996. High frequency of defective nef alleles in a long-term survivor with nonprogressive human immunodeficiency virus type 1 infection. J. Virol. 70:7752-7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mariani, R., and J. Skowronski. 1993. CD4 down-regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc. Natl. Acad. Sci. USA 90:5549-5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marks, M. S., L. Woodruff, H. Ohno, and J. S. Bonifacino. 1996. Protein targeting by tyrosine- and di-leucine-based signals: evidence for distinct saturable components. J. Cell Biol. 135:341-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michel, N., I. Allespach, S. Venzke, O. T. Fackler, and O. T. Keppler. 2005. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr. Biol. 15:714-723. [DOI] [PubMed] [Google Scholar]
- 54.Motley, A., N. A. Bright, M. N. Seaman, and M. S. Robinson. 2003. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162:909-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nesterov, A., R. E. Carter, T. Sorkina, G. N. Gill, and A. Sorkin. 1999. Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J. 18:2489-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohno, H., J. Stewart, M. C. Fournier, H. Bosshart, I. Rhee, S. Miyatake, T. Saito, A. Gallusser, T. Kirchhausen, and J. S. Bonifacino. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269:1872-1875. [DOI] [PubMed] [Google Scholar]
- 57.Philips, J. A., E. J. Rubin, and N. Perrimon. 2005. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science 309:1251-1253. [DOI] [PubMed] [Google Scholar]
- 58.Piguet, V., Y. L. Chen, A. Mangasarian, M. Foti, J. L. Carpentier, and D. Trono. 1998. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J. 17:2472-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piguet, V., F. Gu, M. Foti, N. Demaurex, J. Gruenberg, J. L. Carpentier, and D. Trono. 1999. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 97:63-73. [DOI] [PubMed] [Google Scholar]
- 60.Piguet, V., L. Wan, C. Borel, A. Mangasarian, N. Demaurex, G. Thomas, and D. Trono. 2000. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2:163-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puertollano, R., and J. S. Bonifacino. 2004. Interactions of GGA3 with the ubiquitin sorting machinery. Nat. Cell Biol. 6:244-251. [DOI] [PubMed] [Google Scholar]
- 62.Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot, and R. A. Ezekowitz. 2002. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for Escherichia coli. Nature 416:644-648. [DOI] [PubMed] [Google Scholar]
- 63.Rapoport, I., Y. C. Chen, P. Cupers, S. E. Shoelson, and T. Kirchhausen. 1998. Dileucine-based sorting signals bind to the beta chain of AP-1 at a site distinct and regulated differently from the tyrosine-based motif-binding site. EMBO J. 17:2148-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rhee, S. S., and J. W. Marsh. 1994. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J. Virol. 68:5156-5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riggs, N. L., H. M. Craig, M. W. Pandori, and J. C. Guatelli. 1999. The dileucine-based sorting motif in HIV-1 Nef is not required for down-regulation of class I MHC. Virology 258:203-207. [DOI] [PubMed] [Google Scholar]
- 66.Roeth, J. F., and K. L. Collins. 2006. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol. Mol. Biol. Rev. 70:548-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roeth, J. F., M. Williams, M. R. Kasper, T. M. Filzen, and K. L. Collins. 2004. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 167:903-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rose, J. J., K. Janvier, S. Chandrasekhar, R. P. Sekaly, J. S. Bonifacino, and S. Venkatesan. 2005. CD4 down-regulation by HIV-1 and simian immunodeficiency virus (SIV) Nef proteins involves both internalization and intracellular retention mechanisms. J. Biol. Chem. 280:7413-7426. [DOI] [PubMed] [Google Scholar]
- 69.Ross, T. M., A. E. Oran, and B. R. Cullen. 1999. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. 9:613-621. [DOI] [PubMed] [Google Scholar]
- 70.Rossi, F., A. Gallina, and G. Milanesi. 1996. Nef-CD4 physical interaction sensed with the yeast two-hybrid system. Virology 217:397-403.8599229 [Google Scholar]
- 71.Saksela, K., G. Cheng, and D. Baltimore. 1995. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 14:484-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salvi, R., A. R. Garbuglia, A. Di Caro, S. Pulciani, F. Montella, and A. Benedetto. 1998. Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J. Virol. 72:3646-3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanfridson, A., B. R. Cullen, and C. Doyle. 1994. The simian immunodeficiency virus Nef protein promotes degradation of CD4 in human T cells. J. Biol. Chem. 269:3917-3920. [PubMed] [Google Scholar]
- 74.Schwartz, O., V. Marechal, S. Le Gall, F. Lemonnier, and J. M. Heard. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2:338-342. [DOI] [PubMed] [Google Scholar]
- 75.Sheffield, P., S. Garrard, and Z. Derewenda. 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15:34-39. [DOI] [PubMed] [Google Scholar]
- 76.Shugars, D. C., M. S. Smith, D. H. Glueck, P. V. Nantermet, F. Seillier-Moiseiwitsch, and R. Swanstrom. 1993. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 67:4639-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stoddart, C. A., R. Geleziunas, S. Ferrell, V. Linquist-Stepps, M. E. Moreno, C. Bare, W. Xu, W. Yonemoto, P. A. Bresnahan, J. M. McCune, and W. C. Greene. 2003. Human immunodeficiency virus type 1 Nef-mediated downregulation of CD4 correlates with Nef enhancement of viral pathogenesis. J. Virol. 77:2124-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stroschein-Stevenson, S. L., E. Foley, P. H. O'Farrell, and A. D. Johnson. 2006. Identification of Drosophila gene products required for phagocytosis of Candida albicans. PLoS Biol. 4:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swann, S. A., M. Williams, C. M. Story, K. R. Bobbitt, R. Fleis, and K. L. Collins. 2001. HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology 282:267-277. [DOI] [PubMed] [Google Scholar]
- 80.Whitney, J. A., M. Gomez, D. Sheff, T. E. Kreis, and I. Mellman. 1995. Cytoplasmic coat proteins involved in endosome function. Cell 83:703-713. [DOI] [PubMed] [Google Scholar]
- 81.Williams, M., J. F. Roeth, M. R. Kasper, T. M. Filzen, and K. L. Collins. 2005. Human immunodeficiency virus type 1 Nef domains required for disruption of major histocompatibility complex class I trafficking are also necessary for coprecipitation of Nef with HLA-A2. J. Virol. 79:632-636. [DOI] [PMC free article] [PubMed] [Google Scholar]