

Abstract
Mutations in MECP2, encoding methyl CpG binding protein 2 (MeCP2), cause most cases of Rett syndrome (RTT), an X-linked neurodevelopmental disorder. Both RTT and autism are “pervasive developmental disorders” and share a loss of social, cognitive and language skills and a gain in repetitive stereotyped behavior, following apparently normal perinatal development. Although MECP2 coding mutations are a rare cause of autism, MeCP2 expression defects were previously found in autism brain. To further study the role of MeCP2 in autism spectrum disorders (ASDs), we determined the frequency of MeCP2 expression defects in brain samples from autism and other ASDs. We also tested the hypotheses that MECP2 promoter mutations or aberrant promoter methylation correlate with reduced expression in cases of idiopathic autism. MeCP2 immunofluorescence in autism and other neurodevelopmental disorders was quantified by laser scanning cytometry and compared with control postmortem cerebral cortex samples on a large tissue microarray. A significant reduction in MeCP2 expression compared to age-matched controls was found in 11/14 autism (79%), 9/9 RTT (100%), 4/4 Angelman syndrome (100%), 3/4 Prader-Willi syndrome (75%), 3/5 Down syndrome (60%), and 2/2 attention deficit hyperactivity disorder (100%) frontal cortex samples. One autism female was heterozygous for a rare MECP2 promoter variant that correlated with reduced MeCP2 expression. A more frequent occurrence was significantly increased MECP2 promoter methylation in autism male frontal cortex compared to controls. Furthermore, percent promoter methylation of MECP2 significantly correlated with reduced MeCP2 protein expression. These results suggest that both genetic and epigenetic defects lead to reduced MeCP2 expression and may be important in the complex etiology of autism.
Keywords: autism, DNA methylation, Rett syndrome, neurodevelopmental disorders, MeCP2
INTRODUCTION
Rett syndrome (RTT) and autism are neurodevelopmental disorders within the category of “pervasive developmental disorders” (PDDs). RTT is an X-linked dominant disorder that causes mental retardation and occurs almost exclusively in females. RTT patients exhibit normal early development followed by a loss of social, cognitive, and language skills at around 6–18 months (1). Autism is more common in males and is characterized by deficits in three main areas: social interaction, language and communication, and restricted and repetitive behavior and interests (2–4). Autism and RTT overlap phenotypically and show a similar delayed onset after apparently normal prenatal and early postnatal development. Whereas the majority of RTT cases are caused by mutations in methyl CpG binding protein 2 (MECP2) (5), autism has an unknown but likely complex genetic basis involving multiple genes and environmental contributions.
Interestingly, the phenotypic spectrum of MECP2 coding mutations extends beyond RTT. MECP2 mutations have been reported in individuals with infantile autism, severe neonatal encephalopathy, motor abnormalities, respiratory dysfunction, mental retardation, bipolar disorder, schizophrenia, mild learning disabilities, milder late-onset versions of RTT, and normal phenotype (due to non-random X-chromosome inactivation favoring the wild-type allele) (6, 7). Heterozygous deletions encompassing most or all of MECP2 have been reported in some RTT cases (8–11). MECP2 duplications have been found in RTT syndrome and severe mental retardation, indicating that correct dosage of MeCP2 is critical for proper neurodevelopment (9, 10, 12–14).
Although a few instances of coding MECP2 mutations have been found in autism (15–17), they are not common (18, 19). DNA sequence variants in the non-coding regions of MECP2, such as the promoter, introns, and 3′-untranslated region (3′-UTR), might be present in autism and affect MeCP2 expression. In support of this hypothesis, a higher frequency of sequence variants within the MECP2 3′ UTR has been reported in autism versus controls (20). Furthermore, genetic mapping studies have found modest support for linkage and association for the genomic region containing MECP2 (Xq27–28) in autism (21, 22).
MeCP2 expression is spatially and developmentally regulated and is characterized by heterogeneous expression in subpopulations of neurons in the brain (23–25). The highest expression of MeCP2 is in mature postnatal neurons with some differences between brain regions. Two major MECP2 mRNA transcripts, which differ in 3′ UTR length, are detected in human brain by Northern blotting (26–28). Additionally, two protein isoforms of MeCP2 are generated by alternative splicing (29, 30). Recently, a major Mecp2 transcriptional start site 47 bp upstream of the previously annotated exon 1 was identified in mouse (31). Functional analysis of the mouse Mecp2 promoter identified a 733 bp promoter segment (−677 to +56) that conferred neuron-specific expression in embryonic and postnatal development. The complex developmental and tissue-specific expression of MeCP2 suggests that multiple pathways could alter MeCP2 expression in the developing brain.
We previously reported abnormal MeCP2 protein expression in autism spectrum disorder frontal cortex samples (32). In addition, different patterns of MeCP2 protein isoform expression and MECP2 3′ UTR usage were observed, suggesting that both transcriptional and post-transcriptional mechanisms might be involved in abnormal MeCP2 protein expression in autism spectrum disorders (ASDs). In this study, we have extended the analysis of MeCP2 expression in frontal cortex and fusiform gyrus to determine the frequency, specificity, and possible causes of reduced MeCP2 in autism. Reduced MeCP2 protein expression was frequently observed in autism spectrum disorder brain samples. Although genetic variants within a ~2 kb region upstream of MECP2 were rare, increased MECP2 promoter methylation was frequently found in male autism frontal cortex by bisulfite sequencing. This increased methylation was concentrated at two 5′ CpG sites and significantly correlated with reduced MeCP2 protein expression. These results suggest that an epigenetic defect of aberrant MECP2 promoter methylation may be important in the etiology of autism.
MATERIALS AND METHODS
Tissue samples and microarray construction
Frozen frontal cerebral cortex (Brodmann Area 9) and fusiform gyrus samples were obtained with assistance of the Autism Tissue Program from the University of Maryland Brain and Tissue Bank for Neurodevelopmental Disorders, the Harvard Brain Tissue Resource Center, the University of Miami Brain and Tissue Bank for Neurodevelopmental Disorders, and Drs. Paul Hagerman and Flora Tassone (UC Davis). Brain samples were chosen from human cadavers where death resulted from non-neurological causes and where the post mortem interval (PMI) was short. The cause of death, PMI, and other information is provided in Supplementary Table I. Frozen brain samples were fixed in 10% buffered formalin for 48 h, washed three times in 1X PBS, and embedded in paraffin. A 5 μm section was cut and stained with hematoxalin and eosin (H & E) and examined for tissue histochemistry. Regions containing cerebral cortical layers III–V were circled to guide sampling from the corresponding tissue block. Triplicate 600 μm diameter tissue cores were extracted from cerebral cortical layers III–V from each paraffin-embedded tissue block and inserted into a recipient paraffin block using a Beecher Instruments tissue microarrayer. Serial 5 μm sections were cut from the finished recipient block and every 10th section was stained with H & E to ensure completeness and sampling accuracy. The remaining unstained sections were baked overnight at 56° C. Methods for tissue section deparaffinization and epitope exposure were performed as described previously (23).
Immunofluorescence and Laser Scanning Cytometry (LSC)
A chicken anti-human MeCP2 C-terminus antibody (epitope: RPNREEPVDSRTPVTERVS) was custom made by Aves and used for detection of total MeCP2 protein (both protein isoforms). This antibody was determined to be specific by lack of immunoreactivity in a Mecp2 null mouse (data not shown) and in a human male with a truncating mutation (RTT 1238) (Table 1). Anti-MeCP2 and anti-histone H1 (Upstate Biotechnology) were diluted 1:10000 and 1:100, respectively, in IF stain buffer (PBS/1% FCS/0.5% Tween-20). Two hundred μl of this dilution were added to the tissue microarray slide and incubated for 2 h at 37° in a humidified chamber. As a negative control, a parallel IF was performed using an MeCP2-depleted chicken IgY fraction and a non-specific mouse IgG. Three 5 minute washes in PBS/0.5% Tween-20 preceded incubation with secondary antibodies. A Cy5-labeled donkey anti-chicken IgY (Jackson ImmunoResearch) and a Cascade Blue-labeled goat anti-mouse IgG (Molecular Probes) were each diluted 1:100 in IF stain buffer containing 10 μg/ml RNase and 7 μg/ml propidium iodide. Slides were incubated in 200μl of this secondary antibody solution for 1 hr at 37° in a humidified chamber, followed by three 5 minute washes in PBS/0.5% Tween-20. Slides were then mounted in PBS/50% glycerol with 7 μg/ml propidium iodide, coverslipped, and sealed with nail polish. Laser scanning cytometry was performed to quantify protein expression (as described in (23)). Briefly, slides were scanned with a laser scanning cytometer (CompuCyte, Cambridge, MA) with a 20X microscope objective using argon, violet, and HeNe lasers. Fluorescence data was collected using the software program Wincyte (CompuCyte, Cambridge, MA) in the blue, red, and long red channels as max pixel values. Voltage, PMT, and threshold settings were identical for experimental and control slides. Cell nuclei were contoured in the red channel with propidium iodide fluorescence. Large cell clusters were removed by gating single cells on the area vs. red max pixel scattergram. MeCP2 immunofluorescence was normalized to histone H1 immunofluorescence by dividing the former value by the latter for each core analyzed. MeCP2 percent high cells (MeCP2hi) were determined by setting a colored gate at the right half-max of a histogram of MeCP2 fluorescence of all adult control samples.
Table 1.
MeCP2 protein expression in tissue microarray brain samples Prefrontal cortex (BA9) samples
| Prefrontal cortex (BA9) samples | ||||||
|---|---|---|---|---|---|---|
| phenotype | sample ID | age | sex | normalized MeCP2a,b | % MeCP2hi | known genetic variants |
| control | 125 | 76 days | M | 0.52 ± 0.08 | 6.0 ± 1.6 | |
| control | 1055 | 96 days | M | 0.72 ± 0.09 | 29.4 ± 4.1 | |
| control | 1275 | 2 y | F | 1.11 ± 0.15 | 48.2 ± 2.9 | |
| control | 229 | 4 y | M | 0.90 ± 0.12 | 59.5 ± 4.0 | |
| control | 1377 | 5 y | F | 0.96 ± 0.13 | 57.3 ± 5.3 | |
| control | 3835 | 9 y | F | 1.40 ± 0.21 | 46.6 ± 4.2 | |
| control | 662 | 12 y | F | 1.02 ± 0.14 | 43.7 ± 2.5 | |
| control | 1065 | 15 y | M | 1.32 ± 0.18 | 45.2 ± 5.8 | |
| control | 812 | 18 y | F | 1.51 ± 0.18 | 58.2 ± 5.0 | |
| control | 1027 | 22 y | M | 1.126 ± 0.13 | 45.4 ± 2.4 | |
| control | 602 | 27 y | M | 0.92 ± 0.15 | 30.1 8.3 | |
| control | 1029 | 29 y | M | 1.11 ± 0.15 | 45.3 ± 1.9 | |
| control | 1136 | 33 y | F | 1.32 ± 0.15 | 52.0 ± 5.7 | |
| control | 1104 | 35 y | M | 1.13 ± 0.14 | 51.0 ± 5.2 | |
| control | 1406 | 38 y | F | 1.18 ± 0.09 | 56.8 ± 1.8 | |
| control | 1135 | 42 y | M | 1.23 ± 0.12 | 55.1 ± 2.4 | |
| control | B4192 | 46 y | M | 0.86 ± 0.11 | 35.7 ± 4.9 | |
| control | B4503 | 56 y | M | 1.15 ± 0.19 | 53.3 ± 3.0 | |
| control | 1206 | 57 y | M | 0.64 ± 0.04 | 22.6 ± 4.0 | |
| control mean (n=19)c | 1.06 ± 0.06 | 44.3 ± 3.2 | ||||
|
| ||||||
| RTT | 1238 | 1 y | M | 0.40 ± 0.04 ** | 0.5 ± 0.4 *** | MECP2 c.1154_1185del32 |
| RTT | B4687 | 8 y | F | 0.68 ± 0.09 * | 19.4 ± 2.2 *** | MECP2 p.R255X |
| RTT | B5214 | 10 y | F | 0.69 ± 0.08 * | 8.6 ± 2.1 *** | MECP2 p.R270X |
| RTT | 1815 | 18 y | F | 0.57 ± 0.07 *** | 17.1 ± 1.5 *** | MECP2 c.378-2 A>G |
| RTT | 1420 | 21 y | F | 0.67 ± 0.06 *** | 50.3 ± 2.2 | |
| RTT | 1748 | 22 y | F | 0.38 ± 0.03 *** | 6.2 ± 1.5 *** | |
| RTT | 3381 | 22 y | F | 0.83 ± 0.12 * | 13.1 ± 4.2 *** | |
| RTT | 4321 | 23 y | F | 0.50 ± 0.11 ** | 10.4 ± 3.4 *** | |
| RTT | 4312 | 24 y | F | 0.72 ± 0.11 * | 17.9 ± 4.1 ** | MECP2 p.R168X |
| RTT | B5020 | 24 y | F | 0.60 ± 0.08 ** | 15.7 ± 3.2 *** | MECP2 p.R255X |
| RTT mean (n=10) | 0.60 ± 0.05 *** | 15.9 ± 4.2 *** | ||||
|
| ||||||
| Angelman | 1824 | 4 y | M | 0.50 ± 0.03 *** | 11.6 ± 2.7 *** | 15q11-13 del |
| Angelman | 1754 | 4 y | M | 0.54 ± 0.04 *** | 23.6 ± 5.3 ** | 15q11-13 del |
| Angelman | 293 | 20 y | F | 0.61 ± 0.08 *** | 17.9 ± 3.6 *** | 15q11-13 del |
| Angelman | 1494 | 43 y | F | 0.35 ± 0.02 *** | 4.6 ± 2.2 *** | 15q11-13 del |
| Angelman mean (n=4) | 0.50 ± 0.06 *** | 14.4 ± 4.1 ** | ||||
|
| ||||||
| Prader-Willi | 865 | 43 y | F | 0.65 ± 0.08 ** | 23.5 ± 3.1 *** | 15q11-13 del |
| Prader-Willi | 1290 | 44 y | F | 0.62 ± 0.07 *** | 19.1 ± 3.4 *** | 15q11-13 UPD |
| Prader-Willi | 1447 | 45 y | M | 0.66 ± 0.12 * | 26.8 ± 7.5 * | 15q11-13 UPD |
| Prader-Willi | 1510 | 56 y | M | 0.75 ± 0.08 | 43.3 ± 3.1 | 15q11-13 del |
| Prader-Willi mean (n=4) | 0.67 ± 0.03 *** | 28.2 ± 5.3 * | ||||
|
| ||||||
| autism | 3871 | 5 y | M | 0.80 ± 0.12 | 13.0 ± 2.4 *** | |
| autism | 1174 | 7 y | F | 0.65 ± 0.09 * | 15.7 ± 3.9 *** | |
| autism | B4925 | 9 y | M | 0.71 ± 0.10 * | 30.6 ± 4.0 ** | |
| autism | 797 | 9 y | M | 0.73 ± 0.14 * | 25.7 ± 4.5 * | |
| autism | 1182 | 9 y | F | 0.35 ± 0.04 *** | 3.7 ± 0.0 *** | |
| autism | B5342 | 11 y | F | 0.46 ± 0.09 ** | 17.7 ± 2.9 ** | MECP2 g.-1398 T>C |
| autism (suspected) | 732 | 15 y | M | 0.98 ± 0.13 | 31.8 ± 1.8 *** | |
| autism | 3924 | 16 y | F | 1.15 ± 0.19 | 52.1 ± 2.6 | |
| autism | 1638 | 20 y | F | 0.77 ± 0.12 * | 43.6 ± 2.5 | MECP2 c.1035 A>G |
| autism | B5144 | 20 y | M | 0.71 ± 0.10 * | 34.6 ± 2.6 * | |
| autism | B5000 | 27 y | M | 0.69 ± 0.12 * | 11.3 ± 2.2 *** | |
| autism | B5173 | 30 y | M | 0.59 ± 0.06 *** | 46.4 ± 4.4 | |
| autism (suspected) | 967 | 32 y | M | 0.67 ± 0.08 ** | 37.9 ± 2.1 * | |
| autism | B4498 | 56 y | M | 0.32 ± 0.03 *** | 2.8 ± 0.8 *** | |
| autism mean (n=14) | 0.68 ± 0.06 ** | 26.2 ± 4.2 * | ||||
|
| ||||||
| Down syndrome | 1267 | 10 y | M | 0.88 ± 0.12 | 51.0 ± 3.5 | Trisomy 21 |
| Down syndrome | 1276 | 13 y | M | 0.87 ± 0.15 | 34.4 ± 4.9 | Trisomy 21 |
| Down syndrome | 707 | 22 y | M | 0.74 ± 0.12 * | 23.3 ± 4.5 * | Trisomy 21 |
| Down syndrome | 753 | 23 y | M | 0.73 ± 0.12 * | 48.6 ± 2.5 | Trisomy 21 |
| Down syndrome | 1258 | 44 y | F | 0.63 ± 0.08 ** | 11.1 ± 2.3 *** | Trisomy 21 |
| Down syndrome mean (n=5) | 0.77 ± 0.05 * | 33.7 ± 7.6 | ||||
|
| ||||||
| ADHD | 625 | 6 y | F | 0.49 ± 0.05 *** | 10.5 ± 1.1 *** | |
| ADHD | 1077 | 11 y | M | 0.40 ± 0.04 *** | 1.3 ± 0.4 *** | |
|
| ||||||
| Fragile X | none | 25 y | M | 0.90 ± 0.16 | 35.2 ± 4.3 | FMR1 mutation |
| Fusiform gyrus samples | ||||||
|
| ||||||
| control | 3835 | 9 y | F | 0.70 ± 0.08 | 35.4 ± 1.8 | |
| control | 1065 | 15 y | M | 0.93 ± 0.12 | 50.9 ± 2.1 | |
| control | 1027 | 22 y | M | 1.12 ± 0.15 | 43.1 ± 4.1 | |
| control | 602 | 27 y | M | 1.30 ± 0.16 | 62.3 ± 1.7 | |
| control | 1029 | 29 y | M | 1.20 ± 0.20 | 58.9 ± 3.2 | |
| control | 1104 | 35 y | M | 1.14 ± 0.16 | 57.9 ± 1.8 | |
| control | B4192 | 46 y | M | 0.67 ± 0.07 | 26.3 ± 3.8 | |
| control | B4503 | 56 y | M | 0.95 ± 0.09 | 47.4 ± 3.4 | |
| control mean (n=8) | 1.00 ± 0.08 | 47.8 ± 4.4 | ||||
|
| ||||||
| autism | B4925 | 9 y | M | 0.63 ± 0.08 | 31.6 ± 4.4 * | |
| autism | B5342 | 11 y | F | 0.42 ± 0.05 ** | 24.1 ± 4.8 * | MECP2 g.-1398 T>C |
| autism | B5144 | 20 y | M | 0.70 ± 0.06 ** | 39.7 ± 2.9 * | |
| autism | B5000 | 27 y | M | 0.56 ± 0.07 *** | 10.3 ± 3.2 *** | |
| autism | B5173 | 30 y | M | 0.44 ± 0.04 *** | 18.4 ± 1.2 *** | |
| autism | B4498 | 56 y | M | 0.27 ± 0.03 *** | 2.1 ± 5.6 *** | |
| autism mean (n=6) | 0.50 ± 0.07** | 21.0 * | ||||
MeCP2 protein quantification is shown as the average max pixel fluorescence of three experimental replicates (3 cores per sample per replicate) normalized to histone H1 max pixel fluorescence. Mean ± SEM is shown.
Asterisks indicate significantly lower compared to three closest age-matched controls, except in the case of group means, where asterisks indicate significantly lower expression in a disorder group compared to the control group.
P < 0.05;
P < 0.001;
P < 0.0001
Only age-matched controls were used
Immunoblotting
Protein extracts were isolated from frozen brain samples using TRIzol (Invitrogen) according to manufacturer’s protocol. Ten mg of protein were loaded on a 4–15% Tris–HCl polyacrylamide gel (Biorad) and transferred to a nitrocellulose membrane. After a 5 minute wash in 1X phosphate-buffered saline (PBS), membranes were blocked with 5% non-fat dry milk (Carnation) in 1X PBS/0.05% Tween. Primary antibody incubation was performed overnight at 4°C with a rabbit anti-C-terminal MeCP2 (Affinity) (1:2000 in 1X PBS) or mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Advanced Immunochemical) (1:600 in 1X PBS). Following four 5 minute washes in 1X PBS, secondary antibody incubation was performed for 1 hr at room temperature with 1:2500 of goat anti-rabbit horseradish peroxidase (HRP) (Bio-Rad) or 1:10000 goat anti-mouse HRP (Bio-Rad). After four 5 minute washes in 1X PBS, detection was performed with Supersignal West Pico chemiluminescent substrate (Pierce). Quantification was conducted with Nucleotech Gel Expert Version 2.0.
PCR and Sequencing
Frontal cortex (BA9) genomic DNA was isolated from frozen postmortem human samples using the PureGene Kit (Gentra). PCR primers were designed to amplify all four exons of MECP2, including intron-exon boundaries. All neurodevelopmental disorder samples on the tissue microarray were sequenced to check for MECP2 coding mutations (39 samples). Another group of overlapping PCR primers was designed to amplify the putative promoter and upstream regulatory sequences of MECP2 as well as a short fragment of intron I. PCR primer sequences are shown in Supplementary Table II. A 1.9 kb portion of the MECP2 promoter region was sequenced including 1630 bp upstream of the transcription start site, exon 1, and 254 bp of intron 1. A total of 51.3 kb was analyzed (1.9 kb/27 individuals). For primers amplifying MECP2 exons 2-4, Invitrogen Taq polymerase (2.5 units) was used in a 50 μl reaction containing 1.5 mM MgCl2, 20 mM tris-HCl (pH 8.4), 50 mM KCl, 0.4 mM each dNTP, 0.4 μM each primer, and approximately 10–50 ng genomic DNA. For primers amplifying the CG-rich MECP2 exon 1, promoter, and upstream sequences, 2.5 units of TaKaRa LA Taq were used in a hot start 50 μl reaction with 1X GC buffer 1, 2.5 mM MgCl2 0.4 mM each dNTP, 0.4 μM each primer, and approximately 10–50 ng genomic DNA. PCR products were purified with the Qiaquick Gel Purification kit (Qiagen). DNA sequencing was performed by the UC Davis Division of Biological Sciences Sequencing facility. Sequences were compared to the human genomic clone AF030876.
Genotyping assay for g.-1398T>C variant
DNA from autism and control brain samples was used for genotyping. In addition, 65 autism DNA samples were obtained from the Autism Genetic Resource Exchange (AGRE). These samples were from both males and females diagnosed with idiopathic autism and were all unrelated. A total of 79 autism individuals and 20 controls were genotyped for the g.-1398T>C variant. PCR primers were designed to flank the variant site (Supplementary Table II), producing a 175 bp amplicon. These primers (0.4 μM each) were used in a 50 μl PCR reaction containing 2.5 units Invitrogen Taq, 1.5 mM MgCl2, 20 mM tris-HCl (pH 8.4), 50 mM KCl, 0.4 mM each dNTP, and approximately 10–50 ng genomic DNA. The annealing temperature was 66.8° C. The C allele creates an HpyCH4III restriction enzyme site. The PCR products were purified (Qiaquick Gel Purification kit, Qiagen), and 2 μl of each was digested with 5 units of HpyCH4III (New England Biolabs) in a 25 μl reaction containing 1X NEB buffer 4 at 37° C for 3 hours. Digested PCR products were electrophoresed on a 2% agarose gel for detection.
Single Strand Conformation Polymorphism (SSCP) analysis
The 175 bp PCR product used for HpyCH4III genotyping was used for SSCP with the Genegel/Excel 12.5/24 kit (GE Healthcare/Amersham) following the manufacturer’s instructions. Briefly, 5 μl of purified PCR product was mixed with 5 μl denaturing solution and incubated at 95° for 5 minutes, then placed immediately on ice. Six μl of this mixture was loaded onto a precast polyacrylamide gel pre-cooled to 10° C and run at 25 mA/600 V on a GenePhor electrophoresis unit (GE Healthcare/Amersham). Gels were visualized with silver staining. Seventy-nine autism samples and 20 controls totaling 17.3 kb (175 bp X 99 individuals) were assayed by SSCP.
Bisulfite Sequencing
Frontal cortex (BA9) genomic DNA was isolated from frozen postmortem human samples using the PureGene Kit (Gentra). Genomic DNA was sheared with a syringe needle, extracted with phenol/chloroform/isoamyl alcohol, and precipitated with sodium acetate/ethanol and resuspended in sterile water. Approximately 1 μg of frontal cortex genomic DNA was used for bisulfite conversion using the CpGenome kit (Chemicon) following the manufacturer’s instructions. Briefly, DNA samples were treated overnight at 55° with sodium bisulfite, desalted, desulfonated, and eluted in 20 μl TE. One μl of converted DNA was used for bisulfite PCR. Bisulfite primers were designed using the online tool Methprimer (http://www.urogene.org/methprimer/index.html) (55). The forward (GTTAGGTTTTAGGGTGGGTAATTTT) and reverse (CCCCTCCAACTATTAATTAACTACTTTC) primers (0.4 μM each) were used in a 50 μl volume PCR reaction with 2.5 units Roche FastStart High Fidelity Taq, 1X Roche FastStart High Fidelity Reaction Buffer (with 1.8 mM MgCl2,) 0.2 mM of each dNTP, and 1X DMSO. The cycling conditions were 94° for 1 min., 56.8° for 30 sec., 72° for 1 min, for a total of 40 cycles. PCR products were gel purified with the Qiaquick Gel Purification kit (Qiagen) and ligated overnight at 4° C with the pGEM-T-Easy kit (Promega). Two to five μl of the ligation were used to transform JM109 competent cells (Promega) and bacteria were plated on LB Ampicillin with X-gal and IPTG. White colonies were picked and grown up in overnight LB Amp cultures. Plasmids were purified with the Qiaprep kit (Qiagen) and tested for presence of the correct insert with a Not I digest (New England Biolabs). Positive plasmids were sequenced using the T7 promoter sequencing primer by the UC Davis Division of Biological Sciences Sequencing Facility. A minimum of ten colonies were sequenced for each brain sample.
RESULTS
MeCP2 expression is significantly reduced in a high frequency of autism and autism spectrum disorder brain samples
A tissue microarray was constructed using human postmortem brain tissue from ASDs and controls (Figure 1A). Tissue microarrays allow high-throughput analysis of multiple tissue samples on a single microscope slide (23). We aimed to determine 1) the frequency and specificity of MeCP2 expression defects in autism and related disorders and 2) if MeCP2 expression defects are limited to a single brain region in autism. Frontal cortex Brodmann area 9 (BA9) samples were included because of the importance of frontal cortex in autism and the previous finding of high MeCP2 expression in this region (23). Fusiform gyrus samples were included because of the importance of this brain region in autism, particularly with regards to defects in face processing (33). To compare idiopathic autism cases with those of known neurodevelopmental disorders with partial autism comorbidity, brain samples from Angelman syndrome (AS), Prader-Willi syndrome (PWS), Down syndrome, attention deficit hyperactivity disorder (ADHD), and Fragile X syndrome were included. Brain sample characteristics are provided in Table 1 and Supplementary Table I.
Figure 1.
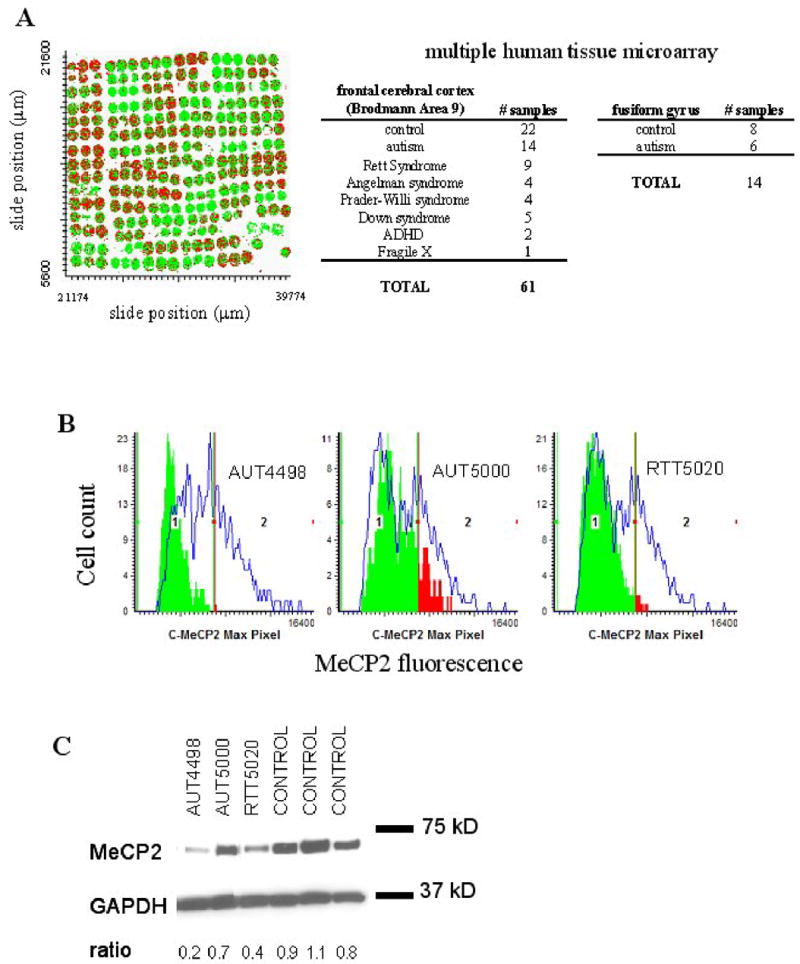
Decreased MeCP2 protein expression in autism samples detected by IF/LSC and immunoblot. A. X,Y scattergram of multiple tissue microarray used for IF/LSC. Each colored circle is a 600 μm core of human cerebral cortex postmortem tissue, containing approximately 200–500 cell nuclei. Cell populations with low and high MeCP2 expression are differentiated by dividing the adult control sample MeCP2 histogram at the right half-max. All cells to the left of the gate are colored green (MeCP2lo), and all cells to the right of the gate are colored red (MeCP2hi) B. IF/LSC analysis of MeCP2 protein expression in three representative cerebral cortex samples. MeCP2 expression for autism and RTT samples are shown as green and red colored solid histograms. RTT 5020 and AUT 4498 show decreased MeCP2 protein expression compared to three closest age-matched controls (blue line overlay). AUT 5000 shows less significantly decreased MeCP2 protein expression compared to controls. C. Immunoblot analysis shows similar results as IF/LSC analysis in B. Anti-GAPDH was used as a loading control and MeCP2 expression is normalized to GAPDH and shown as a ratio below the blot image.
Immunofluorescence (IF) was performed on tissue microarray slides with an anti-MeCP2 C-terminal antibody (which detects both isoforms of MeCP2) normalized to histone H1. Histone H1 was chosen because it is an abundant nuclear protein with genome-wide distribution, and does not exhibit major changes in expression during development (24). MeCP2 and histone H1 nuclear fluorescence intensities were quantified by laser scanning cytometry (LSC) for individual nuclei. Figure 1B shows representative histograms of LSC quantification of MeCP2 expression in three individually gated frontal cortex samples (RTT B5020, AUT B4498, AUT B5000) (filled histograms), each compared to the three closest age-matched controls (open histograms). Immunoblotting analysis of autism frontal cortex samples confirmed the validity of this approach (Figure 1C). The mean normalized MeCP2 fluorescence (representing 200–500 cells) was calculated for each tissue core. Triplicate cores of each neurodevelopmental disorder sample were analyzed for each experimental replicate, and three experimental replicates were performed (n=9). Normalized MeCP2 immunofluorescence for each neurodevelopmental disorder sample were compared to that of the three closest age-matched controls (n=27), and statistical significance was determined by t-test. Table 1 shows that 11/14 (79%) of autism frontal cortex samples showed significantly deficient MeCP2 protein expression (significantly lower than age-matched controls), exhibiting up to a two-fold reduction. The percentage of cells expressing high levels of MeCP2 (MeCP2hi) was significantly lower than age-matched controls in 11/14 (79%) of autism samples.
Six autism brains were represented on the tissue microarray with both frontal cortex and fusiform gyrus samples. Five out of six autism fusiform gyrus samples displayed significantly decreased MeCP2, and these samples also showed significantly decreased MeCP2 in frontal cortex (AUT B5342, AUT B5144, AUT B5000, AUT B5173, and AUT B4498). AUT B4925, which was significantly decreased in frontal cortex, was lower in fusiform gyrus but did not reach statistical significance (Table 1). As a group, autism samples had significantly lower MeCP2 expression than controls in both frontal cortex (P=0.0001) and fusiform gyrus (P=0.0004). The general concordance of frontal cortex and fusiform gyrus data suggests that the decrease in MeCP2 protein is not limited to a single brain region but is present in at least two brain regions with suspected functional relevance to autism.
All RTT samples, including those without MECP2 coding mutations, showed significantly decreased MeCP2. Other neurodevelopmental disorders showed varying frequencies of MeCP2 expression abnormalities. Four out of four Angelman syndrome (AS), 3/4 Prader-Willi syndrome (PWS), 3/5 Down syndrome, and 2/2 attention deficit hyperactivity disorder (ADHD) frontal cortex samples showed significantly reduced MeCP2. A single fragile X frontal cortex sample showed normal MeCP2. These results suggest that most, but not all, autism spectrum disorders share a common pathway of reduced MeCP2 expression.
A rare MECP2 promoter variant correlates with decreased MeCP2 expression in an autistic female
We hypothesized that mutations in DNA regulatory elements of MECP2, such as the promoter, might reduce MeCP2 expression in autism cases without a known genetic cause. First, we sequenced all four MECP2 exons (including exon-intron boundaries) to determine if coding mutations might be present in these ASD cases. We confirmed coding MECP2 mutations in several previously reported RTT cases (RTT 1238, RTT B4687, RTT B5214, RTT 4312, and RTT B5020) (Table 1). RTT 1815 was determined to harbor a rare RTT-causing splice site mutation (c.378-2 A>G) (Figure 2). For four RTT samples (RTT 1420, RTT 1748, RTT 3381, and RTT 4321) no coding mutation could be detected. One female autism sample, AUT 1638, was heterozygous for a silent polymorphism in exon 4 (c.1035 A>G; p.K345K) (Figure 2). No other MECP2 coding variants were found within the remaining neurodevelopmental disorder samples.
Figure 2.
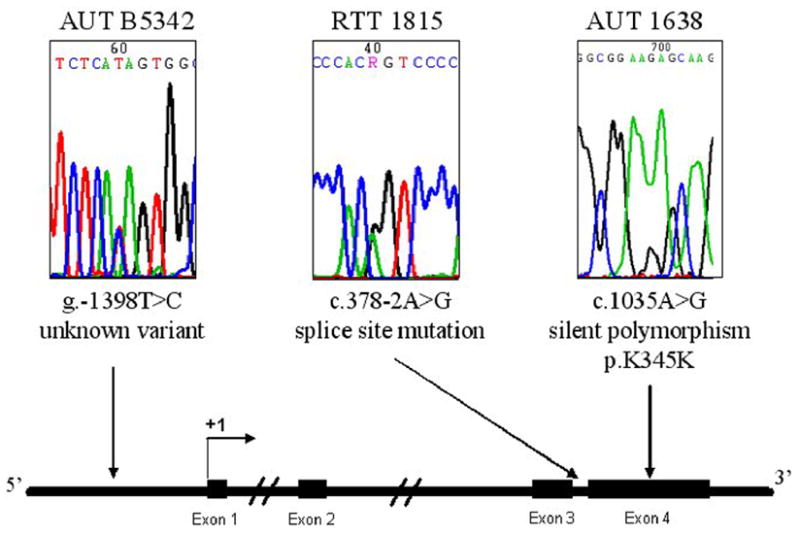
Sequencing of MECP2 coding and promoter sequences in brain genomic DNA. The 4 MECP2 coding exons, including intronexon boundaries, were sequenced in all neurodevelopmental disorder samples on the tissue microarray. For the MECP2 promoter, a 1.9 kb portion of the MECP2 promoter region was sequenced including 1630 bp upstream of the transcription start site, exon 1, and 254 bp of intron 1. For promoter sequencing, a total of 51.3 kb was analyzed (1.9 kb/27 individuals). Sequencing chromatograms for three brain samples with sequence variations (AUT B5342, RTT 1815, and AUT 1638) are shown above a schematic diagram of the MECP2 genomic region. Exons are indicated with filled rectangles and the transcription start site is shown as +1. Arrows underneath each sequencing chromatogram point to the approximate location of the sequence variant.
Next, we sequenced the MECP2 promoter region in non-MECP2 mutant RTT, autism, and other neurodevelopmental disorder samples that had been included on the tissue microarray (Supplementary Table I). All sequences were compared to the MECP2 genomic clone AF030876 reference sequence and the sequence positions reported here were determined using this reference sequence and the transcription start site defined by Adachi et al. (31) as +1. None of the samples analyzed carried the g.-165_166insC insertion variant present in AF030876 (previously reported as g.-219_220insC). This is in agreement with previous work showing that this variant appears to be rare in both RTT and controls (34). A female autism sample (AUT B5342, 11 y) was determined to be heterozygous for a novel promoter variant (g.-1398T>C) (Figure 2). While this variant occurs upstream of the defined human promoter (36), it disrupts a putative Pax4 transcription factor binding site. Furthermore, the wild-type nucleotide (T) is conserved in chimp, rhesus, rabbit, and dog (UCSC Genome Browser).
We hypothesized that this variant might be present in a subset of individuals with autism and might contribute to risk for autism by affecting transcription factor binding and reducing MeCP2 expression. DNA from family members of AUT B5342 was unavailable, so we were unable to determine if this mutation segregates with any ASD phenotype. Alternatively, 79 AGRE and brain autism DNA samples and 20 controls (both males and females) were tested for the g.-1398T>C variant and found negative. To test for allelic heterogeneity, the same samples were tested with single strand conformational polymorphism (SSCP) using PCR primers that spanned 175 bp (94 bp upstream and 80 bp downstream of -1398), but no additional sequence variants were observed, suggesting that MECP2 promoter mutations in this region (−1492 to −1318) are rare in autism.
Increased MECP2 promoter methylation is common in male autism frontal cortex and correlates with reduced MeCP2 expression
Since a common genetic cause of MeCP2 expression deficiency in the 5′ regulatory region could not be found, it was hypothesized that epigenetic events, such as aberrant promoter methylation, might be involved. Bisulfite sequencing was performed on male autism, Down syndrome, and control frontal cortex genomic DNA to determine the pattern of DNA methylation at the MECP2 promoter, which largely overlaps with a CpG island. Since MECP2 is an X-linked gene, only males were analyzed because any promoter methylation observed would be aberrant, as the male X chromosome is actively expressed and unmethylated. Initially, 658 bp (−531 to +127) of the MECP2 regulatory region was analyzed, and it was found that the most 5′ portion showed increased DNA methylation in some autism samples (data not shown). Subsequently, a 289 bp region (−531 to −243) containing 15 CpG sites was analyzed for patterns of DNA methylation by bisulfite sequencing (Figure 3A). Several autism males showed some specific increases in methylation of the MECP2 promoter compared to control males. Representative bisulfite sequencing results are shown in Figure 3B, with all results shown in Supplementary Figure 1. A total of nine autism and nine control frontal cortex male samples were tested for DNA methylation by bisulfite sequencing of at least 10 clones per sample. Percent MECP2 promoter methylation was calculated as the percent methylated CpGs out of the total CpGs assayed for each brain sample. Autism males showed a significant (P=0.0269, t-test) increase in overall MECP2 promoter methylation compared to male controls (Figure 3C). In addition, percent methylation of each CpG site within the sequenced region was compared. Sites #1 (−496) and #3 (−445) showed the highest percent methylation in autism samples, but site #3 was the only site to show a significant increase in autism compared to controls (P=0.0031, t-test) (Figure 3E). This site did not show increased methylation in four male Down syndrome samples, suggesting specificity to autism. Most importantly, normalized MeCP2 protein expression (from Table 1) showed a significant inverse correlation with percent MECP2 promoter methylation (Figure 3D), suggesting that percent MECP2 promoter methylation is a relevant quantitative epigenotype. These results demonstrate that increased MECP2 promoter methylation correlates with reduced MeCP2 expression and is a frequent characteristic of autism male brains.
Figure 3.
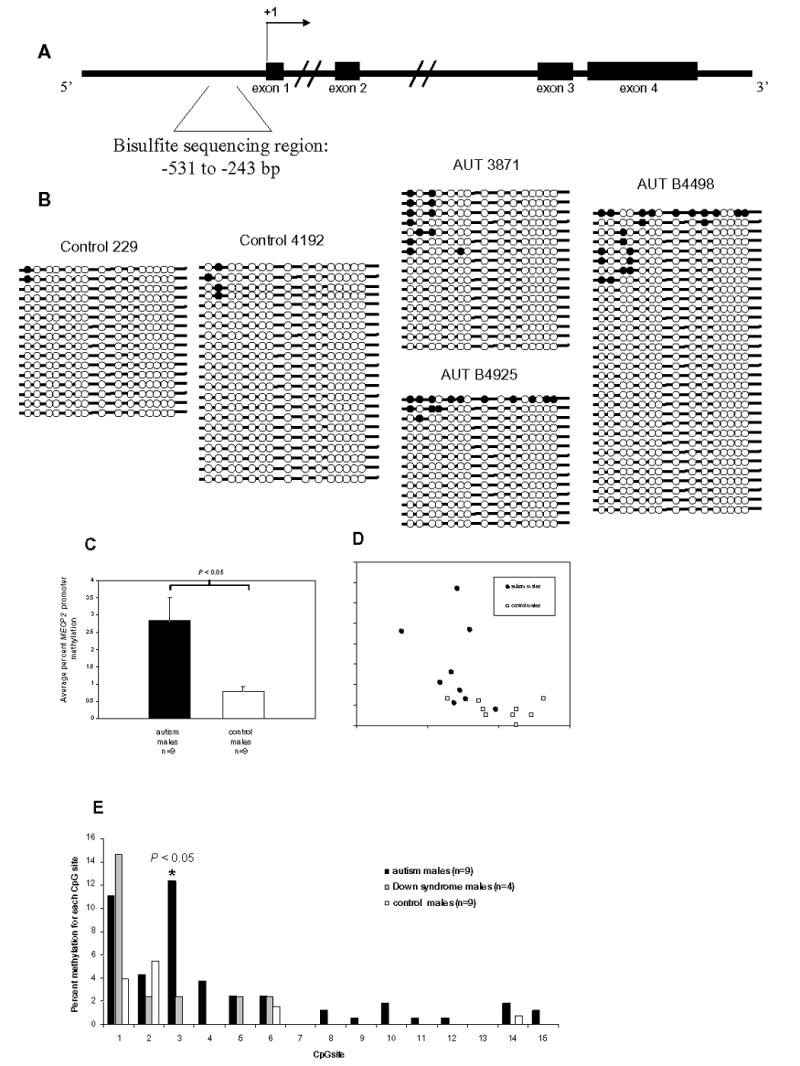
Bisulfite sequencing analysis of MECP2 promoter methylation in frontal cortex DNA from autism and control postmortem brain samples. A. Schematic diagram of the MECP2 genomic region showing exons (filled rectangles), the transcription start site (+1), and the bisulfite sequencing region (bracketed sequence). B. Representative bisulfite sequencing data from two control males (229 and 4192) and three autism males (AUT 3871, AUT B4925, and AUT B4498). Each line with circles represents an individual clone. Filled circles indicate methylated CpG sites, and open circles indicate unmethylated CpG sites. A minimum of 10 clones were analyzed for each brain sample. All bisulfite data are shown in Supplementary Figure I. C. Percent MECP2 methylation for autism (n=9), and control (n=9) males was calculated by dividing the number of methylated CpG sites by the total number of CpG sites assayed for each brain sample. Results are graphed as mean ± SEM, and significance was determined by t-test. A statistically significant difference between autism and controls was also observed by analyzing the percentage of clones with one or more methylated sites out of the total number of clones for each sample (data not shown) D. Scatterplot showing percent MECP2 methylation and normalized MeCP2 expression for each brain sample. Autism samples are shown as filled circles and controls are shown as open squares. Regression was calculated for autism and controls together (R2=0.3235, P<0.05), as well as for autism (R2=0.1334) and control(R2=0.1065) samples alone. E. The percent methylation for each CpG site was compared between autism (n=9), Down syndrome (n=4), and control (n=9) males. CpG sites are shown on the x axis as in B. CpG site #3 showed a significant increase (P=0.0031, t-test) in autism vs. controls.
Since MECP2 methylation was not observed in every clone, the possibility that increased MECP2 methylation occurred in a subset of cells that normally express high levels of MeCP2 (MeCP2hi) was examined. A modest degree of methylation in MeCP2hi cells could have a large impact on overall MeCP2 protein levels in the brain. To examine this possibility, the difference in percent MeCP2hi cells in each autism sample from the mean percent MeCP2hi of the three closest age-matched controls was calculated from the LSC data in Table 1. Table 2 shows the change in the percent MeCP2hi population compared to the percentage of cells with one or more methylated CpG site. Since autism male samples are hemizygous for MECP2, each clone analyzed for bisulfite sequencing represents a single cell. Table 2 shows a striking correspondence between the percentage of cells obtained for LSC and bisulfite analyses for autism males in the juvenile age group (R2 = 0.936, P = 0.032, n = 4), suggesting a connection between MECP2 methylation and a decrease in MeCP2hi cells specific to young samples.
Table 2.
Percent MeCP2hi cells compared to percent methylated MECP2 cells in autism males
| age group | sample ID | age | change in % MeCP2hi from 3 closest age- matched controlsa | % methylated cellsb |
|---|---|---|---|---|
| juvenilec | 3871 | 5 y | 42.0 | 41.0 |
| 4925 | 9 y | 18.6 | 21.0 | |
| 797 | 9 y | 23.6 | 25.0 | |
| 732 | 15 y | 17.3 | 13.0 | |
|
| ||||
| adult | B5144 | 20 y | 6.0 | 16.0 |
| B5000 | 27 y | 31.2 | 8.0 | |
| B5173 | 30 y | −4.0 | 19.0 | |
| 967 | 32 y | 11.5 | 22.0 | |
| B4498 | 56 y | 34.0 | 25.0 | |
Percent MeCP2hi cells (taken from Table 1) were calculated by gating the control adult MeCP2 histogram at the right half max, and designating all cells to the right of the gate as MeCP2 hi (23)
Percent methylated cells were calculated by counting the number of bisulfite clones with one or more methylated CpGs and dividing by the total number of clones for that sample. Since the X-linked MECP2 is hemizygous in males, each clone represents the single allele from a single cell
Regression analysis shows that for juvenile autism samples, R2 = 0.9361, P = 0.032 (n=4)
DISCUSSION
Although autism is a highly heritable disorder, the genetic etiology of autism remains elusive. In contrast, Rett syndrome is a phenotypically overlapping PDD primarily caused by mutations in MECP2. The progress made in studying the function of MeCP2 and its role in Rett syndrome provides an opportunity to apply some of this knowledge to other related neurodevelopmental disorders. We previously found defective MeCP2 expression in 4/4 autism frontal cortex samples (32). Here we expanded this analysis to frontal cortex (BA9) samples from 14 autism patients; six of these patients were represented with both frontal cortex and fusiform gyrus brain samples. Additionally, a greater number of Rett, PWS, and AS samples were included. Down syndrome, ADHD, and Fragile X samples were also included because of some degree of phenotypic overlap or comorbidity between these disorders and autism. Using a tissue microarray containing all of these samples as well as controls, we found that 79% of the autism frontal cortex samples showed significant decreases in MeCP2 total protein expression compared to age-matched controls. In general, there appeared to be good concordance in MeCP2 reduction in autism between the frontal cortex and fusiform gyrus, suggesting that MeCP2 expression defects in autism are not limited to a single brain region.
All Rett syndrome samples showed significantly reduced MeCP2. Since these experiments were performed using a C-terminal MeCP2 antibody that detects both MeCP2 protein isoforms, it is expected that RTT individuals with nonsense MECP2 mutations would show lower expression. In addition, RTT females are heterozygous and mosaic for MECP2 mutations and non-cell autonomous mechanisms result in reduced MeCP2 expression in wild-type expressing cells (35). Reduced expression was also observed for RTT samples without MECP2 coding mutations. This might be because these samples are heterozygous for MECP2 deletions. Alternatively, these samples might harbor non-coding MECP2 mutations that affect MeCP2 expression. Promoter mutations in the MECP2 −1630 to +254 genomic region were not found in these samples, however new cis-regulatory regions have recently been described (36) and could be tested. The reduced MeCP2 expression in RTT samples without MECP2 mutations suggests that altered regulation of a pathway involving MeCP2 is common to all Rett cases.
PWS, AS, ADHD and Down syndrome samples also had high frequencies of MeCP2 reduction, whereas a single Fragile X brain did not. MeCP2 expression defects appeared to increase with age in the Down syndrome samples, suggesting that early changes in brain development caused by trisomy for chromosome 21 might affect MeCP2 expression in adulthood, since high MeCP2 expression is a marker of mature neurons (24, 25). The significantly lower MeCP2 expression in ADHD samples was surprising, but this result may be brain region-specific, and the small sample number limits further interpretation. However, given the phenotypic overlap between ADHD and autism, these results warrant further investigation with additional ADHD samples and different brain regions. Taken together, the tissue microarray data indicate that MeCP2 deficiency is frequent in autism and RTT, observed in at least two brain regions in autism, and not restricted to RTT and autism. MeCP2 expression may therefore be a marker of a shared dysregulated pathway of abnormal brain development in multiple neurodevelopmental disorders.
Reduced MeCP2 expression is hypothesized to have important functional consequences. Expression of GABRB3 and UBE3A were previously shown to be reduced in human MECP2-mutant RTT brain samples, Mecp2-null mouse brain, and human autism brain (37). Expression of other genes regulated by MeCP2, such as BDNF and DLX5, may also be affected by reduced MeCP2 (38–40). The dysregulation of MeCP2 target genes is expected to have several neurological consequences, such as defects in excitatory neurotransmission and long-term potentiation, leading to some of the cognitive and behavioral deficits in ASDs.
As a first step in finding the etiology of reduced MeCP2 in ASDs, the protein-coding region and a 1.9 kb promoter region of MECP2 were sequenced in ASD samples. As expected, coding mutations were found or confirmed in some RTT samples. One female autism sample, AUT 1638, was heterozygous for a silent polymorphism in exon 4 (c.1035 A>G). This polymorphism has been identified previously and causes a synonymous amino acid change (p.K345K) (20). MECP2 promoter sequencing yielded a single sequence variant in an autistic female (AUT B5342) who was heterozygous for a novel T>C transition 5′ of the human MECP2 promoter (g.-1398 T>C). The significance of the −1398 variant remains unclear at this stage and will require further functional studies to determine if it could be a rare mutation. One conclusion drawn from these experiments is that mutations in the MECP2 −1492 to −1318 genomic region are rare in autism and are unlikely to be a major contributor to its etiology. It cannot be excluded that sequence variation in other promoter regions, introns, 3′ UTR, or distant regulatory sequences might affect MeCP2 expression. MECP2 exon 1 and promoter mutations are rare in RTT (34), but there is some evidence for involvement of 3′ UTR variants in autism (20). Recent data suggest that long-range regulation of MECP2 transcription is influenced by sequences up to 130 kb away from the gene itself, presumably exerting their effects through chromatin looping (36).
Since coding or 5′-regulatory genetic variants did not appear to explain most cases of reduced MeCP2 expression, we hypothesized that aberrant MECP2 promoter methylation might contribute to decreased MeCP2 expression and the autism phenotype. MECP2 is an X-linked gene and is subject to X-inactivation (26). MECP2 in males is expected to be unmethylated and expressed, so any promoter methylation in autism males would be aberrant. Bisulfite sequencing of the MECP2 promoter (−531 to −243) in male autism and control cerebral cortex found evidence for significantly increased promoter methylation in autism compared to controls. Although not all CpG sites or alleles were methylated in any clone or sample, the overall level of DNA methylation was statistically significantly higher in autism versus controls. One CpG site out of 15 assayed, site #3, showed significantly increased methylation in autism but not four Down syndrome males. This site is located at position −445 and is part of the functional promoter region (31, 36). Furthermore, ENCODE chromatin immunoprecipitation (ChIP) data show that RNA polymerase II, SP1, SP3, E2F1, and other transcription factors bind at or around CpG site #3, and this region contains a DNase I hypersensitivity site in neuronal cells, strongly suggesting functional relevance (Supplementary Figure II). Female samples were not included because evidence of bias (not the expected 50% methylation) was observed in control samples, likely due to major differences in amplification and cloning efficiency between the highly methylated inactive and unmethylated active alleles following bisulfite conversion. However, 60% of alleles were methylated at site #3 in female control samples, suggesting that methylation of this site corresponds to transcriptional silencing (data not shown).
The relatively low level of total methylation observed in autism brain samples suggests that the affected cells may only be a subpopulation of the cells from which DNA was isolated from the brain. Since LCS analysis collected data from thousands of cells per sample but bisulfite sequencing analysis was limited to 10–20 cells, the LSC data is ultimately a better reflection of actual cell populations. Despite the sampling discrepancies, the two assays showed a remarkable correlation between the percentage of methylated cells and the decrease in MeCP2hi cells, particularly for juvenile autism samples. In contrast, the adult autism samples likely had other indirect factors influencing MeCP2 expression, similar to the observation of reduced MeCP2 in adult but not juvenile Down syndrome samples (Table 1). It would be interesting to determine if changes in MECP2 promoter methylation are detectable and quantifiable in blood in young autism patients with methylation assays that reflect a larger number of cells, as this might facilitate a diagnostic tool for molecular characterization of suspected autism cases.
A mixed oligogenic and epigenetic etiology has been proposed for autism (41). Epigenetic pathways are relevant in autism because they implicate both genetic and environmental factors. For example, dietary methyl supplementation can affect gene expression via DNA methylation (42). The observation of monozygotic twins discordant for autism (43) suggests epigenetic differences, as methylation changes increase over the lifespan of monozygotic twins (44). Abnormal patterns of DNA methylation have been observed in cancer, as both whole genome hypomethylation and tumor-suppressor gene hypermethylation (45). Interestingly, abnormal DNA methylation has also been found in a several psychiatric disorders. Promoter hypermethylation of the autism and schizophrenia candidate gene reelin (RELN) was found in postmortem occipital and frontal cortex schizophrenia samples (46). Interestingly, the methylation increases were not at all RELN CpG sites analyzed, but rather only at a few key sites, similar to the increased methylation of one MECP2 CpG site in autism reported here. It seems likely that the epigenetic defects observed in psychiatric disorders are more subtle that than observed in other conditions, such as cancer.
Increased DNA methylation has been observed in alcoholism, and HERP promoter hypermethylation in blood correlated with elevated homocysteine levels and decreased HERP mRNA (47). In autism, aberrant DNA methylation was found in the gene UBE3A, but did not correlate with reduced expression (41). Mutations in UBE3A cause Angelman syndrome, but UBE3A is also an autism candidate gene based on cytogenetic, positional, epigenetic, and expression data (37, 48, 49). Increased acetylated histone H3 lysine 9, which is a mark of active chromatin, correlated with decreased MeCP2 expression in autism brain (50). These observations indicate that epigenetic pathways are relevant in multiple human neurodevelopmental disorders and open the door for research into epigenetic etiology in autism and other disorders.
What could be the cause of increased MECP2 promoter methylation in autism? One possibility is lack of complete erasure of methylation derived from X chromosome inactivation on the maternally inherited X chromosome. Alternatively, environmental factors, including in utero or postnatal exposure to certain chemicals or pollutants, could result in de novo methylation (51). Proximal genetic variation does not appear to explain increased MECP2 promoter methylation, in contrast to Fragile X syndrome, where 5′ trinucleotide repeats influence FMR1 promoter methylation (52). However, genetic variants in trans could potentially cause aberrant MECP2 methylation. Since male Down syndrome samples with reduced MeCP2 expression did not show increased methylation, a simplistic explanation that abnormal brain development is the cause of aberrant MECP2 methylation is unlikely.
MeCP2 is a key epigenetic regulator which mediates the effects of DNA methylation by binding to methylated DNA and recruiting additional factors that modify chromatin (1). In this study we show that increased MECP2 promoter methylation correlates with reduced expression of MeCP2 in autism brain. While our data suggest that aberrant methylation causes reduced MeCP2 expression, we cannot rule out the possibility that factors that cause reduced MeCP2 expression could also cause increased methylation of the MECP2 promoter. It would be interesting to determine if aberrant MECP2 promoter methylation recruits itself (MeCP2 protein) or other methyl-binding domain (MBD) proteins resulting in reduction of MeCP2 expression. Alternatively, MECP2 promoter methylation at a few key CpG sites may block specific transcription factor binding, independent of MBD protein binding. The latter case seems more likely since one specific CpG site (site #3) showed statistically significantly increased methylation in autism brain. Methylation of this site may affect several putative transcription factor binding sites (CTCF, C/EBP, CAP).
Further understanding of epigenetic pathways in autism will likely lead to a greater understanding of the complex etiology of this disorder. No molecular test currently exists for autism, and diagnosis is based solely on clinical observations. Defining epigenetic abnormalities in autism could lead to a molecular diagnostic test, such as the DNA methylation test used in Angelman and Prader-Willi syndromes (53). Drugs that act on epigenetic pathways, such as methyltransferase or histone deacetylase inhibitors, are in development or clinical trials for the treatment of cancer (54). By defining epigenetic defects in neurodevelopmental disorders, some of these advancements may be useful in the diagnosis and eventual treatment of autism and autism spectrum disorders.
Acknowledgments
We thank Dr. Paul Hagerman for Fragile X brain and critical review of the manuscript. We also thank D. Yasui, S. Swanberg, S. Peddada, K. Thatcher, and M. Baerwald for manuscript review. Human brain tissue samples were generously provided by the Autism Tissue Program, The University of Maryland Brain and Tissue Bank for Developmental Disorders, The University of Miami Brain and Tissue Bank for Developmental Disorders, and the Harvard Brain Tissue Resource Center (supported in part by NIH R24MH-068855). This work was supported in part by NIH 1R01HD048799 and a predoctoral fellowship (RN) from the U.C. Davis M.I.N.D. Institute.
ABBREVIATIONS
- ASD
autism spectrum disorder
- PDD
pervasive developmental disorder
- RTT
Rett syndrome
- MECP2
gene symbol for human methyl-CpG binding protein 2
- Mecp2
gene symbol for mouse methyl-CpG binding protein 2
- MeCP2
methyl-CpG binding protein 2
- MBD
methyl-CpG binding domain protein
- 3′-UTR
3′-untranslated region
- AS
Angelman syndrome
- PWS
Prader-Willi syndrome
- ADHD
attention deficit hyperactivity disorder
- LSC
laser scanning cytometry
- AGRE
Autism Genetic Resource Exchange
- SSCP
single strand conformational polymorphism
- BA9
Brodmann Area 9
References
- 1.Kriaucionis S, Bird A. DNA methylation and Rett syndrome. Hum Mol Genet. 2003;12:R221–227. doi: 10.1093/hmg/ddg286. Spec No 2. [DOI] [PubMed] [Google Scholar]
- 2.Nicolson R, Szatmari P. Genetic neurodevelopmental influences in autistic disorder. Can J Psychiatry. 2003;48:526–537. doi: 10.1177/070674370304800804. [DOI] [PubMed] [Google Scholar]
- 3.Lamb JA, Moore J, Bailey A, Monaco AP. Autism: recent molecular genetic advances. Hum Mol Genet. 2000;9:861–868. doi: 10.1093/hmg/9.6.861. [DOI] [PubMed] [Google Scholar]
- 4.Tager-Flusberg H, Joseph R, Folstein S. Current directions in research on autism. Ment Retard Dev Disabil Res Rev. 2001;7:21–29. doi: 10.1002/1098-2779(200102)7:1<21::AID-MRDD1004>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2 encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, et al. X chromosome inactivation on Rett syndrome phenotypesAnn Neurol 200047670–679. [PubMed] [Google Scholar]
- 7.Hammer S, Dorrani N, Dragich J, Kudo S, Schanen C. The phenotypic consequences of MECP2 mutations extend beyond Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:94–98. doi: 10.1002/mrdd.10023. [DOI] [PubMed] [Google Scholar]
- 8.Laccone F, Junemann I, Whatley S, Morgan R, Butler R, Huppke P, Ravine D. Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum Mutat. 2004;23:234–244. doi: 10.1002/humu.20004. [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Shibayama A, Liu Q, Nguyen VQ, Feng J, Santos M, Temudo T, Maciel P, Sommer SS. Detection of heterozygous deletions and duplications in the MECP2 gene in Rett syndrome by Robust Dosage PCR (RD-PCR) Hum Mutat. 2005;25:505. doi: 10.1002/humu.9338. [DOI] [PubMed] [Google Scholar]
- 10.Ariani F, Mari F, Pescucci C, Longo I, Bruttini M, Meloni I, Hayek G, Rocchi R, Zappella M, Renieri A. Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: Report of one case of MECP2 deletion and one case of MECP2 duplication. Hum Mutat. 2004;24:172–177. doi: 10.1002/humu.20065. [DOI] [PubMed] [Google Scholar]
- 11.Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, Kerr A, Bunyan D, Kerr B, Sweeney E, Davies SJ, et al. Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet. 2006;43:451–456. doi: 10.1136/jmg.2005.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lugtenberg D, de Brouwer AP, Kleefstra T, Oudakker AR, Frints SG, Schrander-Stumpel CT, Fryns JP, Jensen LR, Chelly J, Moraine C, et al. J Med Genet. 2006;43:362–370. doi: 10.1136/jmg.2005.036178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28:205–211. doi: 10.1016/s0887-8994(02)00624-0. [DOI] [PubMed] [Google Scholar]
- 16.Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A. Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet. 2002;111:305–309. doi: 10.1007/s00439-002-0786-3. [DOI] [PubMed] [Google Scholar]
- 17.Lam CW, Yeung WL, Ko CH, Poon PM, Tong SF, Chan KY, Lo IF, Chan LY, Hui J, Wong V, et al. Rett syndrome. J Med Genet. 2000;37:E41. doi: 10.1136/jmg.37.12.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobo-Menendez F, Sossey-Alaoui K, Bell JM, Copeland-Yates SA, Plank SM, Sanford SO, Skinner C, Simensen RJ, Schroer RJ, Michaelis RC. Absence of MeCP2 mutations in patients from the South Carolina autism project. Am J Med Genet. 2003;117B:97–101. doi: 10.1002/ajmg.b.10016. [DOI] [PubMed] [Google Scholar]
- 19.Vourc'h P, Bienvenu T, Beldjord C, Chelly J, Barthelemy C, Muh JP, Andres C. No mutations in the coding region of the Rett syndrome gene MECP2 in 59 autistic patients. Eur J Hum Genet. 2001;9:556–558. doi: 10.1038/sj.ejhg.5200660. [DOI] [PubMed] [Google Scholar]
- 20.Shibayama A, Cook EH, Jr, Feng J, Glanzmann C, Yan J, Craddock N, Jones IR, Goldman D, Heston LL, Sommer SS. MECP2 structural and 3'-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet. 2004;128B:50–53. doi: 10.1002/ajmg.b.30016. [DOI] [PubMed] [Google Scholar]
- 21.Gauthier J, Joober R, Dube MP, St-Onge J, Bonnel A, Gariepy D, Laurent S, Najafee R, Lacasse H, St-Charles L, et al. Psychiatry. 2006;11:206–213. doi: 10.1038/sj.mp.4001756. [DOI] [PubMed] [Google Scholar]
- 22.Vincent JB, Melmer G, Bolton PF, Hodgkinson S, Holmes D, Curtis D, Gurling HM. Genetic linkage analysis of the X chromosome in autism, with emphasis on the fragile X region. Psychiatr Genet. 2005;15:83–90. doi: 10.1097/00041444-200506000-00004. [DOI] [PubMed] [Google Scholar]
- 23.LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- 24.Balmer D, Goldstine J, Rao YM, LaSalle JM. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med. 2003;81:61–68. doi: 10.1007/s00109-002-0396-5. [DOI] [PubMed] [Google Scholar]
- 25.Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- 26.D'Esposito M, Quaderi NA, Ciccodicola A, Bruni P, Esposito T, D'Urso M, Brown SD. Isolation, physical mapping, and northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mamm Genome. 1996;7:533–535. doi: 10.1007/s003359900157. [DOI] [PubMed] [Google Scholar]
- 27.Coy JF, Sedlacek Z, Bachner D, Delius H, Poustka A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3"-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet. 1999;8:1253–1262. doi: 10.1093/hmg/8.7.1253. [DOI] [PubMed] [Google Scholar]
- 28.Reichwald K, Thiesen J, Wiehe T, Weitzel J, Poustka WA, Rosenthal A, Platzer M, Stratling WH, Kioschis P. Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mamm Genome. 2000;11:182–190. doi: 10.1007/s003350010035. [DOI] [PubMed] [Google Scholar]
- 29.Kriaucionis S, Bird A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004;32:1818–1823. doi: 10.1093/nar/gkh349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, et al. Nat Genet. 2004;36:339–341. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- 31.Adachi M, Keefer EW, Jones FS. A Segment of the Mecp2 Promoter Is Sufficient to Drive Expression in Neurons. Hum Mol Genet. 2005;14:3709–3722. doi: 10.1093/hmg/ddi402. [DOI] [PubMed] [Google Scholar]
- 32.Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 33.Pelphrey K, Adolphs R, Morris JP. Neuroanatomical substrates of social cognition dysfunction in autism. Ment Retard Dev Disabil Res Rev. 2004;10:259–271. doi: 10.1002/mrdd.20040. [DOI] [PubMed] [Google Scholar]
- 34.Evans JC, Archer HL, Whatley SD, Kerr A, Clarke A, Butler R. Variation in exon 1 coding region and promoter of MECP2 in Rett syndrome and controls. Eur J Hum Genet. 2005;13:124–126. doi: 10.1038/sj.ejhg.5201270. [DOI] [PubMed] [Google Scholar]
- 35.Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13(12):1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Francke U. Identification of cis-regulatory elements for MECP2 expression. Hum Mol Genet. 2006;15(11):1769–1782. doi: 10.1093/hmg/ddl099. [DOI] [PubMed] [Google Scholar]
- 37.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 39.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 40.Horike S, Cai S, Miyano M, Cheng J, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 41.Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, et al. Am J Med Genet. 2004;131A:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- 42.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 44.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 46.Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res. 2006;30:587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- 48.Veenstra-VanderWeele J, Cook EH. Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9:819–832. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- 49.Thatcher KN, Peddada S, Yasui DH, LaSalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- 50.Thatcher KN, LaSalle JM. Dynamic changes in histone lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics. 2006;1:24–31. doi: 10.4161/epi.1.1.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 53.Kosaki K, McGinniss MJ, Veraksa AN, McGinnis WJ, Jones KL. Prader-Willi and Angelman syndromes: diagnosis with a bisulfite-treated methylation-specific PCR method. Am J Med Genet. 1997;73:308–313. [PubMed] [Google Scholar]
- 54.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 55.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]