

Abstract
Plasmids are units of extrachromosomal genetic inheritance found in all kingdoms of life. They replicate autonomously and undergo stable propagation in their hosts. Despite their small size, plasmid replication and gene expression constitute a metabolic burden that compromises their stable maintenance in host cells. This pressure has driven the evolution of strategies to increase plasmid stability—a process accelerated by the ability of plasmids to transfer horizontally between cells and to exchange genetic material with their host and other resident episomal DNAs. These abilities drive the adaptability and diversity of plasmids and their host cells. Indeed, survival functions found in plasmids have chromosomal homologues that have an essential role in cellular responses to stress. An analysis of these functions in the prokaryotic plasmid R1, and of their intricate interrelationships, reveals remarkable overall similarities with other gene- and cell-survival strategies found within and beyond the prokaryotic world.
Keywords: plasmid R1, stability system, parD, toxin–antitoxin pair, RNA decay
R1 architecture: a winning design for DNA survival
Plasmid R1 is a good example of how the need to be stably maintained in host cells shapes the architecture and building blocks of extrachromosomal elements. Four properties can be distinguished in R1, all of which contribute to plasmid survival: antibiotic-resistance genes, horizontal DNA transfer genes, stability systems and the basic replicon (Fig 1A; Womble & Rownd, 1988).
Figure 1.
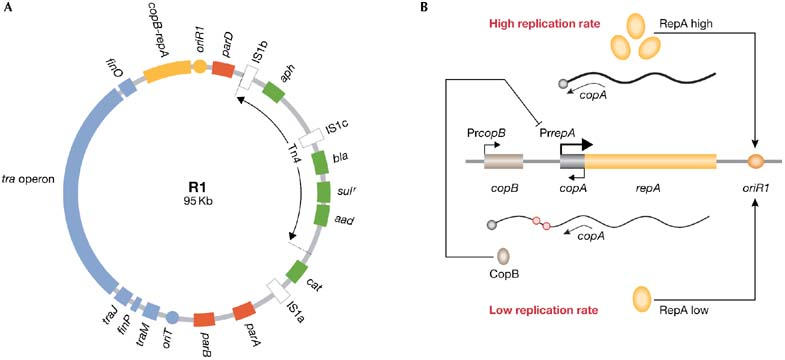
R1 and copy-number control. (A) A map of R1 showing antibiotic resistance genes (green), insertion sequences (white), its basic replicon (yellow), conjugation genes (blue) and stability systems (red). (B) PrcopB produces some RepA as well as CopB, a repressor of PrrepA, which keeps R1 copy number low. In the absence of CopB, stronger PrrepA increases RepA and R1 copy number. Antisense RNA copA limits translation of RepA and is less effective when PrrepA is active. Red circles on RNA denote UUACU sites. Cop, copy-number control gene; ori, origin of replication; Pr, promoter; Rep, replication initiation factor.
R1 carries ampicillin- (bla), chloramphenicol- (cat), kanamycin- (aph), streptomycin/spectinomycin- (aad) and sulfonamide- (sul) resistance genes. These are clustered in the R-determinant, which contains three insertion sequences and a transposon, Tn4. Insertion sequences are minimal transposable elements that encode functions involved in insertion events, and they are found in plasmids and in the chromosomes of prokaryotes, eukaryotes and archaea (Mahillon & Chandler, 1998). They mediate various DNA rearrangements, including the integration of R1 into host chromosomes, which can also influence the transcription of neighbouring genes. Genes flanked by two identical insertion sequences become transposable elements; therefore, antibiotic-resistance genes in R1 can be mobilized as transposons in different combinations.
R1 can also move horizontally into plasmid-free cells by conjugation. To achieve this, R1 induces the synthesis of a pilus that attaches the donor to a recipient cell. One DNA strand of R1 is then cleaved at oriT (origin of transfer) and transferred through the pilus to the recipient cell, in which it is re-circularized and converted into double-stranded DNA. Most genes required for transfer are encoded in the tra operon. In R1, a regulatory network inhibits expression of this operon, limiting the frequency of conjugation to 1×10−3 per R1 copy. However, once transfer occurs, the absence of the limiting regulatory factors in recipient cells allows R1 to spread rapidly in dense bacterial populations (Pölzleitner et al, 1997).
Resistance genes confer growth advantages to R1 hosts in niches in which antibiotics are present, and genetic mobility allows R1 to acquire and evolve these survival strategies (Frost et al, 2005). However, few, if any, of these genes are under strong selection at all times and even if they were, their mobilization into the host chromosome would allow cell survival without the costs of maintaining the plasmid. Therefore, R1 has evolved additional functions to ensure that its survival does not depend on a single strategy, but rather on their additive effects. These functions lie outside the R-determinant. Therefore, loss of antibiotic resistance does not completely compromise plasmid survival.
Low copy number also contributes to R1 survival, minimizing the metabolic burden that the plasmid imposes on host cells. In Escherichia coli, the ratio between R1 and the oriC (origin of chromosomal replication) is approximately 1:1. Chromosomal replication requires the initiator protein DnaA; however, R1 encodes its own initiator factor, RepA. This protein is limiting for the replication of R1, and the plasmid keeps its copy number low by tightly regulating RepA synthesis. Furthermore, RepA uncouples R1 replication from the host cell cycle, a process largely regulated by DnaA (Nordström, 2006).
Apart from the repA gene, the basic replicon of R1 contains oriR1 (origin of replication in plasmid R1) and two copy-number control genes, copA and copB. RepA can be transcribed from two promoters, PrcopB and PrrepA. Weak transcription from PrcopB maintains the low R1 copy number. It generates a bicistronic transcript that encodes both CopB and RepA. CopB represses PrrepA, which then limits the production of RepA and the rate of R1 replication. The other gene, copA, encodes an antisense RNA that limits the translation of RepA, especially when PrrepA is fully repressed (Fig 1B; Nordström, 2006).
Copy number is a quantitative genetic trait of plasmids, but it fluctuates depending on the host and growth conditions. Indeed, the frequency of R1 replication is inversely proportional to its copy number. Control of RepA synthesis allows R1 to correct these fluctuations. This might be essential during conjugation, when a single copy of R1 is transferred into plasmid-free cells and needs to equal the number of oriCs rapidly in order to survive. This is possible because recipient cells lack CopB, allowing de-repression of PrrepA until R1 reaches its normal copy number.
Despite its benefits for plasmid survival, low copy number could reduce the chances of R1 being transmitted to daughter cells. To counteract this, the plasmid has evolved three stability systems: parA, parB and parD. ParA is a partition system that segregates copies of R1 to daughter cells. It is organized as a bicistronic operon that encodes proteins ParM (M for motor) and ParR (R for repressor). ParR binds to parC, an upstream cis-acting centromere-like region that also contains the promoter of parA (PrparA). Binding of ParR to parC generates a partition complex that represses transcription from PrparA in vivo and induces plasmid pairing in vitro (Fig 2A; Ebersbach & Gerdes, 2005).
Figure 2.
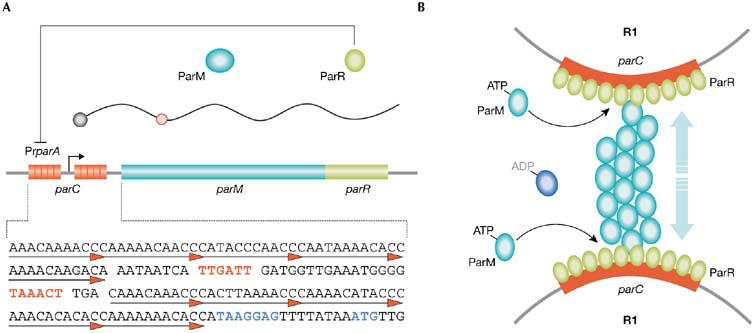
parA and segregation of R1. (A) Transcription from PrparA generates ParM and ParR. Binding of ParR to direct repeats in parC (underlined) represses PrparA. Transcriptional (indicated in red text) and translational (indicated in blue text) signals of parA are embedded in parC. Red circles on RNA denote UUACU sites. (B) parC–ParR stimulates apical growth and increases the stability of ATPParM filaments, allowing segregation of paired copies of R1. Pr, promoter.
ParM is an ATPase. When bound to ATP, it polymerizes into filaments that grow at an equal rate from each end. Hydrolysis of ATP within the filaments destabilizes the polymers, but polymerization and de-polymerization do not occur simultaneously. Filament growth occurs for some time before the process switches to unidirectional decay. This switch seems to be stochastic and, once initiated, disintegration of ParM filaments proceeds to completion and new polymerization requires the rejuvenation of ParM–ADP by nucleotide exchange. Such dynamic instability is important for the search, capture and bipolar alignment of R1 copies. Capture occurs through (parC–ParR)–ParM interactions and requires paired R1 sister copies. It inhibits filament disintegration and stimulates polymer growth at the ParR–ParM interface, promoting constant growth of ParM filaments and pushing plasmids to opposite cell poles. Overall plasmid–filament contacts during partition are sustained by lateral interactions between parallel proto-filaments that grow asynchronously (Fig 2B). Although prolonged stability of ParM filaments requires pairing of sister copies of R1, segregation does not depend on plasmid replication per se, but on its outcome and location: it requires two paired R1 copies positioned at mid-cell. This regulates R1 segregation both temporally and spatially (Ebersbach & Gerdes, 2005, and references therein).
ParB is a post-segregational killing (PSK) system (Franch et al, 1997). It kills host cells that, despite all the efforts made by R1, still lose the plasmid during cell division. ParB encodes Hok (Host killing), a toxin that kills cells by damaging their membrane. Hok translation is coupled to the translation of upstream Mok (Mediator of killing) from a mok–hok messenger RNA. Folding of this mRNA hides the Shine–Dalgarno sequence of mok (SDmok) from ribosomes, which impedes the translation of both genes (Fig 3A). Partial 3′-processing of this transcript induces refolding, exposing SDmok to ribosomes, and enabling translation of Mok and Hok to trigger cell death. However, this is not possible in cells that contain R1 because of the action of sok (suppressor of killing), the third component of parB. sok is an antisense RNA complementary to sokT, a sequence downstream of SDmok. Different folding allows recognition of sokT by sok only in the processed—that is, translatable—mok–hok transcript. This generates a dsRNA structure that is cleaved by RNase III, triggering rapid decay of the mok–hok mRNA and avoiding synthesis of Hok and cell killing. However, antisense sok RNA is unstable and only cells maintaining R1 are able to supply sufficient amounts to escape cell death (Fig 3B; Franch et al, 1997).
Figure 3.
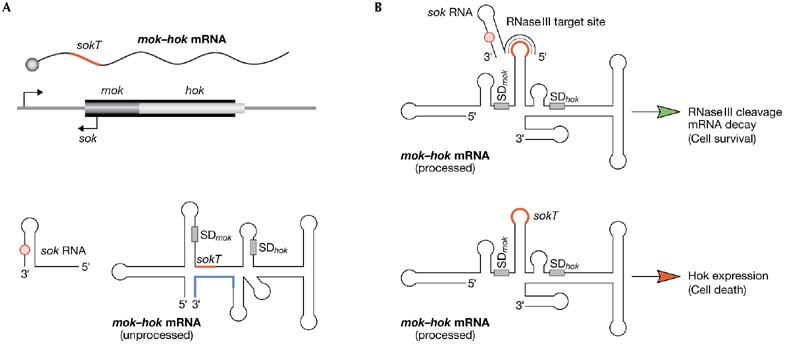
parB and post-segregational killing. (A) parB encodes a messenger RNA-encoding toxin Hok (mok–hok) and a labile antisense RNA, sok. Folding of mok–hok mRNA impedes toxin production and hides the sequence sokT (red) in a double-stranded RNA structure. Partial processing of the 3′ end (blue) of mok–hok mRNA induces refolding. (B) Refolded mRNA can produce Hok and exposes sokT in a single-stranded RNA loop. In cells containing R1, sok-sokT pairing induces rapid mRNA decay by RNase III. Instability of sok makes this impossible in cells that lose R1, which are then killed by Hok. Red circles on RNA denote UUACU sites. Hok, host killing; Mok, mediator of killing; SD, Shine–Dalgarno sequence; sok, suppessor of killing.
parD: 20 years of pre-segregational ‘kidding'
parD is immediately downstream of oriR1. It encodes the toxin Kid (Killing determinant) and the antitoxin Kis (Killing suppressor; Bravo et al, 1988). Cells containing R1 synthesize both proteins, which form a complex that neutralizes Kid toxicity and represses transcription of parD (Fig 4A). Kis is less stable than Kid and, by analogy to parB, it was assumed that a constant supply of neutralizing amounts of Kis was only possible in plasmid-containing cells, suggesting that parD is also a PSK system. Despite this, experimental observations made since the discovery of parD 20 years ago do not fit well with its attributed biological role and mode of action. First, when compared with other PSK systems, parD seemed to be functionally inactive and contributed little to plasmid stability (Jensen et al, 1995). However, for reasons that were not understood at the time, parD seemed to ‘wake' from its ‘dormant' state when the replication of R1 was compromised (Ruiz-Echevarría et al, 1995a). Second, the replicative helicase in E. coli, DnaB, was proposed to be the cellular target of Kid (Ruiz-Echevarria et al, 1995b). Despite this, neither physical interaction between DnaB and Kid nor inhibition of DnaB activity by Kid could be shown. Indeed, this mode of action was difficult to reconcile with the activation of parD that is observed when R1 replication, which depends on DnaB, is compromised. Finally, attempts to eliminate parD + plasmids from E. coli caused cell growth inhibition rather than cell killing. Intriguingly, arrested cells still contained the plasmid (Jensen et al, 1995), which is incompatible with a post-segregational activation of Kid.
Figure 4.
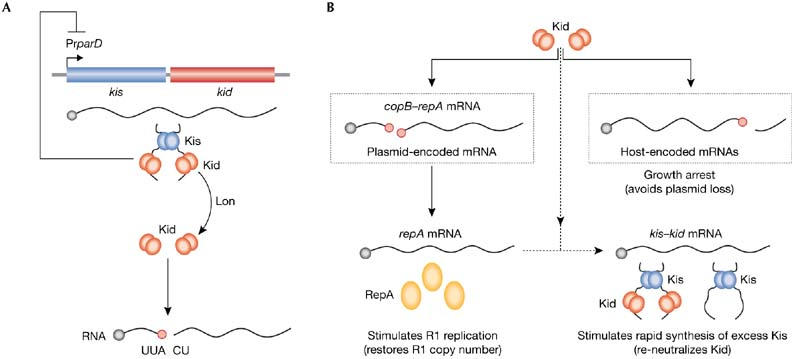
parD and R1 rescue. (A) parD produces the toxin Kid and the antitoxin Kis, which assemble in a 2:1 complex that neutralizes Kid and represses PrparD. Lon degrades Kis, allowing Kid to cleave RNA at UUACU. (B) When R1 copy number decreases, Kid activates pre-segregationally and cleaves host- and plasmid-encoded messenger RNAs. This inhibits cell growth, avoiding plasmid loss, and restores R1 copy number. Kid alone cannot repress PrparD, enabling re-neutralization of Kid when R1 copy number recovers. Red circles on RNA denote UUACU sites. Cop, copy-number control gene; Kid, killing determinant; Kis, killing suppressor; Pr, promoter; Rep, replication initiation factor.
Recently, we showed that Kid cleaves mRNA at 5′-UUACU-3′ sites, which shuts off host gene expression and stops cell growth (Pimentel et al, 2005). Activation of parD occurs when R1 copy number decreases but before it is lost from cells—that is, pre-segregationally—which allows the cleavage of plasmid-encoded copB–repA mRNA at two UUACU sites (Fig 1B). This decreases the levels of CopB, de-represses PrrepA and leads to a higher level of synthesis of RepA. Consequently, the replication rate of R1 increases and plasmid copy number recovers. Simultaneous cleavage of host-encoded mRNA arrests cell growth and avoids plasmid loss. These results explain how parD stabilizes plasmid R1 in bacteria. The stability of Kis, which is degraded by the host protease Lon, and the transcription of parD, which is not repressed by Kid alone, are functionally linked to sense R1 copy-number fluctuations. If a decrease in R1 copy number threatens plasmid stability, pre-segregational activation of Kid occurs. Cell-growth arrest prevents plasmid loss and greater amounts of RepA stimulate replication of R1 to restore its normal copy number. Furthermore, Kid alone cannot repress PrparD and, as one molecule of antitoxin neutralizes two molecules of toxin, synthesis of excess Kis from parD promotes re-neutralization of Kid while R1 recovers. Interestingly, repA and kis lack UUACU sites making them resistant to cleavage, and allowing copy-number rescue and Kid re-neutralization (Fig 4B; Pimentel et al, 2005).
Chromosomal homologues of parD: cell stress managers
ParD is a plasmid stress element. Copy-number fluctuations that threaten R1 stability activate Kid to eliminate the stress rather than killing host cells. Once this is achieved, Kid promotes its own re-neutralization and re-sets the stress element. Archaea and many bacteria encode stress elements that are functionally and structurally similar to parD. E. coli has five such elements, which enable cells to manage stressful growth conditions, such as starvation (Gerdes et al, 2005). Their similarities suggest that plasmid- and chromosomal-stress elements have co-evolved with the assistance of horizontal mobilization and DNA exchange, both of which occur in bacterial plasmids, such as R1, and in the archaeal world.
In E. coli, relBE reduces the global level of translation during amino-acid starvation. This stress activates RelE, which cleaves mRNA stop codons positioned at the ribosomal A site. Release factors (RF) cannot act on mRNA with disrupted stop codons; therefore, RelE traps translating ribosomes, cleaved mRNAs and unfinished polypeptides in inactive complexes. Instead of RFs, a specialized molecule—transfer-messenger RNA (tmRNA)—occupies the A site in these ribosomes. Through a process termed trans-translation, tmRNA promotes tagging of the unfinished proteins with a peptide that marks them for proteolysis. This frees and recycles stalled ribosomes and, by promoting degradation of trapped polypeptides, renews amino-acid pools, helping cells to relieve the stress and survive starvation (Pedersen et al, 2003).
Amino-acid starvation also induces mazEF in E. coli. MazF cleaves mRNA at ACA sites independently of ribosomes. The activities of RelE and MazF inhibit cell growth, and this can be reverted by their antitoxins, RelB and MazE. This suggests that, similar to parD, these systems are switched off after elimination of the stress (Gerdes et al, 2005).
Meta-regulating survival elements: a key to success
The presence of several stability functions in plasmid R1 raises interesting questions about how they coordinate their activities to maximize plasmid survival. The specificity of Kid explains how it interferes with the translation of copB but not of repA or kis, thereby allowing recovery of R1 copy number and re-neutralization of Kid. But parD also ensures that other cellular functions remain intact—for example, transcription, translation, replication and ATP regeneration, which are essential for the correct functioning of the rescue system. Resistance or sensitivity of mRNAs to the action of Kid, as well as the stability of proteins produced before parD is activated, must have a role in this process. This is evident when the activity of Kid is examined in relation to the other stability functions in R1. parA, parB and parD act synergistically to reduce R1 loss to 1×10−7 per cell per generation, and to achieve this they must work in a coordinated and non-interfering manner.
For example, activation of parD must neither impede parB-mediated killing of cells that lose R1, nor induce parB-mediated killing in R1-containing cells. mok–hok mRNA lacks UUACU sites, so it is resistant to Kid and ensures that parB is ready to act if R1 is lost. Of equal importance, Kid does not degrade sok antisense-RNA, and so premature activation of parB is avoided. Interestingly, sok contains one UUACU site; however, it is positioned in a stable double-stranded RNA structure (Franch et al, 1997), and Kid only cleaves single-stranded RNA.
Similarly, parA and parD must function in a coordinated manner because conditions that activate parD are unfavourable for plasmid segregation. First, pairing of sister copies of R1 must be infrequent at low plasmid copy numbers, when Kid is active. Second, Kid inhibits host cell division and this impedes plasmid segregation before normal R1 copy number is restored. Therefore, parA cannot interfere with parD function. But can parD interfere with parA? parA contains one UUACU site and it is unknown whether Kid cleaves at this location. However, binding of ParR to parC, which is essential to generate the partition complex, inhibits transcription from PrparA (Ebersbach & Gerdes, 2005, and references therein). This suggests that ParR and ParM are relatively stable, as effective plasmid segregation does not require—indeed inhibits—their synthesis de novo. All this might help to synchronize parD and parA functions, allowing pairing of new R1 copies as they replicate after Kid activation but segregating them only when R1 copy number is restored and Kid is re-neutralized. It seems likely that a similar relationship contributing to plasmid survival must have evolved between parD and other functions in R1.
Meta-regulation is also important for the correct functioning of chromosomal stress elements. In E. coli, depletion of charged transfer RNAs during amino-acid starvation activates RelA to produce ppGpp (Guanosine-tetraphosphate). This bacterial ‘alarmone' induces stringent control of RNA synthesis, which inhibits transcription of stable RNAs and stimulates that of amino-acid biosynthetic proteins and starvation regulatory factors. ppGpp also activates Lon, which degrades RelB, linking the stringent response to activation of RelE (Gerdes et al, 2005). This protein competes with RFs to occupy the A site in the ribosome to cleave mRNA stop codons. Among these, RelE has a preference for UAG, the stop codon recognized by RF1. Conspicuously, RF1 ends at UAG. This might help to decrease the intracellular concentration of RF1 when RelE is active and favour RelE occupancy of the ribosomal A-site at UAG stop codons during starvation. Another protein, RF2, can terminate translation at UAA and UGA stop codons. Interestingly, the stop codon for RF2, RelB, RelE and Lon is UGA, which is the least sensitive to RelE. Furthermore, UAG is underrepresented in genes that are essential during the stringent response—for example, tmRNA and enzymes involved in amino-acid synthesis. As seen before for kis and repA, which lack UUACU, this feature might have been selected to facilitate the synthesis of essential proteins when RelE is active. This seems to be a general strategy, as MazF sustains transcription, translation—of mRNAs lacking ACA sites—and ATP regeneration in vivo over long periods of time (Suzuki et al, 2005). Clearly, evolution has shaped E. coli so that genes essential for surviving starvation are either resistant to the action of these endoribonucleases or encode stable proteins.
Functionally related survival strategies are physically linked on DNA, such as parD and oriR1, or relA and mazEF in some bacteria. Perhaps evolution has favoured these links, reflecting their meta-regulatory relevance. Interestingly, these types of stress elements are abundant in archaea—for example, 32 in Sulfolobus tokodaii (Gerdes et al, 2005). Such density must have required the establishment of intricate regulatory networks, as well as functional and physical links, between these elements. If this is the case, it is worth noting that these relationships must have evolved in the eukaryotic-like molecular context that is inherent to archaea.
Concluding remarks
The metabolic burden that plasmids impose on host cells drives the evolution of strategies that work in a coordinated, non-interfering manner to maximize plasmid stability. The basic regulatory strategy used by plasmids to achieve this is autogenous control of gene expression. Operons encode at least an autorepressor, which ensures that gene expression is downregulated once a functional level of protein has been produced (Bingle & Thomas, 2001). This strategy has been adopted by R1 to control the expression of its survival functions—copB–repA, parA, parD and the tra operons. An additional level of control might be provided by coordinating the regulation of plasmid replication, transfer and stable maintenance genes, either by grouping them together in one operon or by physically associating their respective operons (Bingle & Thomas, 2001). This could facilitate co-regulation and contribute to the maintenance of physical links during plasmid evolution—for example, their co-acquisition by evolving episomes. Such juxtaposition of functionally related survival roles is present in plasmid R1—that is, parD is located immediately downstream of the plasmid basic replicon. The relevance of this physical link was hidden by the traditional view of parD as a PSK system. Our understanding that Kid acts pre-segregationally and reversibly to rescue R1 copy number unveils the importance of this link and connects parD and the basic replicon of R1.
More sophisticated than simple juxtaposition is the coordinated regulation of survival functions found in some Gram-positive (pSM19035) and Gram-negative (RK2) plasmids (Bingle & Thomas, 2001, and references therein). In pSM19035, a small repressor (ω) is encoded in the same operon as antitoxin ɛ and toxin ζ. Interestingly, ω binds to specific sequences found in the promoters of distant genes involved in plasmid copy number control (copS) and plasmid partition (δ), and regulates their transcription. Furthermore, ω represses its own transcription and therefore that of antitoxin ɛ and toxin ζ, components of the PSK system in pSM19035 (Zielenkiewicz & Cegłowski, 2005). In RK2, the partition proteins IncC and KorB are encoded in a central control operon that also encodes KorA. KorB binds to 12 centromere-like DNA sequences scattered throughout the plasmid backbone. IncC is an ATPase that recognizes KorB–DNA complexes, interacts with them and directs active segregation of RK2 copies during cell division. Six of those centromeric-DNA sequences are located at, or close to, promoters for genes involved in plasmid replication (trfA), stable inheritance (kle and krfA) and conjugative transfer (trbB and traG). Binding of KorB to DNA represses transcription of these genes. To do so, KorB normally cooperates with two other repressors: KorA, also part of the central control operon, and TrbA. This ability to cooperate facilitates strong transcriptional repression without the need to increase repressor concentration and allows the entire system to be finely balanced (Bingle & Thomas, 2001, and references therein).
Plasmid R1 does not seem to follow global regulatory strategies such as those present in pSM19035 and RK2. However, the expression and activities of some of its survival genes are intimately coordinated to sense and correct negative copy-number fluctuations. Activation of parD switches on a programme that influences expression of survival genes both negatively (CopB) and positively (RepA and Kis). Several regulatory loops link the activities of Kid and Kis to plasmid copy number and its control. These involve the transcriptional regulation of survival genes repA by CopB and parD by Kid and Kis; the relative stability of survival proteins—for example, Kis compared with Kid—and its impact on the stoichiometry of the complexes they can form; differential sensitivity of mRNAs transcribed at survival operons to Kid, such as copB–repA, repA, kis–kid; and the consequences of the latter for de novo synthesis of survival proteins, RepA and Kis, and, ultimately, for the replication rate of R1, which is limited by RepA availability (Pimentel et al, 2005). Similar regulatory loops must have evolved between parD and other R1 and host genes, as Kid does not interfere with plasmid stability and cell viability despite its toxic activity.
Our understanding of parD function raises interesting questions about the roles attributed to other plasmid-encoded toxin–antitoxin (TA) protein pairs. The experimental set-up traditionally used to analyse the activities of TA pairs and their potential role in PSK might not be ideal. Promoters and/or replicons different from those naturally associated with the TAs are frequently used (Jensen et al, 1995; Deane & Rawlings, 2004). Clearly, in the case of parD and R1, such an approach would lead to inaccurate conclusions. Therefore, it might be worth re-examining if other TA pairs exert similar roles to parD.
Members of all eight known families of TA pairs can be found in plasmids (Gerdes et al, 2005), and some plasmids encode more than one pair (Moritz & Hergenrother, 2007). Similar to parD, other TA pairs, such as ccdAB (controlled cell death) and pasAB (plasmid addiction system), are physically linked to—and even embedded into—the basic replicon of the plasmids that bear them (Ogura & Hiraga, 1983; Deane & Rawlings, 2004). There is evidence suggesting that the activation of some toxins, such as CcdB, can occur pre-segregationally, exerts a cytostatic effect, forces a large proportion of affected cells to retain plasmids and can be re-neutralized by their cognate antitoxin (Ogura & Hiraga, 1983; Jaffé et al, 1985; Maki et al, 1992; Bahassi et al, 1999). This indicates that these might be important features of the function of these plasmid-encoded TA pairs.
The modes of action of TA pairs are diverse and in many cases unknown (Gerdes et al, 2005). Acting as a plasmid rescue system might not be an exclusive attribute of parD, and might not even be limited to TA pairs encoding ribonucleolytic toxins. For example, CcdB of plasmid F and ParE of RK2 are gyrase inhibitors (Bahassi et al, 1999; Jiang et al, 2002). Interestingly, the activity of CcdB stimulates expression of chaperones DnaK and DnaJ and relaxes plasmid DNA (Maki et al, 1992; Kaneko et al, 1996). The limiting factor for replication of plasmid F—and RK2—is not the availability of the initiator protein but the ratio between its dimeric—inactive for replication—and monomeric—active for replication—forms. Notably, it has been shown that increasing negative superhelicity promotes initiation of this type of replication, and that DnaK and DnaJ are essential for the conversion of dimeric initiators into monomeric ones (Chattoraj, 2000, and references therein; Zzaman & Bastia, 2005). Therefore, it might be possible that the activity of CcdB (and parE) helps to rescue the copy number of plasmid F (and RK2). Indeed, ccd activation seems to occur when plasmid F copy number decreases (Ogura & Hiraga, 1983). If this is the case, it would still be possible that pre- (rescuing) and post-segregational (killing) activities of plasmid-encoded TA pairs are not mutually exclusive.
R1 has evolved different survival functions, each of them operating at specific levels—for example, replication, partition and PSK. Despite their particular levels of action, all of them must function in a coordinated, non-interfering, manner to optimize plasmid, and host, survival. This need for coordination has an important role in the evolution of these systems, and might be even more important in the case of plasmids that contain more than one survival system of the same type (Moritz & Hergenrother, 2007). The same applies to plasmids that share host cells with other resident episomes. Therefore, it would be interesting to analyse whether the specific arsenal of survival functions in one plasmid influences the evolution and function of those found in co-resident plasmids. This might be the case, as at least one example has been reported in which the activity of a plasmid-encoded survival function depends on the presence of another—in this case host-encoded—system of the same type (Hazan et al, 2001).
Despite their prokaryotic origin, it is remarkable how R1 survival strategies resemble others found in eukaryotes. For example, the control of plasmid copy number and DNA mobilization into host chromosomes, and how these two events modulate episomal, and host cell, gene expression, are important for the stability of human papilloma virus, the survival of its infected cells and, of course, the aetiology of human cancer (Longworth & Laimins, 2004).
Similarly, it is remarkable how R1 has evolved systems that—similar to eukaryotic checkpoints—act before (parD), during (parA) and after (parB) cell division to guard DNA stability and integrity. It is even more striking how failure to achieve this activates a programme that triggers bacterial cell death, as with apoptosis in eukaryotes.
Bacterial and archaeal chromosomes encode survival systems that are similar in function and structure to those in R1. They have specialized in cell—rather than DNA—survival under nutritional starvation, and their activity is exquisitely regulated to avoid interference with other metabolic functions that are essential for this process. Their presence in bacteria and archaea suggests that this is possible in metabolic contexts that resemble both prokaryotes and eukaryotes. It is surprising how the overall function and consequences of these bacterial systems resemble those of autophagy—the eukaryotic strategy to survive cell starvation. This resemblance extends to the molecular arena. Some bacterial and archaeal stress elements contain PIN domains (Arcus et al, 2005), which are homologous to those found in eukaryotic ribonucleases that are linked to RNA interference and nonsense-mediated RNA decay—a quality-control system that degrades potentially toxic aberrant mRNAs.
We wonder whether these strategies evolved from common ancestors or whether it is simply that nature can only find a handful of similar solutions to a particular problem. Until these questions are answered, we can at least admire the precision with which evolution has sculpted R1 and its host cells to survive under stress.

Belén Pimentel & Guillermo de la Cueva-Méndez
Acknowledgments
We are grateful to R. Nair, M. Preston, R. Laskey and M. Narita for critical reading of the manuscript. We apologize to colleagues whose work has not been directly cited owing to space limitations. Our work is funded by the UK Medical Research Council.
References
- Arcus VL, Rainey PB, Turner SJ (2005) The PIN-domain toxin–antitoxin array in mycobacteria. Trends Microbiol 13: 360–365 [DOI] [PubMed] [Google Scholar]
- Bahassi EM, O'Dea MH, Allali N, Messens J, Gellert M, Couturier M (1999) Interactions of CcdB with DNA gyrase. Inactivation of GyrA, poisoning of the gyrase–DNA complex, and the antidote action of CcdA. J Biol Chem 274: 10936–10944 [DOI] [PubMed] [Google Scholar]
- Bingle LEH, Thomas CM (2001) Regulatory circuits for plasmid survival. Curr Opin Microbiol 4: 194–200 [DOI] [PubMed] [Google Scholar]
- Bravo A, Ortega S, de Torrontegui G, Díaz R (1988) Killing of Escherichia coli cells modulated by components of the stability system parD of plasmid R1. Mol Gen Genet 215: 146–151 [DOI] [PubMed] [Google Scholar]
- Chattoraj DK (2000) Control of plasmid DNA replication by iterons: no longer paradoxical. Mol Microbiol 37: 467–476 [DOI] [PubMed] [Google Scholar]
- Deane SM, Rawlings DE (2004) Plasmid evolution and interaction between the plasmid addiction stability systems of two related broad-host-range IncQ-like plasmids. J Bact 186: 2123–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersbach G, Gerdes K (2005) Plasmid segregation mechanisms. Annu Rev Genet 39: 453–479 [DOI] [PubMed] [Google Scholar]
- Franch T, Gultyaev AP, Gerdes K (1997) Programmed cell death by hok/sok of plasmid R1: processing at the hok/sok mRNA 3′-end triggers structural rearrangements that allow translation and antisense RNA binding. J Mol Biol 273: 38–51 [DOI] [PubMed] [Google Scholar]
- Frost LS, Leplae R, Summers AO, Toussaint A (2005) Mobile genetic elements: the agents of open source evolution. Nat Rev Micro Biol 3: 722–732 [DOI] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Løbner-Olesen A (2005) Prokaryotic toxin–antitoxin stress response loci. Nat Rev Micro Biol 3: 371–382 [DOI] [PubMed] [Google Scholar]
- Hazan R, Sat B, Reches M, Engelberg-Kulka H (2001) Postsegregational killing mediated by the P1 phage “addiction module” phd-doc requires the Escherichia coli programmed cell death system mazEF . J Bact 183: 2046–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffé A, Ogura T, Hiraga S (1985) Effects of the ccd function of the F plasmid on bacterial growth. J Bact 163: 841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Grohmann E, Schwab H, Díaz-Orejas R, Gerdes K (1995) Comparison of ccd of F, parDE of RP4, and parD of R1 using a novel conditional replication control system of plasmid R1. Mol Microbiol 17: 211–220 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Pogliano J, Helinski DR, Konieczny I (2002) ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44: 971–979 [DOI] [PubMed] [Google Scholar]
- Kaneko T, Mizushima T, Ohtsuka Y, Kurokawa K, Kataoka K, Miki T, Sekimizu K (1996) Co-induction of DNA relaxation and synthesis of DnaK and GroEL proteins in Escherichia coli by expression of LetD (CcdB) protein, an inhibitor of DNA gyrase encoded by the F factor. Mol Gen Genet 250: 593–600 [DOI] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA (2004) Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 68: 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahillon J, Chandler M (1998) Insertion sequences. Microbiol Mol Biol Rev 62: 725–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki S, Takiguchi S, Miki T, Horiuchi T (1992) Modulation of DNA supercoiling activity of Escherichia coli DNA gyrase by F plasmid proteins. Antagonistic actions of LetA (CcdA) and LetD (CcdB) proteins. J Biol Chem 267: 12244–12251 [PubMed] [Google Scholar]
- Moritz EM, Hergenrother PJ (2007) Toxin–antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc Natl Acad Sci USA 104: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström K (2006) Plasmid R1-replication and its control. Plasmid 55: 1–26 [DOI] [PubMed] [Google Scholar]
- Ogura T, Hiraga S (1983) Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci USA 80: 4784–4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M (2003) The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112: 131–140 [DOI] [PubMed] [Google Scholar]
- Pimentel B, Madine M, de la Cueva-Méndez G (2005) Kid cleaves mRNAs at UUACU sites to rescue the copy number of plasmid R1. EMBO J 24: 3459–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pölzleitner E, Zechner EL, Renner W, Fratte R, Jauk B, Högenauer G, Koraimann G (1997) TraM of plasmid R1 controls transfer gene expression as an integrated control element in a complex regulatory network. Mol Microbiol 25: 495–507 [DOI] [PubMed] [Google Scholar]
- Ruiz-Echevarría MJ, de la Torre MA, Díaz-Orejas R (1995a) A mutation that decreases the efficiency of plasmid R1 replication leads to the activation of parD, a killer stability system of the plasmid. FEMS Microbiol Lett 130: 129–136 [DOI] [PubMed] [Google Scholar]
- Ruiz-Echevarría MJ, Gimenez-Gallego G, Sabariegos-Jareno R, Díaz-Orejas R (1995b) Kid, a small protein of the parD stability system of plasmid R1, is an inhibitor of DNA replication acting at the initiation of DNA synthesis. J Mol Biol 247: 568–577 [DOI] [PubMed] [Google Scholar]
- Suzuki M, Zhang J, Liu M, Woychik NA, Inouye M (2005) Single protein production in living cells by an mRNA interferase. Mol Cell 18: 253–261 [DOI] [PubMed] [Google Scholar]
- Womble DD, Rownd R (1988) Genetic and physical map of plasmid NR1: comparison with other IncFII antibiotic resistance plasmids. Microbiol Rev 52: 433–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenkiewicz U, Cegłowski P (2005) The toxin–antitoxin system of the streptococcal plasmid pSM19035. J Bact 187: 6094–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zzaman S, Bastia D (2005) Oligomeric initiator protein-mediated DNA looping negatively regulates plasmid replication in vitro by preventing origin melting. Mol Cell 20: 833–843 [DOI] [PubMed] [Google Scholar]