

Abstract
Neurofibromatosis type I (NFI) is a common genetic disorder that causes nervous system tumors, and learning and memory defects in humans, and animal models. We identify a novel growth factor stimulated adenylyl cyclase (AC) pathway in the Drosophila brain, which is disrupted by mutations in the epidermal growth factor receptor (EGFR), neurofibromin (NF1) and Ras, but not Gαs. This is the first demonstration in a metazoan that a receptor tyrosine kinase (RTK) pathway, acting independently of the heterotrimeric G-protein subunit Gαs, can activate AC. We also show that Gαs is the major Gα isoform in fly brains, and define a second AC pathway stimulated by serotonin and histamine requiring NF1 and Gαs, as well as a third, classical Gαs-dependent AC pathway, which is stimulated by Phe-Met-Arg-Phe-amide (FMRFamide) and dopamine. Using mutations and deletions of the human NF1 protein (hNF1) expressed in Nf1 mutant flies, we show that Ras activation by hNF1 is essential for growth factor stimulation of AC activity. Further, we demonstrate that sequences in the C-terminal region of hNF1 are sufficient for NF1/Gαs-dependent neurotransmitter stimulated AC activity, and for rescue of body size defects in Nf1 mutant flies.
INTRODUCTION
Mutations in the human NFI gene are characterized by benign but disfiguring tumors of the peripheral nervous system, as well as increased incidence of malignant peripheral nerve sheath tumors and central nervous system tumors (1). About 40% of children with NFI exhibit learning deficits (2,3), and mouse models of NFI recapitulate both the tumor and learning phenotypes (4–6). In Drosophila, Nf1 mutations affect circadian rhythms (7), body size (8), responses to neuropeptides (9) and olfactory learning (10). Thus, the NF1 protein is essential for normal neural development and plasticity in both vertebrates and invertebrates.
Gaining insights into the molecular mechanisms of NF1 function requires the identification of cellular signal transduction pathways that are disrupted by NFI mutations. Biochemical and genetic analysis in mammals and Drosophila has revealed that NF1 inhibits Ras activity (4–7), and regulates AC activity and cAMP levels (8–13). The NF1 protein has a central GTPase activating protein (GAP)-related domain (GRD), which catalyzes the intrinsic GTPase activity of Ras (14). Many of the tumor phenotypes and learning deficits observed in NFI patients and animal models have been attributed to hyperactivation of Ras, that is observed, for example, in Schwann cells and mast cells (15–18). However, the NF1-regulated AC/cAMP pathway is important for controlling neuropeptide responses (9) and learning (10) in flies, as well as neuropeptide-stimulated AC activity in both flies and mammals (12,13). The NF1-dependent activation of AC versus downregulation of Ras may therefore have important phenotypic consequences, but the molecular mechanism, whereby NF1 regulates AC activity has not yet been determined.
The product of the Drosophila Ras1 gene is functionally equivalent to vertebrate H-Ras, K-Ras, or N-Ras that are mutated in 30% of human cancers (19). Ras signaling is down-regulated by the activity of GAPs, which catalyze the hydrolysis of Ras-GTP to Ras-GDP. Five genes are reported to encode Ras-specific GAPs in Drosophila (20). The Gap1 and Nf1 genes each encode a GRD that can bind with Ras and catalyze GTPase activity (8,21), however, the Gap1 protein requires regions outside the GRD to achieve full catalytic activity (22). Guanine exchange factors (GEFs) promote the exchange of GDP for GTP to activate Ras, thereby enabling interaction with downstream effectors such as Raf-1 and PI3 kinase (23,24). GEF activation of Ras is controlled by signaling through RTKs such as sevenless and the Drosophila EGFR (25–27). Classical genetic studies in Drosophila identified the sevenless RTK and its GEF son-of-sevenless (SOS) through their effect on eye development (25). Mutations in the Gap1, Ras1, sevenless and EGFR genes also lead to defects in eye development and embryo patterning (21,25,26). The Nf1 gene product does not perform a critical function in either of these pathways, probably owing to redundancy of Gap1 and NF1 activity, as Gap1;Nf1 double mutants are lethal (8).
Our study identifies three distinct AC signaling pathways in the Drosophila brain, including a novel growth factor activated NF1/Ras-dependent AC, that remarkably does not require Gαs, as well as two separate neurotransmitter-stimulated AC pathways, one requiring NF1 and Gαs, whereas the other requires Gαs alone. Analysis of the effect of human NF1 mutations and partial deletions, expressed in flies with no NF1, shows that separate domains of NF1 control the different AC pathways. In particular, we show that RasGAP activity of NF1 is necessary for Ras/ NF1-dependent AC signaling but not NF1/Gαs-dependent AC signaling, whereas part of the C-terminal region is sufficient for NF1/Gαs-dependent AC signaling and regulation of body size.
RESULTS
NF1 and Ras activate AC
The first indication that Ras may activate AC was shown by incubation of human H-Ras with Drosophila head membrane extracts to produce a dose- and time-dependent increase in AC activity, as measured by increases in cAMP levels (Fig. 1A). AC activity was also stimulated by human K-Ras (Fig. 1B), but not Rab3a (Fig. 1C), suggesting that activation is specific to the Ras family of small GTPases, and not because of depletion of GTP or other factors. Secondly, this stimulation was shown to be NF1-dependent, as it was eliminated in Nf1 homozygous null mutant flies, Nf1P1 and Nf1P2 (Fig. 1D), which do not express any detectable NF1 protein (8). Furthermore, acute expression of a wild-type Nf1 transgene in the mutant background, controlled by a heat-shock promoter (hsNf1; Nf1P2), was able to fully restore the H-Ras-stimulated AC activation to wild-type levels (Fig. 1D). The acute nature of the response to NF1 indicates that this is not a developmental effect, and that NF1 is a critical component of the Ras-stimulated AC activity.
Figure 1.
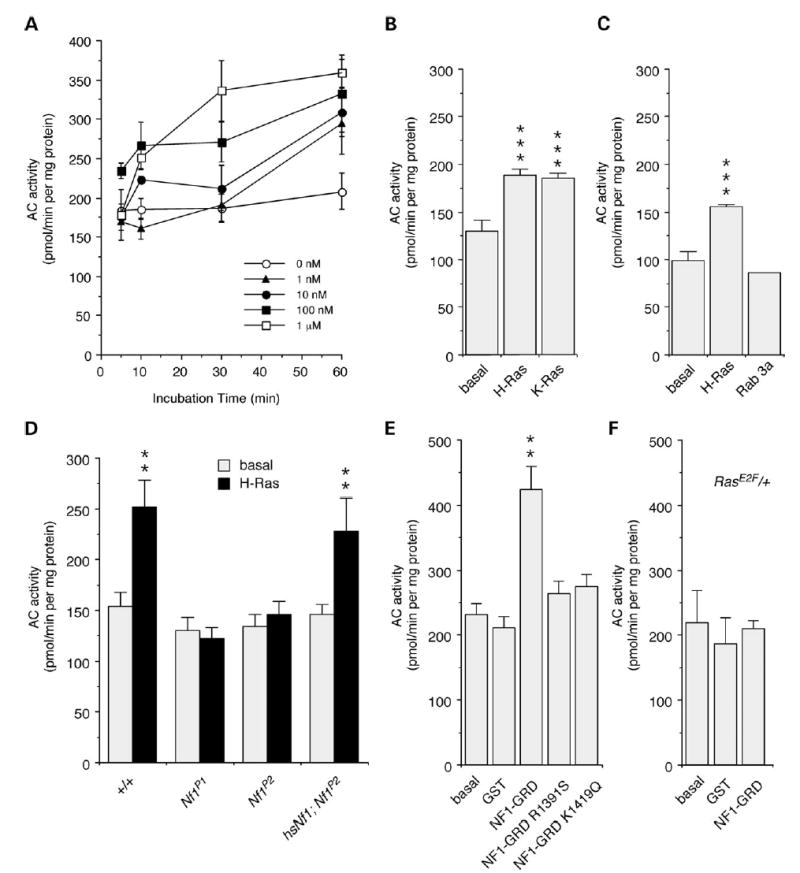
NF1 and Ras activate AC. (A) Significant increases in AC activation were observed after 10 min to 60 min incubation with human H-Ras at different concentrations (P < 0.05; n = 3). (B) Both H-Ras and K-Ras stimulate AC activity (1 mm; t = 30 min; n = 4). (C) Rab3a does not stimulate AC activity (1 mm; t = 30 min; n = 4). (D) H-Ras stimulation of AC was eliminated in Nf1P1 and Nf1P2 mutant flies, and restored by heat-shock induced expression of a fly Nf1 transgene in hsNfI;Nf1P2 flies (1 mm; t = 60 min; n = 8, 8, 8, 3). (E) A human NF1–GRD–GST fusion protein is able to stimulate AC, in the absence of H-Ras. There was no stimulation by GST alone, or by NF1–GRD–GST missense mutants, R1391S and K1419Q, that reduce RasGAP activity (1 mm; t = 30 min; n = 4). (F) Stimulation by human NF1–GRD–GST was abolished in Rase2F/+ heterozygotes (1 mm; t = 30 min; n = 2). (A–F) Values are mean ± SEM (**P < 0.05; ***P < 0.01).
To further define the role of NF1 in Ras-stimulated AC activity we examined the effect of a purified GST-fusion protein containing an NF1-GRD fragment that retains GAP activity (28). Significant increases in AC activity, measured by increased cAMP levels, were shown in wild-type extracts treated with NF1-GRD fusion protein in the absence of H-Ras (Fig. 1E). This effect is specific to the GAP activity of the NF1-GRD fragment, as it is abolished in two NF1-GRD mutants (R1391S; K1419Q; Fig. 1E) with reduced GAP activity, found in NFI patients (28–30). The NF1-GRD fragment was also unable to stimulate AC activity above control levels in Rase2F/+ heterozygotes (Fig. 1F), which have an inactivating mutation in the Switch I region of Ras (25) that normally activates Ras and interacts with downstream effectors. This suggests that levels of active Ras in these heterozygous flies are insufficient to stimulate AC activity, and that endogenous Drosophila Ras can interact with human NF1.
Growth factors stimulate the novel NF1/Ras-dependent AC pathway
To evaluate the functional significance of this novel pathway, we developed an assay to examine effects of neurotransmitters and growth factors on Ras stimulation of AC activity in vivo. Significant stimulation of AC activity was observed in wild-type larval brains treated with EGF or TGFα (Fig. 2A and B). Stimulation of AC activity was abolished in Drosophila EGFR mutants (Fig. 2A and B), including the Egfrt1 hypomorphic mutant and the Df(2R)Egfr18/+ deficiency heterozygote (31), demonstrating that these growth factors are acting directly on the Drosophila EGFR to stimulate AC activity. The stimulation of AC activity by growth factors is also abolished in both Nf1 homozygous null mutants and in Rase1B/+ and Rase2F/+ heterozygotes (Fig. 2A and B). The Rase1B mutation affects the Switch II activator/effector domain of Ras (25) that contacts R1391S of NF1. Again, this demonstrates a requirement for both Ras and NF1 in the stimulation of AC activity.
Figure 2.
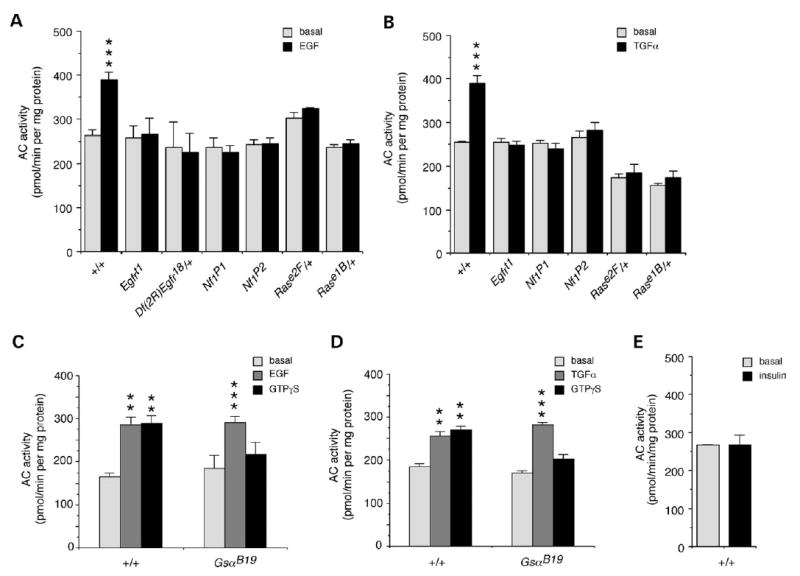
Growth factors stimulate the novel NF1/Ras-dependent AC pathway. (A) AC activity was significantly increased by treatment of larval brains with 2 mm EGF (n = 18). This stimulation was abolished in EGFR mutants, Egfrt1, and heterozygotes, Df(2R)Egfr18/CyO; in Nf1 null mutants, Nf1P1 and Nf1P2; and in Ras heterozygotes, Rase2F/TM3 and Rase1b/TM3 (n = 4). (B) Stimulation of AC by 2 mm TGFα was similarly abolished in the Egfrt1 mutant, Nf1 mutants and Ras heterozygotes (n = 4). Stimulation of AC by 2 mm EGF (C) or TGFα (D) is not affected in a hypomorphic Gαs mutant, GsαB19, whereas stimulation by 20 mm GTPγS is perturbed (n = 3). (E) There was no stimulation of AC by 2 mm insulin (n = 3). (A–E) Values are mean ± SEM (**P < 0.01; ***P < 0.005).
To ensure that there is no crosstalk between EGFR and Gαs, we assayed growth factor stimulation of AC in GsaB19 hypomorphic mutants (32). Normal levels of stimulation of AC activity by both EGF and TGFα growth factors were seen in larval brains of GsaB19 mutants (Fig. 2C and D), consistent with the fact that the Drosophila EGFR does not contain the juxtamembrane domain that facilitates crosstalk in vertebrate EGFRs (33). Stimulation by GTPγS is very low in the GsaB19 mutants (Fig. 2C and D), indicating that Gαs is indeed the major stimulatory G-protein in larval brains. Control treatment of larval brains with insulin did not stimulate AC activity (Fig. 2E). Thus, stimulation of AC by both EGF and TGFα growth factors require EGFR, Ras and NF1, but does not involve Gαs. The identified ligands for the Drosophila EGFR are members of the TGFα family (33). This suggests that stimulation of the Ras/NF1-dependent AC pathway in flies may be activated by binding of endogenous ligands to the EGFR.
Neurotransmitters stimulate two additional AC pathways
We next examined the effects of neurotransmitters and neuromodulators that are ligands for G-protein coupled receptors. Stimulation of AC by the neuropeptide FMRFamide, and by the neurotransmitter dopamine was not affected in Nf1 null mutants or Ras/+ heterozygotes, however, it was abolished in GsaB19 mutants that disturb the classical G-protein signaling pathway (Fig. 3A and B). Thus, alterations in NF1 or Ras that disrupt growth factor-dependent stimulation of AC activity (Fig. 2A and B), do not affect classical G-protein dependent stimulation of AC.
Figure 3.
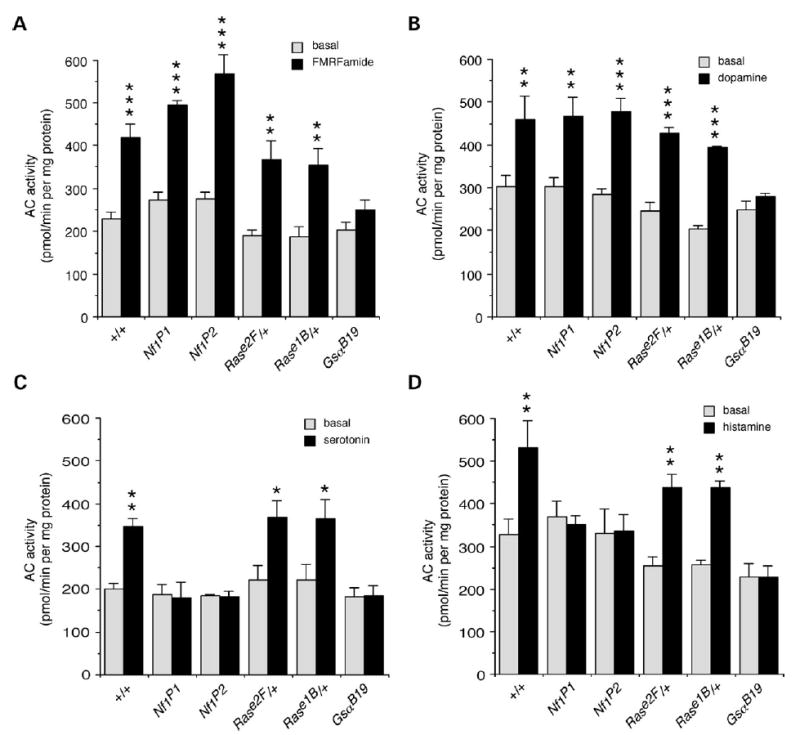
Neurotransmitters and neuromodulators stimulate two additional AC pathways. FMRFamide and dopamine stimulate Gαs-dependent AC: activation of AC by 200 nm FMRFamide (A) and dopamine (B) is disrupted in Gαs mutants, but not in Nf1 mutants or Ras heterozygotes (n = 3–4). Serotonin and histamine however, stimulate NF1/Gαs-dependent AC: activation of AC by 200 nm serotonin (C) and histamine (D) is disrupted in Gαs and Nf1 mutants but not in Ras heterozygotes (n = 4). (A–D) Values are mean ± SEM (*P < 0.05; **P < 0.01; ***P < 0.005).
In contrast, stimulation of AC by the neurotransmitters serotonin and histamine was disrupted in both Nf1 null mutants and GsaB19 mutants but not in Ras/+ heterozygotes (Fig. 3C and D), demonstrating an NF1/Gαs-dependent pathway for stimulation of AC activity that does not require Ras. A number of other neurotransmitters and neuromodulators had no effect on AC activity, including the neuropeptide pituitary AC activating polypeptide neuropeptide (PACAP38) (data not shown), suggesting that there are no receptors for these ligands in the larval brain.
Human NF1 mutations affect MAPK activity in Nf1 mutant flies
To address the possibility that NF1-dependent activation of AC versus downregulation of Ras activity is responsible for the variety of phenotypes seen in NFI patients and animal models, we examined clinically relevant missense mutations from NFI patients that are scattered throughout the length of the hNF1 protein (34–36), as well as deletions of hNF1. We report here the effect of expressing hNF1 containing four different missense mutations and five partial deletions (Fig. 4A) in the Drosophila Nf1 mutant background, which were assayed for their effect on growth factor and neurotransmitter-stimulated AC activity. The mutations chosen for this study occur in multiple patients and affect conserved amino acids (Table 1). When assayed in yeast, the GRD domain mutants R1391S and K1423E drastically reduce GAP activity (29,30,37), whereas the R1276P mutant completely abolishes GAP activity (38). Transcription of UAS-hNF1 transgenes in flies was controlled using Gal4 drivers (39), including the one that is expressed globally (e22c-Gal4); (40) and a nervous system specific driver (elav-Gal4); (41). Assays were performed on flies that carry one copy of the normal or mutant UAS-hNF1 transgene and one copy of the Gal4 driver in the Nf1 mutant background (Fig. 4B and C), showing that hNF1 functions in Drosophila, and defining two separate domains that mediate activation of distinct AC pathways.
Figure 4.
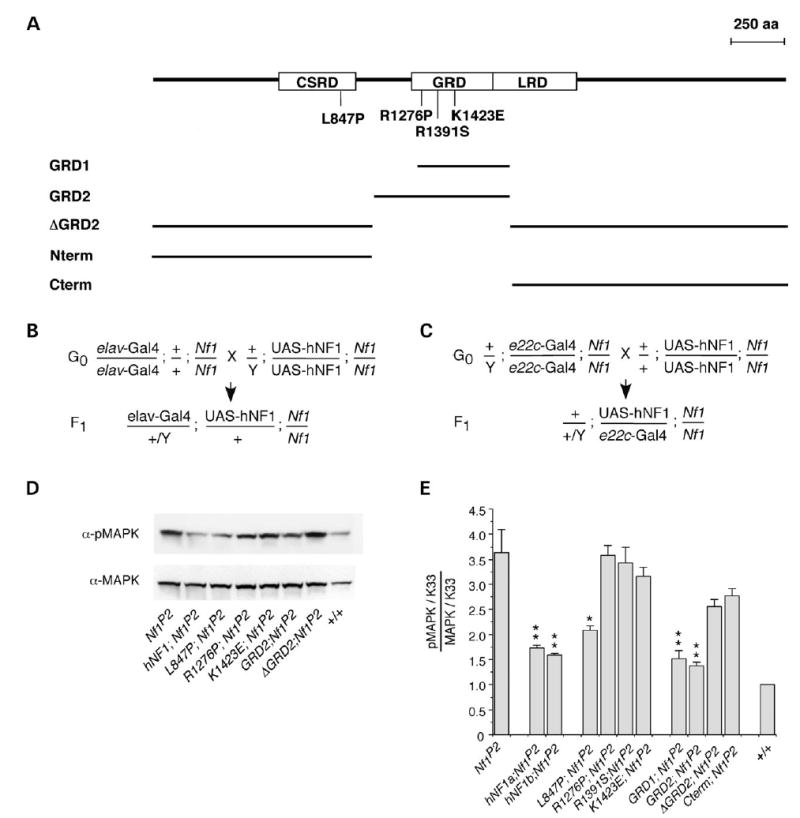
Missense mutations and deletions of human NF1 modulate Drosophila MAPK activity. (A) Position of four hNF1 missense mutations, and size of five hNF1 deletion constructs, that have been expressed and analyzed in Drosophila Nf1 null mutants (CSRD, Cys–Ser-rich domain; GRD, GAP-related domain; LRD, Leu-rich domain). Crosses required to generate F1 progeny expressing UAS-hNF1 mutants or deletion constructs under control of the nervous system specific elav-Gal4 driver (B) on the X chromosome or the globally expressing e22c-Gal4 driver (C) on the second chromosome. (D) Representative western blot of head extracts from flies expressing normal and mutant hNF1s and deletions, probed with anti-phospho-MAPK then stripped and re-probed with anti-MAPK antibodies. (E) Levels of phospho-MAPK versus total MAPK levels in flies expressing hNF1 mutants and deletions, normalized to K33 wild-type (+) control values (see Materials and Methods). (D and E) Expression is under control of the e22c-Gal4 driver. (E) Values are mean ± SEM (*P < 0.05; **P < 0.01; n = 4–6).
Table 1.
Human NF1 missense mutations expressed in Drosophila
| Mutationa | Amino acids conservedb | Effect | Mutagenic primerc | Site added | Number of lines |
|---|---|---|---|---|---|
| Human NF1 | — | Normal | — | — | 4 |
| L847P (2) | gFLcALGGVC | Not known | 5′pTGGAGGCACACTCCCCCAGGTGCACAAAGGAAGCCAGTC3′ | Apa LI | 2 |
| R1276P (1) | MQTLFRGNSL | Abolish GAP activity | 5′pGGCCAAGCTGTTGCCCGGGAA GAGAGTCTGC3′ | Sma I | 1 |
| R1391S (1) | mFLRFINPAI | Reduce GAP activity | 5′pGGCAGGATTGATAAAGCTTAG GAACATGGC3′ | Hind III | 2 |
| K1423E (5) | kLMSKILQsI | Reduce GAP activity | 5′pGATTGGCAATACTCTGCAAGATCTCGGACATTAACTTC3′ | Bgl II | 3 |
Amino acids that are identical in human, mouse and fly are capitalized and the mutated amino acid is underlined.
Restriction site added is underlined and mutated bases are in bold. Primers are complementary to the coding strand of hNF1 and 5′-phosphorylated.
Phosphorylation of mitogen-activated protein kinase (MAPK) is elevated in Drosophila Nf1 mutants because of increased Ras activity (7). We first showed that normal hNF1 is able to inhibit Ras by showing that phospho-MAPK is reduced to wild-type levels when hNF1 (two independent lines; hNF1a and hNF1b) is expressed in Nf1 mutant flies under control of the e22c-Gal4 global driver (Fig. 4D and E). As expected, mutant hNF1s with defective RasGAP activity (R1276P, R1391S, K1423E) or lacking the GRD (ΔGRD2, Cterm) cannot reduce phospho-MAPK levels (Fig. 4E). The GRD fragments alone (GRD1, GRD2) were able to restore phospho-MAPK to wild-type levels, and the L847P mutation did not affect the RasGAP activity of full-length hNF1 (Fig. 4E).
Human NF1 mutations affect AC activity in Nf1 mutant flies
We then demonstrated that the RasGAP activity of hNF1 was required for growth factor-stimulated AC activity, by expressing the mutant hNF1s or deletions under control of the nervous system-specific elav-Gal4 driver for larval brain assays. Mutant hNF1s with defective RasGAP activity, or lacking the GRD, did not respond to EGF stimulation (Fig. 5A and C). However, the L847P mutant and the GRD fragments responded normally to EGF (Fig. 5B and C), indicating that the RasGAP activity of the GRD is indeed required for growth factor-stimulated NF1/Ras-dependent AC activity.
Figure 5.
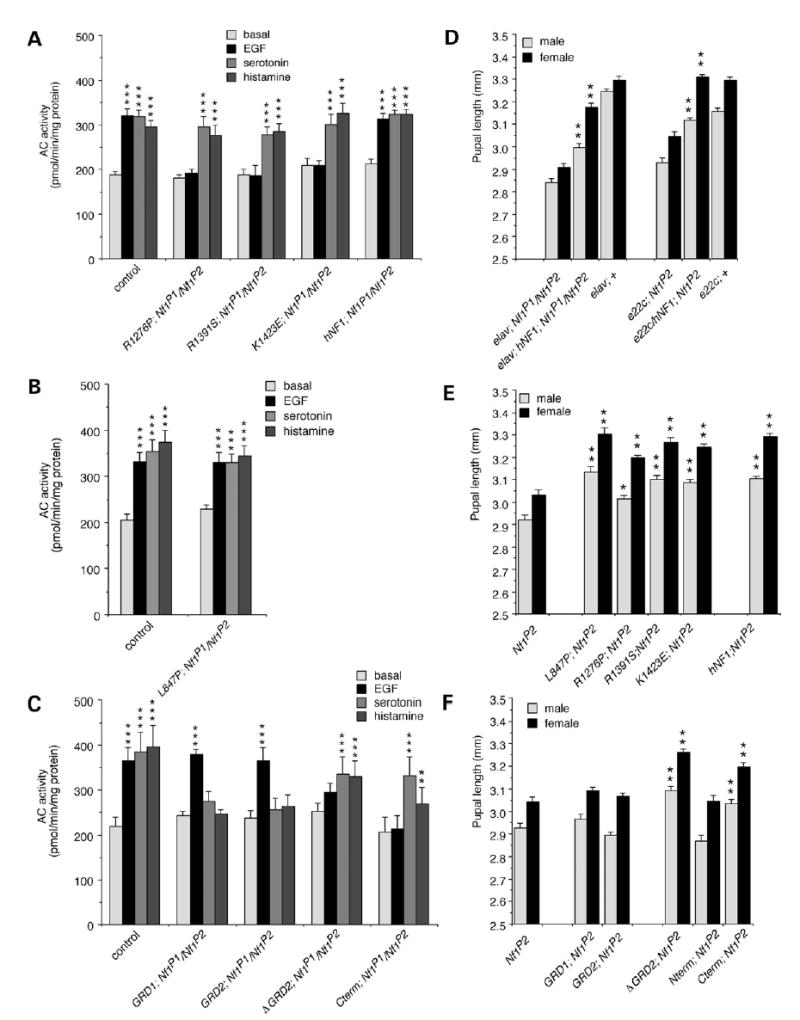
Separate domains of human NF1 mediate activation of different AC pathways. (A) EGF does not stimulate AC activity in flies expressing RasGAP-defective mutant hNF1s (R1276P, R1391S, K1423E), compared with K33 (control) flies or flies expressing normal hNF1, however, serotonin- and histamine-stimulated AC activity is fully restored. (B) Stimulation of AC activity by EGF, serotonin and histamine is restored in flies expressing the L847P hNF1 mutation. (C) EGF stimulated AC activity is restored in lines expressing GRD fragments (GRD1; GRD2), but serotonin- and histamine-stimulated AC activity is absent. Conversely, serotonin and histamine, but not EGF, stimulate AC activity in flies expressing a GRD deletion (ΔGRD2) or a C-terminal fragment (Cterm) alone. (D) Pupal length is increased in flies expressing normal hNF1 using elav-Gal4 or e22c-Gal4 drivers compared with Nf1 mutant and K33 wild-type (+) controls expressing driver alone. (E) Pupal length is also increased in flies expressing all four missense mutations (L847P, R1276P, R1391S or K1423E) compared with Nf1 mutants expressing driver alone. (F) Pupal length is not increased in flies expressing GRD fragments (GRD1; GRD2) or an N-terminal fragment (Nterm), however it is increased in flies expressing a GRD deletion (ΔGRD2) or a C-terminal fragment (Cterm). (A–C) Expression is under control of the elav-Gal4 driver, values are mean ± SEM (**P < 0.01; ***P < 0.001; n = 4). (D–F) Expression is under control of the e22c-Gal4 driver except where otherwise indicated, values are mean ± SEM (*P < 0.01; **P < 0.001; n > 50).
We next examined serotonin- and histamine-stimulated AC activity to see whether RasGAP activity of NF1 was required for the NF1/Gαs-dependent AC pathway. Stimulation of AC was normal for mutant hNF1s with or without RasGAP activity (Fig. 5A and B), indicating that NF1/Gαs-dependent AC activity does not require RasGAP activity. Consistent with this, the GRD fragments alone were not sufficient to restore NF1/Gαs-dependent AC activity (Fig. 5C). We then asked whether any other region of NF1 is required for NF1/Gαs-dependent AC activity. Constructs lacking the GRD (ΔGRD2, Cterm) were able to restore neurotransmitter-stimulated AC activity (Fig. 5C), demonstrating that sequences in the C-terminal region, common to ΔGRD2 and Cterm (Fig. 4A), are essential for NF1/Gαs-dependent AC activity.
Human NF1 mutations also affect body size in Nf1 mutant flies
In order to confirm the physiological relevance of the NF1/Gαs-dependent AC activity, and to verify that RasGAP activity is not required, we examined the effect of expressing the hNF1 mutants and deletions on the small body size phenotype previously seen in adult flies (8). This phenotype can be rescued by supplying cAMP, but not by decreasing Ras activity (8). We first showed that normal hNF1 is able to rescue the small body size of males and females using both elav-Gal4 and e22c-Gal4 drivers (Fig. 5D). All four clinically relevant missense mutants, including those with defective RasGAP activity, are able to rescue body size just as effectively as normal hNF1 (Fig. 5E) and neither of the GRD fragments was able to rescue body size (Fig. 5F). Thus, the RasGAP activity of hNF1 is not required for rescue of body size. Both the GRD deletion and C-terminal fragment were effective at rescuing body size, but not the N-terminal fragment (Fig. 5F). The L847P mutation in the region upstream of the GRD can still rescue MAPK activity (Fig. 4D and E), AC activity (Fig. 5B) and small body size (Fig. 5E). This mutation may affect other aspects of NF1 function such as regulation or localization, rather than activity.
DISCUSSION
Three separate pathways for AC activation defined in this study are depicted in Figure 6. First, a novel pathway for AC activation, downstream of growth factor stimulation of EGFR that requires both Ras and NF1, but not Gαs. Secondly, an NF1/Gαs-dependent AC pathway operating through the Rutabaga-AC (Rut-AC) and stimulated by serotonin and histamine, as observed here in the larval brain. The Rut-AC pathway may also be stimulated by PACAP38 at the larval neuromuscular junction and in adult heads as shown in previous studies (9,10,12). Thirdly, a classical G-protein coupled receptor-stimulated AC pathway (42) operating through Gαs alone. The AC activated by NF1/Ras (AC-X), or Gαs (AC-Y), has not yet been identified.
Figure 6.
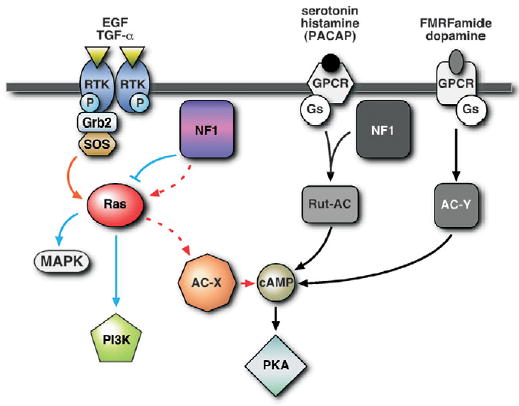
AC can be activated by at least three distinct pathways: First, a novel NF1/Ras-dependent pathway stimulated by growth factors such as EGF and TGFα that activates an unidentified AC (AC-X), and does not involve Gαs; secondly, an NF1/Gαs-dependent pathway, acting through Rutabaga-AC (Rut-AC), stimulated by serotonin and histamine, and possibly PACAP38 (see discussion), that does not require Ras; thirdly, a classical NF1-independent pathway, involving Gαs but not NF1 or Ras, stimulated by FMRFamide and dopamine that activates an unidentified AC (AC-Y).
This study shows for the first time that Ras can stimulate AC in an NF1-dependent manner in higher organisms, via an RTK-coupled pathway that is independent of the Gαs G-protein. The functionality of human NF1 in the fly system, and the high degree of identity between human and fly NF1 (60%); (8), suggests that similar pathways for AC activation may also operate in mammals. Previous studies failed to detect stimulation of AC by Ras in cultured vertebrate cell lines (43), and in Xenopus oocytes (44), however, these cell types may not contain sufficient NF1 to support NF1/ Ras-dependent AC activation. This is consistent with our observation that levels of both Ras and NF1 are critical for stimulation of AC activity in adult head membranes. The reported EGF activation of AC in cardiac myocytes and other tissues requires both Gαs, and the juxtamembrane domain of the EGFR (45,46), which is not present in the Drosophila EGFR (33).
Our experiments with human NF1 mutants show that the GRD domain and the RasGAP activity of NF1 are both necessary and sufficient for growth factor-stimulated NF1/ Ras-dependent AC activity. We also conclude that C-terminal residues downstream of the GRD are critical for both body size regulation and neurotransmitter-stimulated NF1/Gαs-dependent AC activity, thus defining for the first time a region outside the GRD that contributes to this pathway. Interestingly, expression of a human NF1 GRD fragment in Nf1−/− astrocytes results in only partial restoration of NF1-mediated increases in cAMP levels in response to PACAP (13). Thus, regions outside the GRD also seem to be necessary for activation of AC in these mammalian cells.
Thus, NF1, while being a negative regulator of Ras, is also actively involved in stimulation of AC activity. Moreover, it regulates AC activity through at least two different mechanisms, one of which depends on the RasGAP activity of NF1. The multifunctional nature of the NF1 protein illuminates its importance in nervous system development, tumor formation and behavioral plasticity, and may also explain the wide range of clinical manifestations in neurofibromatosis type I.
MATERIALS AND METHODS
Drosophila melanogaster media, strains and heat-shock conditions
Flies were raised at room temperature (22–24°C) on standard cornmeal medium. The Nf1 mutants Nf1P1 and Nf1P2, together with the parental K33 line and hsNf1;Nf1P2 flies were obtained from A. Bernards (Massachusetts General Hospital, Boston, MA, USA). K33 flies used as wild-type controls have a P-element inserted 1.5 kb downstream of the Nf1 locus, that was mobilized to generate the Nf1P1 and Nf1P2 null mutant alleles (8). Nf1P1 deletes most of the Nf1 gene and several downstream genes from the Enhancer of Split locus, whereas Nf1P2 carries a P-element insertion within the first intron of the Nf1 gene, and neither allele produces any detectable NF1 protein (8). Heat-shock induction of NF1 was performed at 35°C for 2 h, then flies were rested at 21–23°C for 1 h. The Rase1B and Rase2F mutants are from the Drosophila Stock Center (Bloomington, IA, USA). Each has an amino acid substitution in either the Switch II or Switch I effector domains, respectively (25). Both affect Ras activation and binding to downstream effectors and are homozygous lethal. The EGFR mutants are also from the Bloomington Stock Center. Egfrt1 is a hypomorph and Df(2R)Egfr18 is a homozygous lethal deficiency (31). Rase1B, Rase2F and Df(2R)Egfr18 heterozygotes carrying a balancer (wild-type) chromosome (TM3 or CyO; 47) were used for all assays. GsαB19 is a hypomorphic mutant (32) provided by M. Forte (Vollum Institute, Portland, OR, USA). Gal4 driver lines: elav-Gal4;Nf1P1 (7) was obtained from A. Sehgal (University of Pennsylvania, Philadelphia, PA, USA); e22c-Gal4 (40) was from N. Perrimon (Harvard Medical School, Boston, MA, USA). White118(isoCJ1) (48) was obtained from T. Tully (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA).
Adult head AC activity assay
The AC activity assay was performed as detailed here, essentially as described by Livingstone (49). About 20 heads/genotype of adult male Drosophila, anaesthetized by chilling, were homogenized in 850 μl lysis buffer (25 mm Tris–acetate buffer at pH 7.5, 1 mm dithiothreitol, 0.01 mg/ml aprotinin and 0.01 mg/ml pepstatin) using a glass pestle. Membranes were recovered by centrifugation at 178 000g for 10 min at 4°C, then re-suspended in 800 μl lysis buffer on ice. Protein concentration (typically 1.5–2.5 mg/ml) was determined using the Bradford Protein Assay (BioRad), and adjusted to 1 mg/ml. Fifty microlitres of 2× assay buffer (50 mm Tris–acetate buffer at pH 7.5, 20 mm MgCl2, 2 mm dithiothreitol, 10 mm creatine phosphate, 200 units/ml creatinine kinase, 0.1 mm cAMP at pH 7.5, 0.2 mg/ml bovine serum albumin, 0.02 mg/ml aprotinin, 0.02 mg/ml pepstatin and fresh 0.2 mg/ml PMSF) was added to 20 μl samples in glass vials on ice. Twenty microlitres Ca2+ solution (2.5 mm CaCl2, 0.25 mg/ml calmodulin and 5 mm EGTA) was then added to samples, bringing the final [Ca2+] to 10−7 m (as calculated using MaxChelator v1.31). Samples were equilibrated at 25°C for 2 min, then H-Ras, K-Ras, Rab3a or GST-NF1-GRD were added (1 nm to 1 μm) to the membrane preparation. After 5–60 min incubation at 25°C, 10 μl radioactive substrate (10 μCi α-P32-ATP, 2 mm ATP) was added, and each sample was incubated at 25°C for a further 10 min. Reactions were stopped by adding 150 μl of stop solution (33 pCi/μl 3H-cAMP, 1.3% SDS, 30 mm ATP, 0.9 mm cAMP at pH 7.5). Then 750 μl water was added and samples were loaded onto 1.5 ml Dowex AG 50W-X4 columns (pre-washed with 10 ml 1 m HCl followed by 10 ml water) and washed with 2 ml of water. Samples were eluted with 4 ml water onto Alumina columns (pre-washed with 10 ml 0.1 m imidazole at pH 7.5) and washed with 1 ml 0.1 m imidazole at pH 7.5. Then samples were eluted with 4 ml 0.1 m imidazole into scintillation vials, and 4 ml scintillation cocktail (Ultima Flow M) was added to each vial. Samples were counted for 5 min in a Beckman LS600IC using two windows 0–400 and 400–1000. Column efficiency was determined by recovery of 3H-cAMP, and the amount of 32P-cAMP produced (pmol/min/mg protein) was calculated taking column efficiency into account. H-Ras and Rab3a were purchased from Sigma. K-Ras was from Merck. Radionucleotides were from Amersham and other chemicals from Sigma. All statistical analyses were performed using the paired student’s t-test. Buffers for all experiments were prepared using Milli-Q purified water (Millipore).
GST fusion protein preparation
Wild-type and mutant NF1-GRD-GST fusion proteins (28) and GST alone were purified using glutathione beads as follows: 1 l cultures of Escherichia coli DH5α cells carrying GST-fusion plasmids were grown in LB plus 100 mg/ml ampicillin at 37°C to log-phase and treated with 1 mm isopropyl-β-d-thiogalactopyranoside for 1 h. Cells were collected and lysed by sonication, at 4°C for six cycles with 20 s each cycle, in 40 ml sonication buffer containing 1 mm EDTA, 1 mm EGTA 0.1% lubrol, 0.1 mm dithiothreitol and protease inhibitor cocktail (Roche). After centrifugation at 15 700g (Beckman JS-13-1) for 30 min at 4°C, ~30 ml of supernatant was added to 1 ml of 50% glutathione beads (Sigma), rotated for 1–2 h at 4°C followed by centrifugation at 735g for 5 min at 4°C. Beads were washed with 10 ml sonication buffer with protease inhibitor cocktail and then washed with 10 ml elution buffer containing 50 mm Tris, 0.5 mm MgCl2 and 0.5 mm dithiothreitol. For elution of the protein, 3 ml of elution buffer was added plus 4.2 mg/ml glutathione (Sigma) and supernatants were collected by centrifugation. Proteins were added to the head membrane extracts, at 1 mm concentration, at different time points as described above for Ras. GST-NF1-GRD fusion constructs were provided by F. Tamanoi (University of California, Los Angeles, CA, USA).
Larval brain AC activity assay
About 30 brains per genotype of third instar larvae were dissected in Drosophila larval saline solution (70 mm NaCl, 5 mm KCl, 10 mm NaHCO3, 115 mm sucrose, 5 mM HEPES, 5 mm trehalose, 1.5 mm CaCl2, 20 mm MgCl2 at pH 7.1) at room temperature. In order to mimic physiological conditions as closely as possible, brains were dissociated manually into individual neurons in 100 μl larval saline, using forceps and pipetting, than separated into control and experimental groups that were kept on ice while other genotypes were dissected. To minimize variability, control and experimental groups in each comparison were always assayed in the same batch. Results generated from such experiments were highly consistent. Experimental groups were incubated at room temperature, with 2 mm growth factors in 10 mm Tris–acetate (pH 7.5) for 5 min, or with 0.2 mm neurotransmitters in 10 mm Tris–acetate for 2 min, as indicated in methods. Controls were treated with 10 mm Tris–acetate alone for 2–5 min. After incubation, cells were centrifuged at low speed (1800g), then re-suspended in 180 μl lysis buffer (see above), and homogenized for 2 min on ice in an Eppendorf tube using a plastic pestle (Kontes). Protein concentration (typically 2–2.5 mg/ml) was determined using the Bradford Protein Assay (BioRad). Fifty microlitres 2× assay buffer (see above) and 20 μl Ca2+ solution (see above) was added to 20 μl samples in glass vials on ice. Samples were equilibrated at 25°C for 2 min, then 10 μl radioactive substrate (see above) was added, and reactions were incubated at 25°C for a further 10 min (growth factors) or 5 min (neurotransmitters). Reactions were stopped, then applied to columns and counted exactly as described for adult heads (see above). Growth factors (mouse EGF, rat TGFα), insulin and neurotransmitters (dopamine, FMRFamide, histamine and serotonin) were purchased from Sigma.
Mutagenesis of hNF1 and cloning of deletion constructs
Clones containing the human NF1 gene were obtained from A. Bernards (Massachusetts General Hospital). The 88:12 clone is a NotI–SalI fragment that contains the entire human NF1 cDNA cloned into NotI–SalI sites of pBluescript (pBSK; Stratagene). The UAS-hNF1 clone contains the NotI–SalI fragment of 88:12 cloned into NotI–XhoI sites of the pUAST vector, destroying both the SalI and XhoI site. For this study, a NotI–XhoI fragment of 88:12 was subcloned into NotI–XhoI cut pBluescript, and a XhoI–KpnI fragment of 88:12 was subcloned into XhoI–KpnI cut pBluescript. Site-directed mutagenesis of the subclones used the Stratagene Chameleon kit with a pBSK-specific phosphorylated selection primer (5′ pCCGCCACCGCGATGTAGCTCCAATTCGC 3′) and mutation-specific mutagenesis primers, which altered a restriction enzyme site in addition to creating the desired clinically identified amino acid mutation (Table 1). Clones were selected by restriction analysis and verified by PCR and sequencing, then mutagenized fragments were digested, gel-purified and ligated into the UAS-hNF1 construct.
Deletion constructs (Fig. 4A) were generated using restriction digests and other enzymes as noted below, and verified by sequencing and PCR. The UAS-GRD2 construct (residues 986–1746, bases 3153–5432) was prepared by subcloning an NheI fragment into the XbaI site of pUAST. The UAS-ΔGRD2 construct (deletion 986–1746) was generated by digesting the UAS-hNF1 clone with NheI to remove bases 3153–5432, then digesting single stranded ends with Mung Bean nuclease (New England Biolabs) and re-ligating to restore the hNF1 reading frame. The UAS-GRD1 construct (residues 1241–1746, bases 3918–5432) was prepared by digesting the UAS-GRD2 construct with XhoI and re-ligating to remove the NheI–XhoI fragment (bases 3153–3917). The UAS-Nterm clone (residues 1–985, bases 198–3152) was prepared by digesting UAS-hNF1 with NheI and XbaI and re-ligating to remove the GRD and C-terminal regions. The UAS-Cterm clone (residues 1748–2843, bases 5433–8717) was prepared by digesting with NotI and NheI, end filling with Klenow and re-ligating the blunt ends to remove the N-terminal and GRD regions.
Transgenic flies
P-element-mediated transformations were performed by injecting the mutated UAS-hNF1 cDNAs and deletion constructs into white118(isoCJ1) (48) Drosophila embryos together with pTURBO as a source of transposase (50). DNA used for injection was prepared using Qiagen kits and checked by PCR and restriction analysis. F1 transformants were identified by eye color and the location of insertions was assayed by crossing to the double balancer line w/Y;CyO/Sp;TM3Ser/Sb (47). Transcription of UAS-hNF1 transgenes in flies was controlled using the global Gal4 driver, e22c-Gal4 and a nervous system-specific X chromosome line, elav-Gal4 (see above). Second chromosome hNF1 insertion lines and Gal4 driver lines were crossed into the NF1P2 mutant background using w/Y;CyO/Sp;TM3Ser/Sb (47) to create doubly homozygous lines with normal or mutant hNF1;Nf1P2 or Gal4 driver;Nf1P2. The crossing schemes designed to generate progeny carrying one copy of the transgene and one copy of the Gal4 driver in the Nf1 mutant background are outlined (Fig. 4B and C). Each of the mutant hNF1s and deletion constructs was tested using multiple Gal4 driver lines (in addition to the two presented here) and multiple insertion lines, except for R1276P for which only one transgenic line could be generated (Table 1).
MAP kinase activity
Flies were collected at the same time each day to minimize circadian differences in phospho-MAPK levels (7). For each genotype, 10 heads were homogenized in 75 μl 1× SDS loading buffer (Invitrogen) plus 0.5 mm dithiothreitol and protease inhibitor cocktail (Roche). Samples were run on precast 10% Tris–glycine gels (Novex, Invitrogen) in 1× Tris–glycine–SDS buffer at 125 V for 2 h. Proteins were transferred to nitrocellulose in 1× Novex buffer plus 20% methanol for 2 h at 25 V. Transfer was verified by Ponceau staining, then blots were blocked in 5% milk/TBST for 1 h at room temperature, rinsed for 3 × 5 min in TBST, then probed with primary antibody diluted 1/500 in 5% milk/ TBST overnight at 4°C. Rabbit polyclonal antibodies to phosphorylated and non-phosphorylated human p44/42 MAPK were obtained from Cell Signaling Technology. Following rinses of 3 × 5 min in TBST, blots were incubated with 1/ 10 000 donkey anti-rabbit HRP-conjugated secondary antibody (Amersham) diluted in 5% milk/TBST for 1 h at room temperature, then rinsed again for 3 × 5 min in TBST, followed by 5 min TBS, prior to detection of signal using the ECL kit (Amersham) and multiple timed exposures to X-ray film. Blots were stripped for re-probing using ReBlot (Chemicon). A representative western blot probed with an anti-phospho-MAPK antibody and then stripped and reprobed with anti-MAPK antibody as shown in Figure 4D. Levels of phospho-MAPK and total MAPK were quantified using the densitometric function of the FluorChem imager (Alpha Inno-tech). After subtraction of in-lane background, levels of phospho-MAPK and total MAPK were normalized relative to control K33 wild-type samples (+) run in parallel on each gel (Fig. 4D). The ratio of phospho-MAPK to MAPK was determined and the results of four to six independent experiments are graphed (Fig. 4E). Expression of full-length hNF1 was confirmed by western blot using a rabbit polyclonal antibody sc-68 (Santa Cruz) directed against the C-terminal domain of human NF1 (data not shown).
Body size measurement
The normal hNF1 gene has been shown to partially rescue AC-dependent small body size defects when expressed in the Nf1 mutant background, using the global Gal4 drivers armadillo-Gal4 and e22c-Gal4 (12). In order to improve the statistical power of our body size analysis, we separated males and females for pupal size measurements in this study, as the large difference in body size between the sexes may mask the effects of the transgenes. Body size was assayed by measuring the length of late stage 10 pupae (eye pigments visible); (51) with a digital micrometer (Mitutoyo). Pupae were placed into a 96-well-plate and their sex determined after eclosion of adults. At least 50 pupae of each sex were measured and statistical significance was assessed using a paired student’s t-test.
Acknowledgments
We thank Andre Bernards for providing Nf1 mutant flies and human NF1 clones. We also thank both Inessa Hakker and Kimberley Weinreich for their excellent technical assistance in the generation of transgenic Drosophila lines and mutant hNF1s. This work was supported by a National Neurofibromatosis Foundation Young Investigator Award (F.H.), and by grants from the National Institutes of Health to Y.Z. (NIH 2R01 NS34779-06) and from the US Army Neurofibromatosis Research Program to F.H. and Y.Z. (DAMD17-99-1-9500), and to F.H. (W81XWH-04-1-025).
Conflict of Interest statement. The authors declare that they have no conflict of interest regarding this manuscript.
References
- 1.Riccardi VM. Genotype, malleotype, phenotype, and randomness: lessons from neurofibromatosis-1 (NF-1) Am J Hum Genet. 1993;5:301–304. [PMC free article] [PubMed] [Google Scholar]
- 2.North KN, Riccardi V, Samango-Sprouse C, Ferner R, Moore B, Legius E, Ratner N, Denckla MB. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121–1127. doi: 10.1212/wnl.48.4.1121. [DOI] [PubMed] [Google Scholar]
- 3.Gutmann DH. Learning disabilities in neurofibromatosis 1: sizing up the brain. Arch Neurol. 1999;56:1322–1323. doi: 10.1001/archneur.56.11.1322. [DOI] [PubMed] [Google Scholar]
- 4.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 6.Costa RM, Silva AJ. Mouse models of neurofibromatosis type I: bridging the GAP. Trends Mol Med. 2003;9:19–23. doi: 10.1016/s1471-4914(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 7.Williams JA, Su HS, Bernards A, Field J, Sehgal A. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science. 2001;293:2251–2256. doi: 10.1126/science.1063097. [DOI] [PubMed] [Google Scholar]
- 8.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 9.Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 10.Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. 2000;403:895–898. doi: 10.1038/35002593. [DOI] [PubMed] [Google Scholar]
- 11.Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci. 2001;15:1110–1116. doi: 10.1523/JNEUROSCI.21-04-01110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- 13.Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–8954. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viskochil D, White R, Cawthon R. The neurofibromatosis type 1 gene. Annu Rev Neurosci. 1993;16:183–205. doi: 10.1146/annurev.ne.16.030193.001151. [DOI] [PubMed] [Google Scholar]
- 15.DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 16.Ingram DA, Hiatt K, King AJ, Fisher L, Shivakumar R, Derstine C, Wenning MJ, Diaz B, Travers JB, Hood A, et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med. 2001;194:57–69. doi: 10.1084/jem.194.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa RM, Yang T, Huynh DP, Pulst SM, Viskochil DH, Silva AJ, Brannan CI. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat Genet. 2001;27:399–405. doi: 10.1038/86898. [DOI] [PubMed] [Google Scholar]
- 18.Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- 19.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1990;49:4682–4689. [PubMed] [Google Scholar]
- 20.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim Biophys Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 21.Gaul U, Mardon G, Rubin GM. A putative Ras GTPase activating protein acts as a negative regulator of signaling by the Sevenless receptor tyrosine kinase. Cell. 1992;68:1007–1019. doi: 10.1016/0092-8674(92)90073-l. [DOI] [PubMed] [Google Scholar]
- 22.Powe AC, Jr, Strathdee D, Cutforth T, D’Souza-Correia T, Gaines P, Thackeray J, Carlson J, Gaul U. In vivo functional analysis of Drosophila Gap1: involvement of Ca2+ and IP4 regulation. Mech Dev. 1999;81:89–101. doi: 10.1016/s0925-4773(98)00230-5. [DOI] [PubMed] [Google Scholar]
- 23.Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog Nucl Acid Res Mol Biol. 2002;71:391–444. doi: 10.1016/s0079-6603(02)71047-7. [DOI] [PubMed] [Google Scholar]
- 24.Rommel C, Hafen E. Ras–a versatile cellular switch. Curr Opin Genet Dev. 1998;8:412–418. doi: 10.1016/s0959-437x(98)80111-1. [DOI] [PubMed] [Google Scholar]
- 25.Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell. 1991;67:701–716. doi: 10.1016/0092-8674(91)90065-7. [DOI] [PubMed] [Google Scholar]
- 26.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 27.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 28.Kim, M.R. and Tamanoi, F. (1998) Neurofibromatosis 1 GTPase activating protein-related domain and its functional significance. In Upadhyaya, M. and Cooper, D.N. (eds.), Neurofibromatosis Type 1 from Genotype to Phenotype Bios Scientific Publishers, Oxford, UK, Washington, DC, pp. 89–112.
- 29.Gutmann DH, Boguski M, Marchuk D, Wigler M, Collins FS, Ballester R. Analysis of the neurofibromatosis type 1 (NF1) GAP-related domain by site-directed mutagenesis. Oncogene. 1993;8:761–769. [PubMed] [Google Scholar]
- 30.Upadhyaya M, Osborn MJ, Maynard J, Kim MR, Tamanoi F, Cooper DN. Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet. 1997;99:88–92. doi: 10.1007/s004390050317. [DOI] [PubMed] [Google Scholar]
- 31.Clifford R, Schupbach T. Molecular analysis of the Drosophila EGF receptor homolog reveals that several genetically defined classes of alleles cluster in subdomains of the receptor protein. Genetics. 1994;137:531–550. doi: 10.1093/genetics/137.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfgang WJ, Hoskote A, Roberts IJ, Jackson S, Forte M. Genetic analysis of the Drosophila Gs(alpha) gene. Genetics. 2001;158:1189–1201. doi: 10.1093/genetics/158.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogdan S, Klambt C. Epidermal growth factor receptor signaling. Curr Biol. 2001;11:R292–R295. doi: 10.1016/s0960-9822(01)00167-1. [DOI] [PubMed] [Google Scholar]
- 34.Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000;66:790–818. doi: 10.1086/302809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–555. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Serra E, Ars E, Ravella A, Sanchez A, Puig S, Rosenbaum T, Estivill X, Lazaro C. Somatic NF1 mutational spectrum in benign neurofibromas: mRNA splice defects are common among point mutations. Hum Genet. 2001;108:416–429. doi: 10.1007/s004390100514. [DOI] [PubMed] [Google Scholar]
- 37.Poullet P, Lin B, Esson K, Tamanoi F. Functional significance of lysine 1423 of neurofibromin and characterization of a second site suppressor which rescues mutations at this residue and suppresses RAS2Val-19-activated phenotypes. Mol Cell Biol. 1994;14:815–821. doi: 10.1128/mcb.14.1.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klose A, Ahmadian MR, Schuelke M, Scheffzek K, Hoffmeyer S, Gewies A, Schmitz F, Kaufmann D, Peters H, Wittinghofer A, Nurnberg P. Selective disactivation of neurofibromin GAP activity in neurofibromatosis type 1. Hum Mol Genet. 1998;7:1261–1268. doi: 10.1093/hmg/7.8.1261. [DOI] [PubMed] [Google Scholar]
- 39.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 40.Duffy JB, Harrison DA, Perrimon N. Identifying loci required for follicular patterning using directed mosaics. Development. 1998;125:2263–2271. doi: 10.1242/dev.125.12.2263. [DOI] [PubMed] [Google Scholar]
- 41.Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 42.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 43.Beckner SK, Hattori S, Shih TY. The ras oncogene product p21 is not a regulatory component of adenylate cyclase. Nature. 1985;317:71–72. doi: 10.1038/317071a0. [DOI] [PubMed] [Google Scholar]
- 44.Birchmeier C, Broek D, Toda T, Powers S, Kataoka T, Wigler M. Conservation and divergence of RAS protein function during evolution. Cold Spring Harbor Symp Quant Biol. 1985;50:721–725. doi: 10.1101/sqb.1985.050.01.089. [DOI] [PubMed] [Google Scholar]
- 45.Sun H, Seyer JM, Patel TB. A region in the cytosolic domain of the epidermal growth factor receptor antithetically regulates the stimulatory and inhibitory guanine nucleotide-binding regulatory proteins of adenylyl cyclase. Proc Natl Acad Sci USA. 1995;92:2229–2233. doi: 10.1073/pnas.92.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stryjek-Kaminska D, Piiper A, Zeuzem S. Epidermal growth factor regulates adenylate cyclase activity via Gs and Gi1-2 proteins in pancreatic acinar membranes. Biochem J. 1996;316:87–91. doi: 10.1042/bj3160087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindsley, D.L. and Zimm, G.G. (1992) The Genome of Drosophila melanogaster. Academic Press, San Diego, CA, USA.
- 48.Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 49.Livingstone MS. Genetic dissection of Drosophila adenylate cyclase. Proc Natl Acad Sci USA. 1985;82:5992–5996. doi: 10.1073/pnas.82.17.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 51.Bainbridge SP, Bownes M. Staging the metamorphosis of Drosophila melanogaster. J Embryol Exp Morphol. 1981;66:57–80. [PubMed] [Google Scholar]