

Summary
Individuals with sickle cell disease (SCD) may have oxyhaemoglobin desaturation during the steady-state, the causes of which are incompletely known. We studied a cohort of 585 children who have sickle cell anaemia (SS), sickle β0-thalassaemia (Sβ0), sickle-haemoglobin C disease (SC), or sickle β +-thalassaemia (Sβ+) to determine the relationships between steadystate oxyhaemoglobin saturation (SpO2) and SCD genotype, age, gender, steady-state haemoglobin (Hb) and reticulocyte count, and rate of acute chest syndrome (ACS). The SS/Sβ0group (n = 390) had lower mean SpO2 than the SC/Sβ+ group (n = 195) (96.3% vs. 98.7%, P < 0.001). Among SS/Sβ0subjects, a decrease in steady-state SpO2 correlated with a decrease in Hb, an increase in reticulocytes, older age and male gender. These correlations were not found in the SC/Sβ+ group. Prior ACS did not correlate with steady-state SpO2. A multivariate model explained 45% of the variability in SpO2, but only 5% of the variation in SpO2 was explained by Hb. We conclude that steady-state desaturation is common in individuals with SCD, but it appears to be unrelated to prior episodes of ACS and largely unexplained by chronic anaemia.
Keywords: sickle cell disease, pulse oximetry, hypoxaemia, children
Many individuals with sickle cell disease (SCD) have arterial oxyhaemoglobin desaturation during the steady-state in the absence of overt cardiopulmonary illness (Homi et al, 1997). Steady-state desaturation in SCD is partly caused by a rightward shift of the oxyhaemoglobin dissociation curve because of the properties of sickle haemoglobin (Hb S) in solution (Seakins et al, 1973; Ueda et al, 1979) and the effects of chronic anaemia mediated through 2,3-bisphosphoglycerate (Milner, 1974). Desaturation is not a universal finding among individuals who have homozygous sickle cell anaemia (SS), however, and the causes of inter-patient variability are incompletely known.
Prior studies have consistently shown a direct correlation between steady-state haemoglobin (Hb) concentration and steady-state oxyhaemoglobin saturation measured by pulse oximetry (SpO2) (Rackoff et al, 1993; Homi et al, 1997; Setty et al, 2003). Past studies also concur that SpO2 is lower, on average, among individuals who have SS compared with those who have sickle-haemoglobin C disease (SC) (Rackoff et al, 1993; Homi et al, 1997; Setty et al, 2003). Another cause of steady-state desaturation could be chronic cardiopulmonary disease caused by recurrent episodes of acute chest syndrome (ACS), yet prior studies provide contradictory evidence for such an association (Rackoff et al, 1993; Homi et al, 1997). Reported correlations between SpO2 and age, gender, and reticulocyte count have also been contradictory or not corroborated by all investigators (Rackoff et al, 1993; Homi et al, 1997; Setty et al, 2003).
Therefore, we aimed to resolve the discrepancies in the literature by using multivariable statistical techniques in a large cohort of children with SCD. We developed a model to determine the relative contributions to steady-state SpO2 of multiple factors, namely a history of ACS, steady-state Hb, steady-state reticulocyte count, age, gender and SCD genotype. We hypothesised that steady-state Hb concentration did not explain most of the variation in SpO2 and that a history of ACS could explain some of this variability.
Patients and methods
This study is a retrospective analysis of our centre’s existing comprehensive SCD database. We identified every subject in this database who: (i) was less than 20 years of age; (ii) had a diagnosis of either SS, SC, sickle-β+-thalassaemia (Sβ+), or sickle-β0-thalassaemia (Sβ0); (iii) had at least one outpatient clinical evaluation during the preceding 5 years; and (iv) had a documented steady-state Hb concentration, reticulocyte count (%), and SpO2. The Institutional Review Board of UT Southwestern Medical Center approved the use of the database for this project. The majority of these subjects were members of the previously described Dallas Newborn Cohort (Quinn et al, 2004). Individuals who were currently receiving chronic red blood cell transfusions were excluded from the analysis. All steady-state values were calculated as rolling averages of at least three measurements obtained during routine ‘well’ or ‘steadystate’ clinic visits. Steady-state SpO2 was measured in room air by trained nurses and technicians using the Nellcor N-395 pulse oximeter (Nellcor Puritan Bennett Inc., Pleasanton, CA, USA).
We searched the database for the clinical diagnoses, ‘acute chest syndrome’, ‘pneumonia’ and abbreviations or permutations thereof that were recorded at the time of the events. In our clinical practice, we defined ACS as an acute pulmonary illness in a person who has SCD that is characterised by a new radiographic pulmonary infiltrate and some combination of fever, hypoxaemia, thoracic pain, and signs and symptoms of respiratory illness. Lifetime rates of ACS were calculated for a large subset of subjects with SS and Sβ0 (n = 183). This subset included only the children who were ≥5 years of age, and rates were calculated using only episodes of ACS that occurred after the third birthday. This level of stringency was used because ACS in the infant or very young child is typically mild, transient and related to viral respiratory infections (Vichinsky et al, 1997, 2000). The period of follow-up for the rate calculation was the interval between the third birthday and the last documented clinical encounter or the date of initiation of a disease-modifying therapy (hydroxyurea or stem cell transplantation). Thus, all rates were calculated for a minimum of 2 years of follow-up in children who were 5 years of age or older.
We combined subjects into two groups for study: (i) SS or Sβ0 and (ii) SC or Sβ+. We combined the genotypes in this way because of the known clinical similarity of the grouped diseases, the very small number in the Sβ0 subgroup (n = 9), and because exploratory analyses showed the subjects within each group to be similar. For descriptive statistics, we calculated mean values, standard deviations and frequency histograms for continuous variables and proportions for categorical variables. We used the t-test, chi-squared test or Fisher’s exact test, where appropriate, to compare groups of subjects (SS/Sβ0 vs. SC/Sβ+; male vs. female). Steady-state SpO2 was defined as the dependent variable and steady-state Hb, steady-state reticulocyte count, age, gender, SCD genotype and ACS rate as independent or potential predictor variables. Pearson correlation was used to investigate relationships between SpO2 and the potential predictor variables and to assess for multi-collinearity between variables. Independent variables for multivariate (regression) analysis were selected based on clinical importance and statistical significance found by bivariate analyses. A linear regression model was used to determine the significant predictors of steady-state SpO2 and their adjusted associations. Partial correlation coefficients were computed to describe the linear relationship between SpO2 and Hb while controlling for the effects of other variables simultaneously. All data were analysed using spss 11.0 statistical software (SPSS Inc., Chicago, IL, USA). P-values <0.05 were considered significant.
Results
Descriptive and bivariate analysis
There were 585 subjects for analysis (Table I). Fig 1 depicts the distributions of steady-state SpO2 by SCD genotype. The results of bivariate analyses are presented in Table II and Fig 2. Among SS/Sβ0 subjects, there were significant correlations between SpO2 and Hb (Fig 2, panel A), reticulocyte count and age (Fig 2, panel C). On the contrary, none of these correlations was found to be significant among SC/Sβ+ subjects (Table II; Fig 2, Panel B). Among the subset of subjects with SS/Sβ0 whose lifetime ACS rates were calculated, no significant correlation was found between ACS rate and SpO2 (Fig 2, panel D). The mean SpO2 of SS/Sβ0 males was lower than females (96.0% vs. 96.7%, P = 0.014), while the SpO2 of SC/Sβ+ subjects did not differ significantly between genders (P = 0.770).
Table I.
Characteristics of subjects.
| SS/Sβ0 | SC/Sβ+ | P-value | |
|---|---|---|---|
| Number | 390 | 195 | – |
| Number of males (%) | 216 (55.4) | 96 (49.2) | 0.16 |
| Mean age, years (SD) | 9.5 (5.7) | 9.2 (5.2) | 0.476 |
| Mean Hb, g/dl (SD) | 8.2 (1.2) | 10.7 (0.9) | <0.001 |
| Mean SpO2 in % (SD) | 96.3 (3.0) | 98.7 (1.7) | <0.001 |
| Percentage with SpO2 <96% | 33.1 | 3.6 | <0.001 |
| Percentage with SpO2 <90% | 2.8 | 0.5 | 0.070 |
SS/Sβ0, sickle cell anaemia/sickle β0-thalassaemia; SC/Sβ+, sickle-haemoglobin C/sickle β+-thalassaemia; SpO2, steady-state oxyhaemoglobin saturation; SD, standard deviation.
Fig 1.
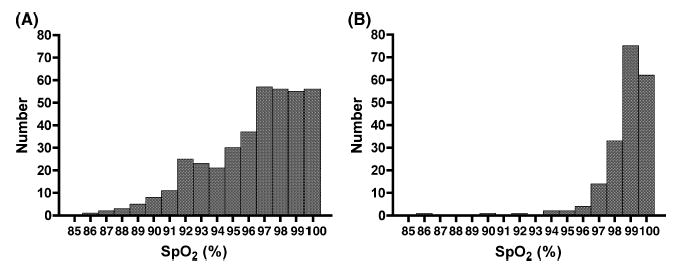
Histograms of steady-state oxyhaemoglobin saturation, SpO2 by sickle cell disease genotype. Panel (A) depicts subjects with either sickle cell anaemia or sickle β0-thalassaemia. Panel (B) depicts subjects with either sickle-haemoglobin C or sickle β+-thalassaemia.
Table II.
Bivariate correlations between SpO2 and other variables by genotype.
| SS/Sβ0 |
SC/Sβ+ |
|||
|---|---|---|---|---|
| Pearson R | P-value | PearsonR | P-value | |
| Steady-state Hb | 0.517 | <0.001 | 0.024 | 0.736 |
| Age | −0.550 | <0.001 | −0.068 | 0.346 |
| Reticulocyte count | −0.443 | <0.001 | −0.055 | 0.445 |
| ACS rate | −0.043 | 0.559 | – | – |
| Mean SpO2
|
P-value
|
Mean SpO2
|
P-value
|
|
| Gender (m/f) | 95.99/96.74 | 0.014 | 98.67/98.74 | 0.770 |
SS/Sβ0, sickle cell anaemia/sickle β0-thalassaemia; SC/Sβ+, sickle-haemoglobin C/sickle β+-thalassaemia; SpO2, steady-state oxyhaemoglobin saturation; ACS, acute chest syndrome.
Fig 2.
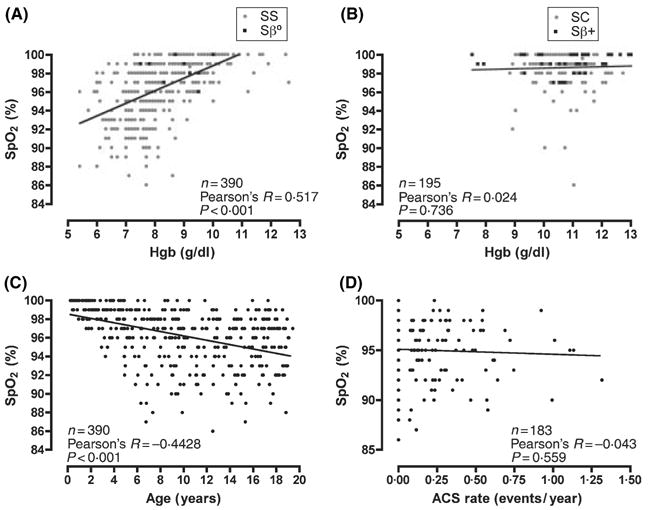
Bivariate correlations between steady-state oxyhaemoglobin saturation (SpO2) and other variables by sickle cell disease genotypes. Panel (A) shows the correlation between Hb (Hgb) and SpO2 among sickle cell anaemia (SS, grey circles) and sickle β0-thalassaemia (Sβ0, black squares) subjects. Panel (B) shows the correlation between Hb (Hgb) and SpO2 among sickle-haemoglobin C (grey circles) and sickle β+-thalassaemia (Sβ+, black squares) subjects. Panel (C) illustrates the correlation between SpO2 and age among SS and Sβ0 subjects. Panel (D) depicts the lack of correlation between SpO2 and rate of acute chest syndrome among SS and Sβ0 subjects.
Multivariate analysis
After testing for the required assumptions, standard multiple regression analysis was performed between SpO2 as the dependent variable, and Hb, age, gender, and the reticulocyte count as independent variables (Table III). The residual analysis suggested that the distribution was reasonably close to normal, linear and homoscedastic. ACS rate was not included in the model as an independent variable because of a lack of linear relationship with the outcome variable. Both Hb and reticulocyte count were included as independent variables in the model because they did not correlate strongly enough with each other to qualify for the multi-collinearity assumption (R = −0.587).
Table III.
Predictors of SpO2 from standard multiple regression analysis by genotype.
| SS/Sβ0 |
SC/Sβ+ |
|||||
|---|---|---|---|---|---|---|
| β | Partial R2 | P-value | β | Partial R2 | P-value | |
| Intercept | 94.244 | – | <0.001 | 98.532 | – | <0.001 |
| Hb (g/dl) | 0.584 | 0.053 | <0.001 | 0.032 | <0.001 | 0.837 |
| Age (years) | −0.156 | 0.125 | <0.001 | −0.019 | 0.003 | 0.458 |
| Gender (female) | 0.643 | 0.019 | 0.006 | 0.125 | 0.001 | 0.615 |
| Reticulocytes (%) | −0.196 | 0.122 | <0.001 | 0.062 | 0.003 | 0.476 |
| n = 390,model R2 = 0.448 | n = 195, model R2 = 0.009 | |||||
The multivariate analysis performed on SC/Sβ+ subjects did not yield any significant results (Table III). Among SS/Sβ0 subjects, all four of the independent variables contributed significantly to prediction of SpO2 (Table III). A multiple correlation coefficient (R = 0.67) showed a significant linear relationship between independent variables and steady-state SpO2 (F = 78.07, P < 0.001). The estimated model for SS/Sβ0 subjects could be given as:
Considering ‘gender’ in the equation, females were assigned a value of 1 and males a value of 0; thus, the multivariate model estimated that females, on average, had an SpO2 that was 0.64% higher (absolute increase) than males. This small difference between males and females was statistically signifi- cant in both bivariate (P = 0.014) and multivariate (P = 0.006) analyses.
In summary, among SS/Sβ0 subjects, a decrease in steadystate SpO2 was related to a decrease in Hb concentration, increase in reticulocyte count, older age, and male gender. Altogether, about 45% (adjusted 44%) of the variability in SpO2 could be explained by these independent variables in the model. Notably, only 5% of the variation in SpO2 was explained byHb while controlling for other variables (Table III).
Discussion
In this largest study to date of pulse oximetry measurements in a cohort of children with SCD, we showed that steady-state desaturation is common among individuals who have SS or Sβ0 and relatively uncommon among those who have SC or Sβ+, consistent with prior studies (Rackoff et al, 1993; Homi et al, 1997; Setty et al, 2003). We also confirmed the observation of a direct relationship between Hb and SpO2 (Homi et al, 1997; Setty et al, 2003). However, we clearly showed that only 5% of the variation in steady-state SpO2 of subjects with SS or Sβ0 is explained by steady-state Hb concentration alone. This new observation indicates that the effects of chronic anaemia do not primarily explain steady-state desaturation in SCD. Setty et al 2003 found a correlation between SpO2 and reticulocytosis. Likewise, we have now shown that 12% of the variation in SpO2 is explained by the reticulocyte count. Hb and reticulocyte count inversely correlated with each other, of course, although not strongly enough in this analysis to qualify for the multicollinearity assumption. Thus, reticulocytosis exerts an effect on SpO2 independent of Hb concentration, perhaps indicating a difference in the shapes of the oxyhaemoglobin dissociation curves between Hb within reticulocytes and Hb within mature erythrocytes.
Another possible explanation for steady-state desaturation is subclinical or chronic cardiopulmonary disease. Recurrent episodes of ACS have been associated with chronic lung disease (Powars et al, 1988). As such, steady-state desaturation might be an indicator of pulmonary injury, and individuals who have suffered more frequent episodes of ACS might be expected to be more hypoxaemic in the steady-state. Rackoff et al 1993 studied 86 children with SS and found that a history of ACS was associated with desaturation after the age of 5 years; however, Homi et al 1997 studied 220 children with SS and found no association between ACS and SpO2 after correction for Hb. We have shown here, in 183 subjects that the rate of ACS, at least during childhood, is not associated with SpO2 (Fig 2, panel D). Thus, reasons other than cumulative ACSrelated pulmonary injury must explain steady-state desaturation. This conclusion may be limited because it is possible that the rate of ACS is an insensitive predictor of chronic lung disease, and rather that the age of onset or the severity of individual episodes of ACS might be fundamental. We did not test this hypothesis because no reliable instruments to grade the severity of ACS have been reported. Also, because we studied episodes of ACS that were diagnosed according to the expert clinical practice at a single centre and not a protocolspecific definition, the ascertainment of ACS cases may have been incomplete or inaccurate. However, the crude incidence of ACS in this study, 18.4 episodes per 100 patient-years, is similar to the overall incidence of 12.8 per 100 patients reported for SS patients in the Cooperative Study of Sickle Cell Disease (Castro et al, 1994).
Here, we demonstrated that males are more likely to have a lower SpO2 than females, a finding not reported in past studies (Rackoff et al, 1993; Homi et al, 1997; Setty et al, 2003). The effect of gender on SpO2 was small and probably not clinically significant, but the difference was statistically significant in both bivariate (P = 0.014) and multivariate analysis (P = 0.006). Although the frequency of some complications of SCD may differ between sexes, we did not investigate any potential biological explanations for this interesting finding.
We also showed that the degree of steady-state desaturation increased with age. Much of the age-related decline in SpO2 occurs in the first 5 years of life (Fig 2, panel C). Because foetal Hb (Hb F) has higher O2 affinity than Hb S, and because the decline of Hb F to its steady-state level may take 2–5 years (Serjeant & Serjeant, 2001), the effect of age on SpO2 could be explained by the normal developmental decline in Hb F concentration. Indeed, Rackoff et al 1993 found a similar association with age, but it was not significant when Hb F was also included in the model. We did not systematically measure the Hb F level in our subjects to test this hypothesis, but there was no statistically significant association between SpO2 and age among children 5 years of age and older (data not shown). Other investigators (Homi et al, 1997; Setty et al, 2003) found no association between age and SpO2, perhaps because they studied individuals with SCD who were 9–18 and 3–19 years of age, respectively, and did not include the youngest children in whom we found the strongest correlation with age.
It is tempting to speculate that slowly worsening steady-state desaturation could be a marker of developing pulmonary hypertension, which is increasingly recognised as a common and life-threatening complication of young adults with SS (Gladwin et al, 2004). The development of pulmonary hypertension is thought to be related to the degree of haemolysis and secondary endothelial dysfunction (Morris et al, 2005). We found that both Hb and reticulocyte count, indicators of haemolysis, were associated with desaturation, but we did not investigate any relationship between pulmonary hypertension and desaturation in this study.
Whatever its cause, desaturation is increasingly recognised as a marker or predictor of certain vaso-occlusive complications of SCD. For example, nocturnal hypoxaemia is associated with higher rates of pain in childhood (Hargrave et al, 2003) and an increased likelihood of central nervous system events (strokes, transient ischemic attacks and seizures) in children and young adults (Kirkham et al, 2001). Setty et al 2003 studied biologic correlates of oxyhaemoglobin saturation to explain these associations (Setty et al, 2003). They found an inverse relationship between saturation and both the degree of erythrocyte– endothelial adhesion and the expression of markers of white blood cell, platelet and endothelial activation. Thus, desaturation may promote vaso-occlusive complications through hypoxia-mediated pathways. Several authors have suggested that screening for and appropriate management of nocturnal hypoxaemia might decrease the frequency of pain and stroke (Kirkham et al, 2001; Hargrave et al, 2003). It is not known whether daytime desaturation, studied here, has similar prognostic significance or if any intervention is needed.
It has long been assumed that cumulative ACS-related pulmonary injury and the effects of chronic anaemia on the oxyhaemoglobin dissociation curve are the causes of steadystate desaturation in SCD. Although we showed that steadystate Hb and SpO2 were directly correlated, we found that only a small fraction (5%) of the variation in SpO2 was explained by Hb concentration alone. Notably, we also demonstrated that past ACS is not associated with steady-state SpO2. Steady-state desaturation is clearly a complex phenomenon with multiple causes. Nevertheless, our multivariate model can explain nearly half of the variation in SpO2. This model provides mechanistic insights to steady-state desaturation and the ability to predict its occurrence in individuals who have SCD. Because of the association of hypoxaemia with vaso-occlusive complications, further study of the role of desaturation as a cause or consequence of SCD-related morbidity is needed.
Acknowledgments
We wish to thank Drs Zora R. Rogers and George R. Buchanan for their critical review of the manuscript. This work was supported by a grant from the National Institutes of Health (U54 HL 70588) (C.T.Q.).
References
- Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette P, Vera JC, Levy PS. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643–649. [PubMed] [Google Scholar]
- Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- Hargrave DR, Wade A, Evans JP, Hewes DK, Kirkham FJ. Nocturnal oxygen saturation and painful sickle cell crises in children. Blood. 2003;101:846–848. doi: 10.1182/blood-2002-05-1392. [DOI] [PubMed] [Google Scholar]
- Homi J, Levee L, Higgs D, Thomas P, Serjeant G. Pulse oximetry in a cohort study of sickle cell disease. Clinical and Laboratory Haematology. 1997;19:17–22. doi: 10.1046/j.1365-2257.1997.00215.x. [DOI] [PubMed] [Google Scholar]
- Kirkham FJ, Hewes DK, Prengler M, Wade A, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet. 2001;357:1656–1659. doi: 10.1016/s0140-6736(00)04821-2. [DOI] [PubMed] [Google Scholar]
- Milner PF. Oxygen transport in sickle cell anemia. Archives of Internal Medicine. 1974;133:565–572. [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysisassociated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powars D, Weidman JA, Odom-Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine. 1988;67:66–76. [PubMed] [Google Scholar]
- Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease. Blood. 2004;103:4023–4027. doi: 10.1182/blood-2003-11-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackoff WR, Kunkel N, Silber JH, Asakura T, Ohene-Frempong K. Pulse oximetry and factors associated with hemoglobin oxygen desaturation in children with sickle cell disease. Blood. 1993;81:3422–3427. [PubMed] [Google Scholar]
- Seakins M, Gibbs WN, Milner PF, Bertles JF. Erythrocyte Hb-S concentration. An important factor in the low oxygen affinity of blood in sickle cell anemia. Journal of Clinical Investigation. 1973;52:422–432. doi: 10.1172/JCI107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serjeant GR, Serjeant BE. Sickle Cell Disease. Oxford University Press; New York, NY: 2001. [Google Scholar]
- Setty BN, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. Lancet. 2003;362:1450–1455. doi: 10.1016/S0140-6736(03)14689-2. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Nagel RL, Bookchin RM. An increased Bohr effect in sickle cell anemia. Blood. 1979;53:472–480. [PubMed] [Google Scholar]
- Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787–1792. [PubMed] [Google Scholar]
- Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, Nickerson B, Orringer E, McKie V, Bellevue R, Daeschner C, Manci EA. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. New England Journal of Medicine. 2000;342:1855–1865. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]