

Abstract
In order to identify small regions of the genome whose specific copy number alteration is associated with high genomic instability in the form of overall genome-wide copy number aberrations, we have analyzed array-based comparative genomic hybridization (aCGH) data from 33 sporadic colorectal carcinomas. Copy number changes of a small number of specific regions were significantly correlated with elevated overall amplifications and deletions scattered throughout the entire genome. One significant region at 9q34 includes the c-ABL gene Another region spanning 22q11–13 includes the breakpoint cluster region (BCR) of the Philadelphia chromosome Coordinate 22q11–13 alterations were observed in nine of eleven tumors with the 9q34 alteration Additional regions on 1q and 14q were associated with overall genome-wide copy number changes, while copy number aberrations on chromosome 7p, 7q, and 13q21.1–31.3 were found associated with this instability only in tumors from patients with a smoking history Our analysis demonstrates there are a small number of regions of the genome where gain or loss is commonly associated with a tumor’s overall level of copy number aberrations Our finding BCR and ABL located within two of the instability-associated regions, and the involvement of these two regions occurring coordinately, suggests a system akin to the BCR-ABL translocation of CML may be involved in genomic instability in about one-third of human colorectal carcinomas.
Keywords: genomic instability, colorectal cancer, array-based CGH
1. Introduction
Genomic instability is widely observed in colorectal carcinomas, occurring in diverse independent forms and generating various degrees of genomic damage [1–3]. Genomic instability arising from endogenous or exogenous sources is generally viewed as essential to the development of solid tumors, generating a wide variety of different genetic events of which a few tend to be selected for during the course of tumor progression [1,4,5] Reports of instances unable to detect genomic instability in tissue culture and animal cancer models of cancer, or in individual human tumor cases, have led to the suggestion that such instability may not be necessary for tumor development, although such failures to detect instability alternatively may reflect incompleteness of the types of instability measurements utilized in the particular studies [6,7].
Microsatellite instability and aneuploidy are now relatively well understood, while the genetic and molecular mechanisms that generate intrachromosomal genomic instability in sporadic cancers remain largely unclear Wang et al have reported a large scale screening of 100 candidate genes; about ten percent of the colorectal tumors examined contained mutations in one of five genes, from three different cellular pathways [8] In family cancer syndromes, the underlying defective genes function principally through destabilization of the genome, yet outside of APC, p53 and mismatch repair genes, these same genes are rarely found mutated in sporadic cancers [9] In sporadic colorectal cancer, a patient’s smoking history affects both the nature and extent of genomic damage within the tumor [10].
Comparative genomic hybridization (CGH) on ordered BAC-Arrays provides a high throughput, high resolution screening method that can be used to quantitatively measure DNA copy number aberrations in solid tumors, and map such alterations directly on the genome This method of genome scanning has been used to search for genomic regions frequently involved in producing solid tumors including breast, colon, brain, esophagus, and kidney [11–18].
BAC array CGH excels for rapid genome-wide characterization of amplifications and deletions; by quantifying the overall extent of these amplifications and deletions, we have been able to measure that one form of genomic instability, and then identify specific regions whose alteration is associated with the overall instability This approach also has some limitations in the types of instability detected, in that it is unable to sense instability events significantly smaller than the ~150 kilobase size of an individual BAC clone, and is unable to detect inversions, translocations and other events not impacting copy number Here we report the results of a CGH BAC-array analysis of 33 sporadic colorectal tumors in various stages, and center our analyses using multiple statistical methods to identify specific regions whose copy number alteration is correlated with the quantitative trait of overall genome-wide genomic instability in each tumor.
2 Materials and Methods
2.1. Tumor tissues and DNA Extraction
The patient set and colorectal tumors used for these studies are described in Table 1 With the exception of excluding from analysis those tumors falling in the MSI-H category, the tumors were randomly selected from a larger set described previously which had been collected consecutively from patients presenting to the gastrointestinal surgery service at the Roswell Park Cancer Institute during the period 1991–1999 [19] (One MSI tumor was examined, and showed mininal alterations on array-CGH.) For the patient set analyzed in the present study, the following parameters apply: 18 male, 14 female; 18 current or former cigarette smokers, 14 patients never smoked; the mean age was 62.4 (SD 12.3) Tumor stages were: stage 0, 1; stage I, 6; stage II, 8; stage III, 7; stage IV, 10 Tissue processing and DNA extraction methods have been described in detail [20]
Table 1.
Characteristics of Patients and Tumors
| Tumor # | Patient Age | Gender | Smoking History** | Tumor Stage |
|---|---|---|---|---|
| 3023 | 63 | M | 0 | 1 |
| 3031 | 58 | F | 2 | 1 |
| 3033 | 61 | M | 1 | 4 |
| 3043 | 69 | M | 1 | 3 |
| 3118 | 65 | F | 0 | 1 |
| 3120Δ | 36 | F | 2 | 1 |
| 3125 | 49 | F | 1 | 4 |
| 3127 | 80 | M | 1 | 2 |
| 3137 | 71 | F | 0 | 0 |
| 3141 | 72 | F | 1 | 4 |
| 3143 | 70 | M | 0 | 3 |
| 3147 | 77 | F | 0 | 4 |
| 3153 | 66 | M | 1 | 3 |
| 3155 | 71 | M | 0 | 2 |
| 3157 | 76 | F | 0 | 1 |
| 3177 | 76 | F | 0 | 4 |
| 3180 | 59 | F | 1 | 2 |
| 3182 | 61 | M | 1 | 3 |
| 3187 | 54 | F | 0 | 3 |
| 3199 | 44 | M | 1 | 2 |
| 3213 | 59 | M | 1 | 3 |
| 3936 | 79 | M | 0 | 2 |
| 6374 | 83 | F | 0 | 2 |
| 6376 | 72 | F | 0 | 1 |
| 6378 | 72 | M | 0 | 1 |
| 6386 | 45 | M | 0 | 4 |
| 6388 | 60 | M | 2 | 2 |
| 6392 | 66 | F | 1 | 4 |
| 12008 | 38 | M | 0 | 2 |
| 12018 | 41 | F | 0 | 4 |
| 12020 | 48 | M | 2 | 4 |
| 12024 | 48 | M | 0 | 4 |
| 12026 | 45 | M | 0 | 3 |
0, never smoked; 1, former smoker; 2, current smoker
Microsatellite unstable tumor, excluded from further analysis.
2.2. BAC-Array Comparative Genomic Hybridization
DNA labeling, hybridization and subsequent image analysis were carried out exactly according to the Nowak method detailed previously [21].
2.3 Statistical Analysis of microarray data
A loess corrected log2 ratio of the background subtracted test/control was calculated for each clone to compensate for non-linear raw aCGH profiles in each sample BAC clones with less than two replicates passing standard quality control steps were excluded from the analysis The mode adjusted median log2 test/control (ALR, short for adjusted log ratio ALR) value was calculated for each BAC clone by subtracting the mode of a non-parametric density estimate of all spots in that array from the median of the loess corrected log2 ratios for the spots that passed the quality control steps Regions with common copy number means were identified by segmenting the genome, using the DNAcopy software [22,23] The ALR median absolute deviations (MAD) were then calculated for the BACs on each segment, and the median of the MAD score (MMAD) was taken across all segments All BACs with an ALR median absolute deviation a MAD value greater than 4 were identified as outliers and assigned a call of gain or loss only if the ALR value were was either greater than 5 x MMAD or less than –5 x MMAD, respectively Segments with a median ALR value greater (less) than 1.0 (−1.0) x MMAD were then assigned a status of gain (loss) BACs that were not assigned a status of gain or loss in this step were assigned a status of “normal” and the standard deviation of the ALR values (SD-ALR) associated with these BACs was calculated.
The relationship between the genome wide percentage of BACs exhibiting copy number aberrations and BAC specific copy number aberrations crossed with smoking status was assessed using ANOVA based F-statistics For the hthBAC the following model was fit:
where αCNA,hi ∈{0,1,2} for the hth BAC in the ith patient exhibiting normal, loss, or gain in relative copy number, respectively, and βS / NS, hi ∈{0,1} depending on the smoking status of the ith patient. For each BAC with a maximum of 5 missing observations across patients, two models were fit: (model 1) yhi was set equal to the percentage of BACs across the balance of the genome exhibiting a relative gain in copy number for the ith patient, and (model 2) yhi was set equal to the percentage of BACs across the balance of the genome exhibiting a relative loss in copy number for the ith patient For each model the ‘balance of the genome’ was taken to be all autosomes excluding the chromosome containing the hth BAC A single step MaxT algorithm [22] was implemented to control the family wise error rate (FWER) associated with the genome scans corresponding the fitting of each model to the eligible BACs (i.e., BACs with a maximum of 5 missing observations) Specifically, 10,000 datasets were generated wherein the mapping of yhi the values were permuted with respect to the patient IDs For each of the permuted datasets, the specified models were fit for all eligible BACs and the maximum F statistics corresponding to each model were recorded Empirical 95% thresholds were calculated using the 9500th ordered maximum F- statistic for each model Observed F-statistics exceeding the thresholds were considered to be significant.
The following reduced models were fit for BACs that were deemed to be significant:
The significance of interaction and, if warranted, main effect terms were assessed via the comparison of the appropriate sums of squares across nested models.
3. Results
We scanned the genome for regions that were correlated with a tumor’s overall level of genomic instability as revealed by array-CGH, in order to help us learn where the gene(s) underlying this form of genomic instability might be located To accomplish this, we: (i) utilized array CGH (aCGH) to identify regions of relative gain and loss in copy number, (ii) fit a series of linear models to detect relationships between local copy number aberrations (CNAs) and the relative amount of copy number aberrations occurring on the balance of the genome, and (iii) interrogated the statistically significant results in the previous step for biological meaningfulness
Table 2 provides a listing of BACs correlated with overall genomic instability in the form of copy number aberrations at an adjusted p-value <0.10 Of the BACs listed, only those located on 7p yielded model fits which contained interaction terms that were significant with a type I error rate (marginal) of 0.05 Models associated with BACs located on 7q, 13q, and 14q contained main effect terms for smoking status which were significant at a marginal level of 0.05 A smoking status effect was significant at a marginal level of 0.05 for a single BAC located on chromosome 22, (RP11-1113I2), but was significant at a marginal level of 0.10 for several other BACs on that chromosome The BAC CNA (i.e., αCNA,hi ) main effect was significant at a marginal level of 0.05 for all BACs listed in Table 2
Table 2.
Individual BAC clones whose loss or gain are significantly correlated to genome-wide amplification, deletion, or the patient's smoking history*
| RP11- clone | chromosome | F statistic | Adjusted p-values | Interaction p-values | Smoking main p-values | Gain/Loss/Either | Chromosome Region (bp) | candidate genes in region |
|---|---|---|---|---|---|---|---|---|
| 422P24 | 1q21.3 | 7.806 | 0.060 | 0.400 | 0.373 | Either | 148,810,010–150,938,155 | S100 protein family |
|
| ||||||||
| 525G13 | 1q23.3 | 12.997 | 0.008 | 0.998 | 0.115 | Gain | 162,588,522–162,784,903 | UCK2 |
|
| ||||||||
| 544B14 | 1q32.1 | 13.209 | 0.007 | 0.105 | 0.199 | Gain/Either | 203,329,259–203,409,712 | IL10, IL19 |
|
| ||||||||
| 115P16 | 8.086 | 0.084 | 0.651 | 0.231 | ||||
| 520H14 | 1q42.1-q43 | 7.404 | 0.122 | 0.698 | 0.280 | Gain | 222,516,739–228,034,708 | RHOU, ADPRT, TSNAX, CAPN9, WNT3A, WNT9A |
| 12H2 | 8.086 | 0.084 | 0.651 | 0.231 | ||||
|
| ||||||||
| 375H24 | 1q42.13 | 7.608 | 0.109 | 0.619 | 0.253 | Gain/Either | 223,281,925–224,613,365 | WNT3A, WNT9A |
|
| ||||||||
| 334N15 | 2q24.3 | 8.741 | 0.059 | 0.450 | 0.757 | Gain | 167,814,597–169,454,975 | STK39 |
|
| ||||||||
| 204J18 | 3p14.2 | 7.920 | 0.091 | 0.124 | 0.404 | Gain | 60,927,060–62,456,006 | FHIT, PTPRG, ID2B |
|
| ||||||||
| 90K6 | 3q13.2 | 8.520 | 0.066 | 0.322 | 0.234 | Gain | 113,221,207–114,199,281 | CD200 |
|
| ||||||||
| 240H8 | 10.016 | 0.029 | 0.064 | 0.029 | ||||
| 160L16 | 10.016 | 0.029 | 0.064 | 0.029 | ||||
| 66O14 | 10.144 | 0.028 | 0.049 | 0.041 | ||||
| 627P22 | 10.016 | 0.029 | 0.064 | 0.029 | ||||
| 600K23 | 10.016 | 0.029 | 0.064 | 0.029 | ||||
| 54N19 | 10.016 | 0.029 | 0.064 | 0.029 | ||||
| 16G1 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 95N10 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 115G23 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 609L3 | 8.742 | 0.059 | 0.040 | 0.068 | ||||
| 138E20 | 8.398 | 0.071 | 0.043 | 0.064 | ||||
| 87M15 | 7p21.1–p11.2 | 9.033 | 0.051 | 0.040 | 0.060 | Gain/Either | 19,394,202–55,274,090 | EGFR, IL6, HUS1, INHBA, GLI3, VRAL, MYCLK1, IGFBP3 |
| 85E16 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 75O22 | 7.523 | 0.114 | 0.044 | 0.082 | ||||
| 121A8 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 462D19 | 8.457 | 0.069 | 0.133 | 0.050 | ||||
| 100C21 | 9.146 | 0.048 | 0.031 | 0.078 | ||||
| 111K18 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 137P13 | 9.146 | 0.048 | 0.031 | 0.078 | ||||
| 84O18 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 832D10 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 81B20 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 339F13 | 9.033 | 0.051 | 0.040 | 0.060 | ||||
| 97P11 | 7.167 | 0.138 | 0.071 | 0.030 | ||||
|
| ||||||||
| 251I15 | 7p11.2 | 10.772 | 0.011 | 0.036 | 0.047 | Either | 55,274,090–55,781,496 | |
|
| ||||||||
| 86P18 | 7.957 | 0.090 | 0.203 | 0.048 | ||||
| 92J13 | 7.957 | 0.090 | 0.203 | 0.048 | ||||
| 78C11 | 7q31.1-q31.32 | 7.957 | 0.090 | 0.203 | 0.048 | Gain | 106,955,503–121,128,048 | MET, WNT2, CYR61, ING3, DOCK4, CBLL1 |
| 110C11 | 7.957 | 0.090 | 0.203 | 0.048 | ||||
| 95L16 | 7.957 | 0.090 | 0.203 | 0.048 | ||||
| 140O21 | 7.957 | 0.090 | 0.203 | 0.048 | ||||
|
| ||||||||
| 247A12 | 8.307 | 0.049 | 0.660 | 0.129 | ||||
| 409K20 | 9q34.11-q34.13 | 8.307 | 0.049 | 0.660 | 0.129 | Loss | 128,543,247–130,681,562 | ABL1 |
| 618A20 | 8.307 | 0.049 | 0.660 | 0.129 | ||||
|
| ||||||||
| 173B14 | 7.316 | 0.082 | 0.440 | 0.006 | ||||
| 318G21 | 13q21.1-q31.3 | 7.316 | 0.082 | 0.440 | 0.006 | Either | 73,769,921–81,136,723 | MYCBP2 |
| 600P1 | 8.348 | 0.043 | 0.548 | 0.002 | ||||
|
| ||||||||
| 238I24 | 14q24.3-qter | 10.815 | 0.012 | 0.708 | 0.052 | Loss | 101,461,362–106,368,585 | XRCC3, MARK3, ASPP1, AKT1, HSPCA |
| 815P21 | 10.815 | 0.012 | 0.708 | 0.052 | ||||
|
| ||||||||
| 1057H19 | 11.557 | 0.008 | 0.376 | 0.085 | ||||
| 22M5 | 11.680 | 0.007 | 0.291 | 0.150 | ||||
| 297B9 | 11.680 | 0.007 | 0.291 | 0.150 | ||||
| 389H23 | 11.680 | 0.007 | 0.291 | 0.150 | ||||
| 540P6 | chr22 | 11.680 | 0.007 | 0.291 | 0.150 | Loss | CHK2, BCR, TOP3B, p300, NF2, ST13, TIMP3 | |
| 1078O11 | 13.257 | 0.003 | 0.266 | 0.063 | ||||
| 395B24 | 13.257 | 0.003 | 0.266 | 0.063 | ||||
| 1021O19 | 13.211 | 0.003 | 0.266 | 0.063 | ||||
| 1113I2 | 9.728 | 0.021 | 0.402 | 0.034 | ||||
Gain = associated with genome-wide copy number amplification
Loss = associated with genome-wide copy number deletion
Either = associated with genome-wide copy number aberration
BACs correlated with overall genomic instability (estimated proportions of copy number aberrations) at an adjusted p-value <0.10. F-statistics correspond to the full model fit described in the methods section. The adjusted p-values are calculated with respect to the adjusted distribution (obtained via permutation) of the maximum F statistic. The p-values listed for smoking status by BAC interactions and smoking status main effects are marginal. Values significant at p<0.05 are in bold. The adjusted p-values are calculated with respect to the permutation distribution discussed in the methods section. Interaction and smoking main effect p-values correspond to the un-adjusted p-values associated with the f-tests for the comparison of the full model to reduced model 1 and the comparison of reduced model 1 to reduced model 2, respectively.
We also found five cases where altered clones were associated with the total number of genome-wide aberrations, be it gain or loss. Three of the regions (on chromosomes 1 and 7) were associated with genome-wide amplifications One of the other regions, chromosome 13q21.1-q31.3, has been seen as a common site for somatic deletions in a variety of malignant tumors Interestingly, of the 12 tumors showing alteration of this region, only two showed deletions The other clone (RP11-422P24) is located on chromosome 1q21.3, an early-replicating region that is highly amplified in human sarcomas and contains genes frequently amplified or overexpressed in gastroesophageal junction carcinomas [24] Of the thirteen tumors showing altered copy numbers of this clone, four were amplified and nine were deleted Both cases suggest that the association may not be causative, but rather the region itself is highly susceptible to change after an instability phenotype is established and may include genes necessary for tumor progression but not for the actual induction of genome instability The S100A proteins located at 1q21.3 are overexpressed in the majority of gastric cancers, independently indicating that they may be important for gastric tumorigenesis [25].
Regions were defined using the nearest clones proximal and distal to the clones of interest For example, on our array chip, the nearest clones to the chromosome 9 set of three clones were RP11-550J21, located 391Kb upstream of RP11-247A12, and RP11-83J21, located 429Kb downstream of RP11-618A20 Since these flanking clones did not show the significant level of deletion, they became the boundaries, defining our region of interest as 2.14Mb total.
Chromosomes 9 and 22
Figure 1 shows a graphical representation of significant BACs on chromosome 9 (figure 1a) and chromosome 22 (figure 1b). The dashed horizontal lines correspond to the empirical 95% thresholds calculated as described in the methods section; bold points above the dashed line correspond to statistically significant BACs (adjusted p-value <0.05) Plots located in the right column of Figure 1 contain the % CNA values segregated by smoking status (points offset to the left for non-smokers and to the right for smokers) and by relative copy number state for a given BAC.
Figure 1.
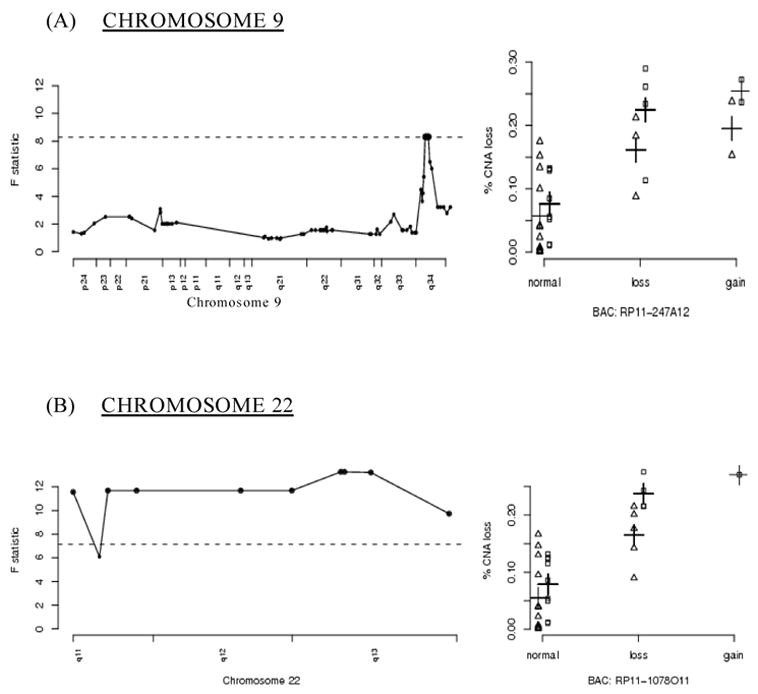
A graphical representation of significant BACs located on chromosomes 9 and 22 The x-axes for figures located in the left column represent the genomic location of BACs in that particular chromosome; the y-axes for these figures correspond to the F-statistic values for the overall model The x-axes for the figures located in the right column represent the relative copy number state of the identified BACs; the y-axes for these figures correspond to the estimated rates of overall copy number aberration (% CNA) on the balance of the genome Horizontal dashed lines correspond to the empirical 95% thresholds; highlighted points correspond to statistically significant BACs The BACs in the right-hand columns are representative of the significant regions of the corresponding chromosomes to their left (e.g. RP11-247A12 for chromosome 9q34) Points in the right hand column are offset according to smoking status: non-smokers (Δ) and smokers ([]) The crosses (+) indicate the fitted means.
The small deleted region encompassing three highly significant BAC clones on chromosome 9q, and three adjoining clones that nearly reach statistical significance, includes the c-ABL gene ABL is an attractive candidate for being a major factor in genomic instability, in that it forms a complex with ATM following genotoxic stress, resulting in the blockade of DNA repair, cell cycle arrest, and/or the induction of apoptosis [26] ABL also phosphorylates the DNA repair proteins DNA-PK and Rad51 [27].
The association of genomic instability with the 22q11 to 22q13 region may be due to its structure, and not by encoding a particular gene product This region is known to contain many low copy repeat sequences prone to rearrange and recombine, and constitutional rearrangements involving this breakpoint cluster region are known to be associated with a number of genomic disorders [28] Rearrangements in this region are associated with other cancers; the best known is the Philadelphia chromosome of CML (and ALL), where 9q34 is translocated with 22q11 [29] Gains and losses of these regions of chromosomes 9 and 22 for each colorectal tumor are listed in Table 3.
Table 3.
Coordinate involvement of 9q34 and 22q11-13 in colorectal carcinomas, listed by assigned tumor number*.
| chromosome 9q34 | chromosome 22q | ||||
|---|---|---|---|---|---|
| copy loss | copy gain | normal | copy loss | copy gain | normal |
| 3023 | 3127 | 3033 | 3023 | 3199 | 3033 |
| 3031 | 3147 | 3118 | 3031 | 3118 | |
| 3043 | 3199 | 3125 | 3043 | 3125 | |
| 3141 | 12026 | 3137 | 3127 | 3137 | |
| 3157 | 3143 | 3141 | 3143 | ||
| 3182 | 3153 | 3157 | 3153 | ||
| 3187 | 3155 | 3187 | 3155 | ||
| 3177 | 12026 | 3177 | |||
| 3180 | 3180 | ||||
| 3213 | 3182 | ||||
| 3936 | 3213 | ||||
| 6374 | 3936 | ||||
| 6376 | 6374 | ||||
| 6378 | 6378 | ||||
| 6388 | 6388 | ||||
| 6392 | 6392 | ||||
| 12008 | 12008 | ||||
| 12018 | 12018 | ||||
| 12020 | 12020 | ||||
| 12024 | 12024 | ||||
Copy number changes of 9q34 and 22q11–13 were determined as gains or losses using the statistical criteria defined in the Methods Tumor numbers are as in table 1.
We investigated if something akin to the rearrangement in the Philadelphia chromosome might be occurring in colorectal cancers, not necessarily as a simple translocation per se, but in more general terms of coordinate involvement of the two regions Of the ten colorectal tumors that showed copy number aberrations of the region on 9q34, nine also showed coordinate aberration on chromosome 22q11–13, and none of the twenty tumor DNAs with no copy number aberration of the 9q34 reqion showed any alteration in the 22q11–13 region (table 4) This association was highly significant, with a Fisher’s exact test p< 0.000001; this result indicates that some form of coordinate involvement of the two regions is occurring.
Table 4.
Coordinate arrayCGH Events on Chromosomes 9q and 22q*
| altered 9q34 | normal 9q34 | |
|---|---|---|
| altered 22q | 9 | 0 |
| normal 22q | 1 | 19 |
This table summarizes the results presented in Table 3.
The general features of the cases described in Table 4 were very similar to the patient set of all cases studied The mean age was 64.1 (12.1), compared with 62.4 (12.3) for the whole set Tumor stages I-II-III-IV partitioned as 3/2/3/3, compared with 6/8/7/10. Gender was 7 male and 4 female, compared with 18 male and 14 female Smoking status of never-former-current was 6/5/0, compared with 18/11/3. Patient outcomes did not significantly differ for tumors with the involvement of 9 and 22.
Chromosome 7
Amplification of a relatively large region of chromosome 7p was also correlated with genome-wide amplification (figure 2a) If amplification of the entire region were advantageous in the progression to malignancy, then one would expect to see these regions amplified in the majority of tumors, not just those with elevated genome-wide amplification rates Nearly two-thirds of the tumors assayed showed amplification of this region of 7p (20/31 tumors). Of these twenty showing the amplified region, fifteen were from patients with a smoking history.
Figure 2.
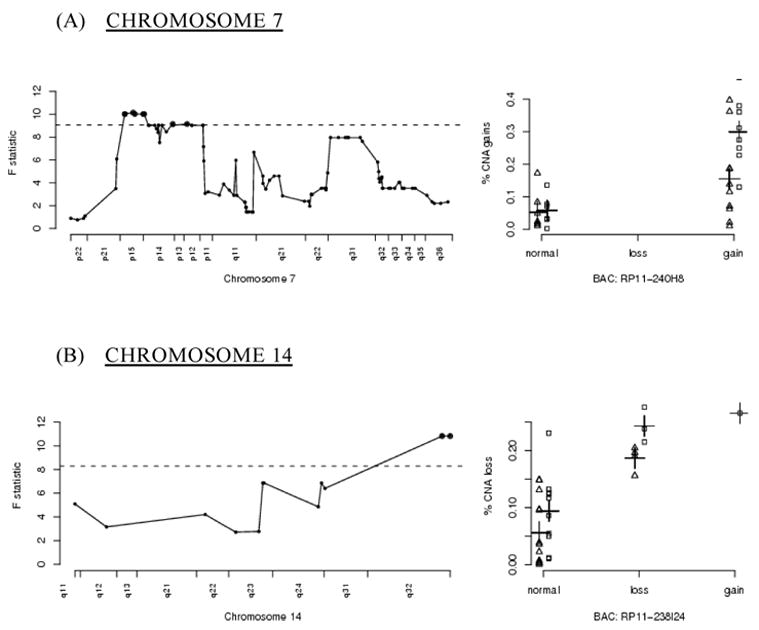
A graphical representation of significant BACs located on chromosomes 7 and 14, otherwise as in figure 1 As seen in the right hand panel for chromosome 7, the patients’ smoking histories particularly impact the selection of amplifications and deletions on chromosome 7.
Aberrant expression of several genes simultaneously can allow for a more efficient progression to malignancy Frequent amplification of this region could be due to the selection of an event advantageous for progression to malignancy, reflecting targeted instability This model’s fit for the BACs in this region on 7p contain statistically significant (at level 0.05) interaction terms comprised of smoking and aberration state of 7p The models show that tumors with copy number gains of the 7p BACs exhibit an increase in genomic gains when compared to tumors that do not contain gains of those BACs; the increase in genomic gains is a function of smoking status, as smokers exhibit a higher rate of genome-wide instability [10].
HUS1 lies within the amplified region at 7p13; considerable evidence ties HUS1 with genomic instability This gene is essential for a functional DNA damage checkpoint response, and cells lacking HUS1 are hypersensitive to ionizing radiation and UV [30,31]. Weiss et al have reported how inactivation of HUS1 in mice results in increased chromosomal abnormalities [32] Hus1 forms a checkpoint complex with rad9 and rad1 that is involved in DNA repair itself [33].
A second region on 7q came close to statistical significance for being associated with genomic instability WNT2 is within this region, and may be involved through association with the APC pathway Mutations in APC can promote chromosomal anomalies through involvement with kinetochores, but no direct evidence associates WNT2 with genomic instability [34].
Chromosome 14
A significant region on chromosome 14q32 includes the potentially relevant cell regulatory genes XRCC3 and PACS2 (figure 2b) XRCC3 is a RecA/Rad51-related protein family member that participates in homologous recombinational DNA repair Mutations in the apoptotic regulatory gene PACS2 have been found in colorectal cancer; its loss causing defective apoptosis could enable extensive DNA damage to accumulate without triggering apoptosis [35].
Regions associated with elevated overall fractional allelic loss rates
BAC-array comparative genomic hybridization compares hybridization signal intensity for each arrayed BAC to the mean hybridization signal intensities for all BACs analyzed for the sample Thus in predominantly aneuploid colorectal tumors, reduced signal intensity cannot be interpreted as reduction to a single allele for a given region.
We compared data from our previous determinations of genome wide loss of heterozygosity (the fractional allelic loss rate, FAL) utilizing 348 markers for each tumor [20], with our BAC-array analyses of these same tumors First we identified those BAC clones that showed frequent significant copy number aberrations, using the criteria of greater than three standard deviation change in array signal intensity, and where this was seen in twenty percent or more of the tumors examined on the BAC-arrays Thirteen BACs met these criteria Then we examined the FAL values of tumors which had significant copy number aberrations for these BAC clones One clone on 7q and six clones on 20q were associated with elevated fractional allelic loss rates The region on 20q includes the Aurora A kinase STK6 associated with centrosomal function, and which has been previously implicated in the aneuploidy form of genomic instability [36].
4. Discussion
Our analysis of BAC-array CGH data for colorectal cancers has identified several regions associated with elevated overall rates of amplifications or reductions in copy numbers Since array CGH data is normalized to the mean hybridization signal intensity for each particular tumor DNA, and non-MSI colorectal tumors tend to be heterogeneous and polyploid [2], reductions in copy number may still leave the tumors diploid (or greater) for the particular region showing copy number reduction.
The 9q34 and 22q11–13 regions were coordinately altered in approximately one-third of the sporadic colorectal carcinomas examined The coordinate alteration of these particular regions is suggestive of the BCR-ABL translocations of CML and ALL; the overall abundance of genomic alterations and other translocations in colorectal tumors may have heretofore obscured the involvement of these two particular regions [2,19]. Several findings are consistent with these two regions together being major factors in genomic instability Engineered expression of BCR-ABL fusion gene gives rise to intrachromosomal genomic instability when introduced into cell lines and transgenic mice [37–39] Imatinib (Gleevec) reduces the rate of genomic instability in cell lines expressing BCR-ABL [40] These results together may reflect that it is BCR-ABL itself that fosters the further evolution of chronic myelogeneous leukemia to the more aggressive and genomically damaged acute lymphocytic leukemias [41]. Our results with colorectal cancers showing that coordinate alterations in the BCR and ABL regions are associated with elevated instability would appear consistent with this same system being involved with genomic instability of a solid tumor system Further investigation of this possibility is appropriate.
Coordinate involvement of two genes in solid tumors, arising from a specific translocation, has a precedent in the recent report by Tomlins et al [42] In this case fusion of TMPRRSS2 and ETS was found to be a recurrent event in prostate cancer, although amidst the similar high background genomic noise of this tumor system, the translocation only became evident upon detailed analysis of expression array data. Translocations of 9q34 with 22q11–13 are known to be not limited to CML and ALL, and have been reported in several solid tumors where it often a frequent, recurring event These include oral squamous cell carcinona [43], lung carcinoma [44], lipoma [45], retinoblastoma [46], osteosarcoma [47], chondrosarcoma [48], and Ewing’s sarcoma [49] Li-Fraumeni cells passaged in tissue culture recurrently select for translocations of the same regions after long term passage [50]. With the highly complex karyotypes of most solid tumors, little attention has been paid to the 9;22 translocation in these other systems.
An alternative explanation for our observation of coordinate involvement of the 9q34 and 22q11–13 regions might arise from the presence of a 76 kilobase duplicon present 1.4 Mb 5’ of ABL and 150 Kb 3’ of BCR [51] FISH studies using a BAC clone derived from the region of chromosome 9 containing the duplicon showed hybridization to both chromosomes 9 and 22 However this possibility is very difficult to reconcile with our observed involvement of regions larger than a single BAC clone, particularly on chromosome 22, and with those cases where copy number gain was observed at 9q34 but loss was seen at 22q11–13.
The regions we report here reflect statistically significant associations with instability as revealed by comparative genomic hybridization on ordered BAC-arrays Additional molecular studies are now essential to evaluate the roles of the identified candidate regions and genes Testing the hypothesis that translocation of 9q34 and 22q11–13 often occurs in colorectal cancer is more complex than it might first appear Translocation currently is generally established with spectral karyotyping or multi-probe FISH, and then verified with PCR using two primers near the translocation point for each of the participating chromosomes The tumors used for this study were all stored frozen at –70C for more than seven years, precluding use of this approach on these specimens This issue is now being examined in detail in further studies utilizing freshly collected tissues and the approaches outlined, along with direct examination of the ABL gene product and RNA.
Acknowledgments
This work was supported by NIH Grant R01-CA74127 to GRA and NIH Center Grant P30-CA16056 to RPCI Drs. Bartos and Gaile contributed equally to this work, and should be regarded as co-first authors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson GR, Stoler DL, Brenner BM. Cancer: the evolved consequence of a destabilized genome. BioEssays. 2001;23:1037–1046. doi: 10.1002/bies.1149. [DOI] [PubMed] [Google Scholar]
- 2.Bartos JD, Stoler DL, Matsui S, Swede H, Willmott LJ, Sait SN, Petrelli NJ, Anderson GR. Genomic heterogeneity and instability in colorectal cancer: spectral karyotyping, glutathione transferase and ras. Mutat Res. 2004;568:283–292. doi: 10.1016/j.mrfmmm.2004.06.052. [DOI] [PubMed] [Google Scholar]
- 3.Davies H, Hunter C, Smith R, Stratton MR, Futreal PA, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:75911–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- 4.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair and induction of cancer. Proc Natl Acad Sci USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diep CB, Kleivi K, Ribiero FR, Teixeira MR, Lindgjaerde OC, Lothe RA. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosomes Cancer. 2005;45:31–41. doi: 10.1002/gcc.20261. [DOI] [PubMed] [Google Scholar]
- 6.Guda K, Upender MB, Belinsky G, Flynn C, Nakanishi M, Marino JN, Ried T, Rosenberg DW. Carcinogen-induced colon tumors in mice are chromosomally stable and are characterized by low-level microsatellite instability. Oncogene. 2004;23:3813–3821. doi: 10.1038/sj.onc.1207489. [DOI] [PubMed] [Google Scholar]
- 7.Tang R, Changchien CR, Wu MC, Fan CW, Liu KW, Chen JS, Chien HT, Hsieh LL. Colorectal cancer without high microsatellite instability and chromosomal instability - - an alternative genetic pathway to human colorectal cancer. Carcinogenesis. 2004;25:841–846. doi: 10.1093/carcin/bgh074. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Cummins JM, Shen D, Cahill DP, Jallepalli PV, Wang TL, Parsons DW, Traverso G, Awad M, Silliman N, Ptak J, Szabo S, Willson JK, Markowitz SD, Goldberg ML, Karess R, Kinzler KW, Vogelstein B, Velculescu VE, Lengauer C. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004;64:2998–3001. doi: 10.1158/0008-5472.can-04-0587. [DOI] [PubMed] [Google Scholar]
- 9.Fishel R, Lescoe MK, Rau MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 10.Swede H, Bartos JD, Chen N, Shaukat A, Dutt SS, McQuaid DA, Natarajan N, Rodriguez-Bigas MA, Nowak NJ, Wiseman SM, Alrawi S, Brenner BM, Petrelli NJ, Cummings KM, Stoler DL, Anderson GR. Genomic profiles of colorectal cancers differ based on patient smoking status Cancer Genet Cytogenet. 2006 doi: 10.1016/j.cancergencyto.2006.02.009. in press. [DOI] [PubMed] [Google Scholar]
- 11.Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo WL, Gray JW, Pinkel D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25:144–146. doi: 10.1038/75985. [DOI] [PubMed] [Google Scholar]
- 12.Hui AB, Lo KW, Yin XL, Poon WS, Ng HK. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Lab Invest. 2001;81:717–723. doi: 10.1038/labinvest.3780280. [DOI] [PubMed] [Google Scholar]
- 13.Ishizuka T, Tanabe C, Sakamoto H, Aoyagi K, Maekawa M, Matsukura N, Tokunaga A, Tajiri T, Yoshida T, Terada M, Sasaki H. Gene amplification profiling of esophageal squamous cell carcinomas by DNA array CGH. Biochem Biophys Res Commun. 2002;296:152–155. doi: 10.1016/s0006-291x(02)00836-7. [DOI] [PubMed] [Google Scholar]
- 14.Wilhelm M, Veltman JA, Olshen AB, Jain AN, Moore DH, Presti JC, Kovacs G, Waldman FM. Array-based comparative genomic hybridization for the differential diagnosis of renal cell cancer. Cancer Res. 2002;62:957–960. [PubMed] [Google Scholar]
- 15.Nakao K, Mehta KR, Fridlyand J, Moore DH, Jain AN, Lafuente A, Wiencke JW, Terdiman JP, Waldman FM. High-resolution analysis of DNA copy number alterations in colorectal cancer by array-based comparative genomic hybridization. Carcinogenesis. 2004;25:1345–1357. doi: 10.1093/carcin/bgh134. [DOI] [PubMed] [Google Scholar]
- 16.Man TK, Lu XY, Jaewon K, Perlaky L, Harris CP, Shah S, Ladanyi M, Gorlick R, Rau PH. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer. 2004;4:45. doi: 10.1186/1471-2407-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, Segraves R, Dairkee S, Tokuyasu T, Ljung BM, Jain AN, McLennan J, Ziegler J, Chin K, Devries S, Feiler H, Gray JW, Waldman F, Pinkel D, Albertson DG. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006;6:96. doi: 10.1186/1471-2407-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han W, Han MR, Kang JJ, Bae JY, Lee JE, Shin HJ, Hwang KT, Hwang SE, Kim SW, Noh DY. Genomic alterations identified by array comparative genomic hybridization as prognostic markers in tamoxifen-treated estrogen receptor-positive breast cancer. BMC Cancer. 2006;6:92. doi: 10.1186/1471-2407-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez-Bigas MA, Petrelli NJ, Anderson GR. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson GR, Brenner BM, Swede H, Chen N, Henry WM, Conroy JM, Karpenko MJ, Issa J-P, Bartos JD, Brunelle JK, Jahreis GP, Kahlenberg MS, Basik M, Sait S, Rodriguez-Bigas MA, Nowak NJ, Petrelli NJ, Shows TB, Stoler DL. Intrachromosomal genomic instability in human sporadic colorectal cancer measured by genome-wide allelotyping and inter-(simple sequence repeat) PCR. Cancer Res. 2001;61:8274–8283. [PubMed] [Google Scholar]
- 21.Cowell JK, Nowak NJ. High resolution analysis of genetic events in cancer cells using BAC arrays and CGHa Adv. Cancer Res. 2003;90:91–125. doi: 10.1016/s0065-230x(03)90003-0. [DOI] [PubMed] [Google Scholar]
- 22.Westfall PH, Young SS. Resampling-based multiple testing: Examples and methods for p-value adjustment. New York: John Wiley & Sons, Inc; 1993. p. 46. [Google Scholar]
- 23.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 24.Koon N, Zaika A, Moskaluk CA, Frierson HF, Knuutilas S, Powell SM, El-Rifai W. Clustering of molecular alterations in gastroesophageal carcinomas. Neoplasia. 2004;6:143–149. doi: 10.1593/neo.03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Rifai W, Moskaluk CA, Abdrabbo MK, Harper J, Yoshida C, Riggins GJ, Frierson HF, Powell SM. Gastric cancers overexpress S100A calcium- binding proteins. Cancer Res. 2002;62:6823–6826. [PubMed] [Google Scholar]
- 26.Yuan ZM, Utsugisawa T, Huang Y, Ishiko T, Nakada S, Kharbanda S, Weichselbaum R, Kufe D. Inhibition of phosphatidylinositol 3-kinase by c- Abl in the genotoxic stress response. J Biol Chem. 1997;272:23485–23488. doi: 10.1074/jbc.272.38.23485. [DOI] [PubMed] [Google Scholar]
- 27.Kharbanda S, Pandey P, Jin S, Inoue S, Bharti A, Yuan ZM, Weichselbaum R, Weaver R, Kufe D. Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature. 1997;386:732–735. doi: 10.1038/386732a0. [DOI] [PubMed] [Google Scholar]
- 28.Shaikh TH, Kurahashi H, Emanuel BS. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genome instability: an update and literature review. Genet Med. 2001;3:6–13. doi: 10.1097/00125817-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol Cell. 2002;10:387–395. doi: 10.1016/s1097-2765(02)00595-6. [DOI] [PubMed] [Google Scholar]
- 30.Weiss RS, Leder P, Vaziri C. Critical role for mouse Hus1 in an S-phase DNA damage and cell cycle checkpoint. Mol Cell Biol. 2003;23:791–803. doi: 10.1128/MCB.23.3.791-803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Hu B, Weiss RS, Wang Y. The effect of Hus1 on ionizing radiation sensitivity is associated with homologous recombination repair but is independent of nonhomologous end-joining. Oncogene. 2006;25:1980–1983. doi: 10.1038/sj.onc.1209212. [DOI] [PubMed] [Google Scholar]
- 32.Levitt PS, Liu H, Manning C, Weiss RS. Conditional inactivation of the mouse Hus1 cell cycle checkpoint gene. Genomics. 2005;86:212–224. doi: 10.1016/j.ygeno.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Lindsey-Boltz LA, Sancar A, Bambara RA. Mechanism of stimulation of human DNA ligase 1 by the Rad9-Rad1-Hus1 checkpoint complex. J Biol Chem. 2006;281:20865–20872. doi: 10.1074/jbc.M602289200. [DOI] [PubMed] [Google Scholar]
- 34.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nature Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 35.Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:1–13. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marumoto T, Zhang D, Sava H. Aurora-A: a guardian of the poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 37.Canitrot Y, Lautier D, Laurent G, Frechet M, Ahmed A, Turhan AG, Salles B, Cazaux C, Hoffman JS. Mutator phenotype of BCR-ABL transfected BaF3 cell lines and its association with enhanced expression of DNA polymerase beta. Oncogene. 1999;18:2676–2680. doi: 10.1038/sj.onc.1202619. [DOI] [PubMed] [Google Scholar]
- 38.Salloukh HF, Laneuville P. Increase in mutant frequencies in mice expressing the BCR-ABL activated tyrosine kinase. Leukemia. 2000;14:1401–1404. doi: 10.1038/sj.leu.2401855. [DOI] [PubMed] [Google Scholar]
- 39.Brain JM, Goodyer N, Laneuville P. Measurement of genomic instability in preleukemic P190 BCR/ABL transgenic mice using inter-simple sequence repeat polymerase chain reaction. Cancer Res. 2003;63:4895–4898. [PubMed] [Google Scholar]
- 40.van der Kuip H, Moehring A, Wohlbold L, Miething C, Duyster J, Aulitzky WE. Imatinib mesylate (STI571) prevents the mutator phenotype of Bcr-Abl in hematopoietic cell lines. Leuk Res. 2004;28:405–408. doi: 10.1016/j.leukres.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 41.Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, Nieborowska-Skorska M, Blasiak J, Skorski T. BCR/ABL oncogenic kinase promotes the unfaithful repair of the reactive oxygen species-dependent double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 42.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montle JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 43.Jin C, Jin Y, Wennerberg J, Annertz K, Enoksson J, Mertens E. Cytogenetic abnormalities of 106 oral squamous cell carcinomas. Cancer Genet Cytogenet. 2006;164:44–53. doi: 10.1016/j.cancergencyto.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Lukeis R, Irving L, Garson M, Hasthorpe S. Cytogenetics of non-small cell lung cancer: analysis of consistent non-random abnormalities. Genes Chromosomes Cancer. 1990;2:116–124. doi: 10.1002/gcc.2870020207. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson M, Meza-Zepeda LA, Mertens F, Forus A, Myklebost O, Mandahl N. Amplification of chromosome 1 sequences in lipomatous tumors and other sarcomas. Int J Cancer. 2004;109:363–369. doi: 10.1002/ijc.11716. [DOI] [PubMed] [Google Scholar]
- 46.Amare Kadam PS, Ghule P, Jose J, Bamne M, Kukure P, Banavali S, Sarin R, Advani S. Constitutional genomic instability, chromosome aberrations in tumor cells and retinoblastoma. Cancer Genet Cytogenet. 2004;150:33–43. doi: 10.1016/j.cancergencyto.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Lau CC, Harris CP, Lu X-Y, Perlaky L, Gogineni S, Chintagumpala M, Hicks J, Johnson ME, Davino NA, Huvos AG, Meyers PA, Healy JH, Gorlick R, Rao PH. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer. 2004;39:11–21. doi: 10.1002/gcc.10291. [DOI] [PubMed] [Google Scholar]
- 48.Tarkkanen M, Wiklund T, Virolainen M, Elomaa I, Knuutiila S. Dedifferentiated chondrosarcoma with t(9;22)(q34;q11–12) Genes Chromosomes Cancer. 1994;9:136–140. doi: 10.1002/gcc.2870090210. [DOI] [PubMed] [Google Scholar]
- 49.Aurias A, Rimbaut C, Buffe D, Zucker J-M, Mazabraud A. Translocation involving chromosome 22 in Ewing’s sarcoma A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984;12:21–25. doi: 10.1016/0165-4608(84)90003-7. [DOI] [PubMed] [Google Scholar]
- 50.Heng HHQ, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy P, Wu GS, Wang YA, Tainsky MA, Ye CJ. Stochastic cancer progression driven by non-clonal chromosome aberrations. J Cell Phys. 2006;208:461–472. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- 51.Saglio G, Storlazzi CT, Giugliano E, Surace C, Anelli L, Rege-Cambrin G, Zagaria A, Velasco AJ, Heiniger A, Scaravaglio P, Gomez AT, Gomez JR, Archidiacono N, Banfi S, Rocchi M. A 76-kb duplicon maps close to the BCR gene on chromosome 22 and the ABL gene on chromosome 9: Possible involvement in the genesis of the Philadelphia chromosome translocation. Proc Natl Acad Sci USA. 2002;99:9882–9887. doi: 10.1073/pnas.152171299. [DOI] [PMC free article] [PubMed] [Google Scholar]