

Abstract
Receptor targeting is an effective method of enhancing fluorescence signal in tumors for optical imaging. We previously used EGF conjugated to IRDye® 800CW to detect and track orthotopic prostate tumors in mice. In this study, our goal was to identify a reliable assay for targeting agent integrity in vitro that correlated with signal strength in vivo. Binding of IRDye® 800CW EGF to intact A431 human epidermoid carcinoma cells was quantified in a microplate assay. Specificity was confirmed by competition with unlabeled EGF or monoclonal antibody blocking. Biological activity of intact and damaged targeting agents relative to unlabeled EGF was determined by binding and stimulation of ERK phosphorylation. Both assays indicated a reduction of up to 60% of the fluorescence intensity with damaged agents. Using a research prototype imaging system optimized for IRDye® 800CW detection, we compared the efficacy of intact and damaged targeting agents for imaging subcutaneous tumors in mice. In live animal images and in sections of the excised tumors, damaged targeting agents consistently yielded diminished fluorescence signals corresponding to the reduction observed in microplate assays. This is the first study to directly correlate targeting agent signal strength in whole cell binding, in-cell western and in vivo near-infrared imaging.
Keywords: EGF receptor, IRDye® 800CW EGF, In-Cell Western, binding affinity, noninvasive, whole animal imaging
Introduction
Studies of tumor growth and metastasis in small animal models have greatly benefited from the expanded availability of optical imaging techniques that specifically detect tumor tissue in live animals (1, 2). A major advantage of optical imaging is the cost effective accessibility of the requisite instrumentation. Several promising methods utilize molecular approaches to enhance the specificity of optical detection. Frequently chosen techniques include measurement of bioluminescence, in which tumor cell lines bearing a luciferase reporter gene are detected by systemic delivery of a luminescent substrate, or gene-encoded fluorescence of tumor cell lines expressing green or red fluorescent proteins (3, 4). Although both tumor detection methods are powerful and sensitive, their major limitation is a requirement for specific cell lines. An alternative approach is the exploitation of high affinity receptor-ligand interactions to target molecular features of the tumor cell with coupled fluorophores.
Targeting agents that have been used successfully for optical noninvasive detection of tumor xenografts in mice include monoclonal antibodies (5, 6), small polypeptide growth factors (7, 8), or peptide analogues of extracellular ligands such as endostatin (9) and somatostatin (10). The most effective coupled fluorophores are those with fluorescence emission maxima in the near infrared (NIR) range of 780-850 nm. Due to low absorbance spectra of bodily fluids and tissues, imaging in the NIR offers the advantages of relatively deep tissue penetration and low autofluorescence (11, 12). Examples include targeting of the integrin αvβ3 using variations of RGD peptides linked to Cy5.5 (13-16), Alexa Fluor® 680 (13), and IRDye® 800CW (16). Recently, EGF labeled with Cy5.5 was used to quantify therapeutic efficacy of anti-EGF receptor antibodies for treating mammary carcinoma in mice (8).
To optimize receptor targeted signal strength in NIR optical imaging, a detailed understanding of the targeting mechanism and fluorophore function is critical. The EGF receptor is a potential biomarker for numerous preclinical models of human cancer because its expression is elevated an order of magnitude or more relative to normal cells in cancers of the breast (17), prostate (18), lung (19), ovary(20), and brain (21). Furthermore, its signaling functions are well characterized and accessible to convenient high throughput in vitro validation assays to ensure bioactivity of the ligand is maintained despite chemical coupling. For example, EGF binds to a cell surface transmembrane receptor tyrosine kinase, which induces dimerization of the receptors, triggering a phosphorylation cascade through numerous extracellular-regulated kinases (22). Since antibodies are available to quantify specifically the phosphorylated form of several kinases, activity of EGF may thus be verified directly by receptor binding and indirectly by stimulated kinase activity.
We previously conjugated IRDye® 800CW to EGF to track longitudinal growth of orthotopic prostate tumors in mice (23) and found the conjugate was a sensitive tumor targeting agent with excellent tissue penetration. We hypothesized that an aspect of the normal function of EGF contributing to the strong signal obtained from this agent could be receptor-mediated endocytosis of the ligand-receptor complex. This would imply that initial high affinity binding of the targeting agent is essential for maximal fluorescence signal strength in animals. Thus, animal numbers could be minimized in imaging experiments if suboptimal conjugate binding in vitro was found to correlate with reduced tumor targeting efficacy in vivo. Furthermore, it would be very powerful to have a straightforward in vitro assay that could directly predict relative efficiency of two different drugs or ligands with the same in vivo target. In this report, we used IRDye® 800CW EGF to target A431 human epidermoid carcinoma cells in vitro and in vivo. Specifically, we compared whole cell binding, ERK phosphorylation, and subcutaneous tumor targeting of optimal and damaged conjugates. Our characterization confirms that poor quality of a receptor targeting agent can be detected by measuring in vitro binding equilibria and that reduced bioactivity is detrimental to imaging sensitivity.
Materials and Methods
Cell Culture, Materials, and Reagents
A431 human epithelial squamous carcinoma cells were purchased from ATCC and maintained in DMEM medium supplemented with 10% fetal bovine serum. PC3M-LN4 human prostate adenocarcinoma cells were kindly provided by Dr. Isaiah J. Fidler (MD Anderson Cancer Center, Houston, TX) and maintained in minimal essential medium containing 10% fetal bovine serum, sodium pyruvate, and nonessential amino acids. A purified mouse diet (AIN-93M) was obtained from Harlan Teklad (Madison, WI). Monoclonal antibody C225 (Cetuximab; Imclone) was graciously provided by Dr. Chun Li (MD Anderson, Houston, TX). TO-PRO®-3 was obtained from Molecular Probes, Inc. (Eugene, OR). Antibodies specific for β-tubulin (H-235) and p-ERK (E-4) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The IRDye® 800CW EGF targeting agent, as well as the Odyssey® Infrared Imaging and Aerius® Automated Infrared Imaging Systems were provided by LI-COR Biosciences (Lincoln, NE).
Characterization of IRDye®800CW EGF binding specificity in vitro
The binding specificity of the IRDye® 800CW EGF compound was evaluated on A431 and PC3M-LN4 cells by an in-cell western assay (23, 24). Briefly, cells were grown to approximately 90% confluency in a microtiter plate (NUNC, Roberts, WI), and starved for 2 hours in serum-free media. For binding assays, starvation media were replaced with media containing increasing concentrations of IRDye® 800CW only (0.002-3.3 μM) or IRDye® 800CW EGF (0.01-70 nM). Specificity of the labeled compound was evaluated by competition assays in which starvation media were replaced with media containing unlabeled EGF (in increasing concentrations from 0.01-2.14 μM)+IRDye® 800CW EGF (70 nM) or C225 (0.5-200 μg/mL) + IRDye® 800CW EGF (70 nM). Cells were incubated at room temperature (25°C) for 2 min and fixed with 4% formaldehyde solution for 20 min. Four washes in 1xPBS+0.1% TritonX-100 were done to remove unbound dye and permeabilize the cells. The plates were blocked in Odyssey® Blocking Buffer for 1.5 hours, then incubated with TO-PRO®-3 (1:5000) for normalization of cell number using the 700nm-channel. Washing steps were repeated and the plate was scanned on Aerius®. Quantifications were normalized by ratiometric analysis of the 700nm values applied to the 800nm values.
Preparation of targeting agents
High quality targeting agents were prepared by standard FPLC methods. All separations were performed on an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA). Sample detection employed a diode array detector set at 260 and 780nm and purifications were carried out at 5°C. Standard FPLC conditions were achieved with this instrument using an isocratic method suitable for size exclusion chromatography. Following conjugation, the coupling mixture was applied to a 35 × 250 mm Omnifit column (Western Analytical Products, Wildomar, CA) packed with Superdex 30 gel filtration media (GE Healthcare/Amersham Biosciences). This column was equilibrated and eluted at 0.5 mL/min (6 bar pressure) with isocratic 1x PBS, pH 7.4. The eluant was filter sterilized by passage through a 0.2 μm syringe filter into a sterile container. Absorbance at 280 nm was used to calculate EGF concentration from the published molar extinction coefficient. Dual wavelength absorbances were used to determine dye:protein ratio, which was maintained consistently across all coupling reactions. Dual wavelength absorbance traces from size exclusion chromatography were also used to determine the relative amounts of coupled and unreacted dye in all preparations.
Poor quality targeting agents were prepared by subjecting unlabeled EGF or IRDye® 800CW EGF to HPLC purification, which is detrimental to protein activity because of the harsh, denaturing solvent conditions (25). A reverse-phase C18 Zorbax column (250× 9.6mm, 300 Å pore size, 5 μm particle size) was used. Solvents included aqueous 50 mM triethylammonium acetate (TEAA), pH 5.9 (A) and acetonitrile (B) in a gradient of 20% B to 31% B in 15 min. The purification was performed at a flow rate of 4.7 mL/min (100 bar pressure). Sample was protected from light, dried overnight under vacuum, reconstituted in 1x PBS, and applied to a cation (Na+ resin) exchange column equilibrated with 1x PBS. Following elution from the column with 1x PBS, the sample was lyophilized. Protein concentration, dye:protein ratio, and unreacted dye quantity was measured as above.
Quantification of downstream signaling by IRDye®800CW EGF
The ability of the targeting agent to stimulate ERK phosphorylation in A431 cells was compared to that of unlabeled EGF. Cells at 90% confluence in a 96-well plate were serum starved for 2 h. Starvation media were replaced with stimulation media containing 0.03-14 nM IRDye® 800CW EGF (FPLC), IRDye® 800CW EGF (HPLC), unlabeled EGF (FPLC), or unlabeled EGF (HPLC). Cells were incubated for 2 min at room temperature followed by fixation and permeabilization as above. An initial scan on Aerius® was done to obtain the level of signal from the conjugated EGF bound to receptors. Plates were then blocked for 1.5 h in Odyssey® Blocking Buffer followed by incubation with anti-β-tubulin (1:100) and anti-p-ERK (1:100) for 1.5 h at room temperature. Plates were washed 4X in 1xPBS+0.1% Tween-20 prior to incubation with IRDye® 800CW goat anti-rabbit (1:800) and IRDye® 680 goat anti-mouse (1:200) antibody conjugates for 1h. Cells were washed again and the plate was scanned on the Aerius®. The final ratiometric analysis was adjusted for the initial binding signal and p-ERK signal was normalized to β-tubulin.
Subcutaneous injections
Male NOD/SCID (Jackson Labs, Bar Harbor, ME) and athymic Nu/Nu mice (Charles River, Wilmington, MA) used in these experiments were maintained under the supervision and guidelines of the University of Nebraska-Lincoln IACUC Committee. PC3M-LN4 cells were trypsinized, washed and resuspended at 5x106 cells/ml in serum free MEM (26). Each NOD/SCID mouse was injected subcutaneously in the flank with 100 μl of cell suspension (0.5x106 cells) using a 1 ml insulin syringe. Tumors reached ∼0.5 cm in size at 4 weeks. A431 cells were trypsinized, washed and resuspended at 2×107 cells/ml in serum free medium for subcutaneous injections in the flank (2×106 cells) of male athymic Nu/Nu mice. Animals were carried for approximately two weeks until tumors reached ∼0.5 cm in size.
In vivo imaging of IRDye®800CW EGF
NIR fluorescence imaging in live animals was performed with a research prototype LI-COR Biosciences small-animal imager. The LI-COR instrument is a light tight chamber equipped with a cooled CCD camera, area illumination via diode lasers, and a selection of excitation and detection optical fluorescent filters tuned specifically for IRDye® 800CW. Images were acquired and analyzed with Wasabi software from Hamamatsu Photonics (Hamamatsu City, Japan) or Adobe Photoshop (Adobe Systems Inc., San Jose, CA). NOD/SCID mice were shaved prior to image collection. All mice were anesthetized with 2% isoflurane throughout procedures.
To establish initial conditions, IRDye® 800CW EGF (1nmol) was injected via the tail vein into a tumor-negative mouse of each strain and evaluated for systemic clearance by NIR imaging at intervals of 1-24 hours over a period of three days, after which time >90% of the signal had cleared. Mice bearing subcutaneous PC3M-LN4 or A431 tumors (diameter ≈0.5 cm) were then injected with IRDye® 800CW EGF (1 nmol) and evaluated for tumor-specific retention of the agent by NIR imaging at 24 hour intervals for up to 8 days. Signal-to-noise ratios calculated as described below were used to determine an optimal time point of 72-96 hours for imaging tumors. Subsequently, an additional eight athymic mice with A431 tumors were injected to compare signal strength of the targeting agents prepared by different methods. One mouse was injected with 0.9% saline, one received unconjugated IRDye® 800CW, two were given IRDye® 800CW EGF (1 nmol) from sources A and B purified by FPLC, and four were injected with HPLC-damaged IRDye® 800CW EGF (1 nmol) purified by HPLC. Animals were imaged 3 days post injection and euthanized. Tumors were excised, weighed, measured, and paraffin embedded for section scan analysis on Odyssey®
Statistical Analysis
Images for each mouse were normalized using the same intensity scale with a common minimum and maximum value. Signal-to-noise ratio (SNR) was calculated using the following formula:
Regions of interest (ROI) with identical areas were used for both tumor and background. ROIs were quantified for total pixel and mean pixel values. The standard deviation of mean backgrounds was calculated using 3-5 ROIs. This calculation yields the number of standard deviations over background represented by the suspect tumor/signal. Due to tumor size differences between the animals receiving quality targeting agents, tumor signal minus background signal of similar size ROI was then corrected for area (pixels). The % difference from FPLC purified agent was determined for HPLC purified agents.
Tissue Section Analysis
After final imaging (day 3 or 4 post-injection), animals were sacrificed. Tumors were excised, scanned on Odyssey®, weighed, measured, and preserved in Z-fix (Anatech Ltd., Battle Creek, MI) followed by paraffin embedding and tissue sectioning. Sections (5 μm thickness) were scanned in both the 700 nm channel, for tissue autofluorescence, and the 800 nm channel, for the targeted IRDye® 800CW EGF fluorescence signal using Odyssey®. The area-weighted fluorescence signal from the 800 nm channel was used to compare targeting agent specificity among experimental conditions.
Results and Discussion
In vitro specificity of IRDye® 800CW EGF for tumor cell lines
Effective use of whole animal NIR optical imaging for sensitive detection of tumor tissue requires highly specific biomarkers that can be labeled to illuminate tumor cells within live mice. Maximal emission of IRDye® 800CW is 789 nm, so it falls in an optimal region of the spectrum to obtain superior signal strength. We previously coupled EGF to IRDye® 800CW and found this targeting agent to be highly sensitive and specific due to its strong signal (23). Our goal in this study was to determine an aspect of EGF biological function quantifiable in vitro that reflects maximal tumor targeting potential.
We first characterized binding of the targeting agent to A431 cells by a microplate NIR fluorescence assay as we had previously done for PC3M-LN4 human prostate tumor cells (23). Confluent monolayer cultures of A431 cells were established in 96-well plates and treated with increasing concentrations of IRDye® 800CW EGF to establish a dose response curve for binding of the targeting agent to EGF receptors on the cells. Fluorescence signal became significant at 1 nM concentrations and increased exponentially in a concentration dependent fashion (Figure 1A). To verify receptor targeting, we performed the same measurements with unconjugated dye, which showed negligible increases in fluorescence over a similar concentration course (Figure 1A, circles). The specificity of the conjugate for the EGF receptor on the cells was evaluated by competitions with unlabeled EGF (Figure 1B) and with C225, a monoclonal antibody that blocks ligand binding by the EGF receptor (27) (Figure 1C). Unlabeled EGF reduced fluorescence in a concentration dependent manner with IC50 of ≈ 0.1 μM. The antibody competition was also dose dependent with IC50 of ≈ 10 μg/ml. These results are comparable to those obtained with prostate tumor cells. Importantly, saturation of binding to whole cells occurred above 0.1 μM. The value was ≈0.16 μM for PC3M-LN4 cells, which shows a half maximal binding constant of ≈70-80 nM, in good agreement with the inhibition constant for unlabeled EGF. Thus, coupling of EGF to the fluorophore did not affect receptor affinity.
Figure 1.
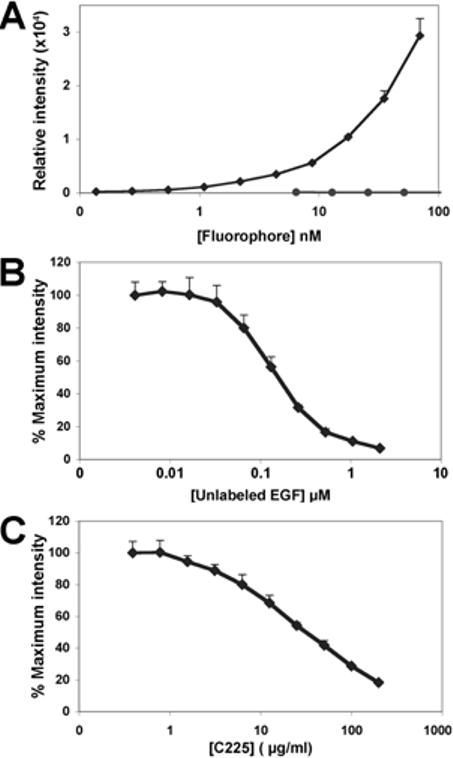
In vitro binding specificity of IRDye® 800CW EGF for tumor cells in culture.A431 cells cultured to 90% confluence in 96-well plates were incubated with increasing concentrations of IRDye® 800CW EGF (diamonds) or unconjugated, non-reactive IRDye®800CW (circles) for 2 min, then washed, fixed, permeabilized and stained with TO-PRO-3 (A). For competitions, cells were incubated with a fixed concentration of IRDye® 800CW EGF (70 nM) containing the indicated concentrations of unlabeled EGF (B), or C225, an EGF receptor blocking antibody (C). All plates were scanned at two wavelengths on the Aerius® near infrared scanner. The 800 nm signal, normalized to the 700 nm control, is plotted as the mean ± SD of three replicate wells.
To confirm that bioactivity of the targeting agent was accurately indicated by binding affinity, we used an in-cell western assay, which was previously developed for the quantification of kinase phosphorylation in whole cells (24). No significant decrease in ERK phosphorylation was noted between cells treated with equimolar concentrations of unlabeled EGF and IRDye® 800CW EGF (not shown). This is consistent with results from other investigators who have shown that both FITC-EGF (7) and fluorescent quantum dot-EGF (28) conjugates are effectively internalized by ligand-induced endocytosis of the bound EGF receptor complex, also a downstream effect of biologically intact EGF.
Binding of EGF and EGF-stimulated ERK phosphorylation are impaired by HPLC purification
We next evaluated whether the microplate binding and in-cell western assays could effectively distinguish between ligands with differential binding affinity. To obtain low affinity ligands for comparisons, we conjugated IRDye® 800CW to EGF from four independent commercial sources and subjected each conjugate to HPLC purification. Binding of these four targeting agents (denoted A, B, C and D in Figure 2) to A431 cells was compared by NIR fluorescence intensity to the binding of standard maximally active FPLC-purified conjugate from sources A and B. All four preparations demonstrated significant reductions in binding (up to 60%) at all concentrations (Figure 2A). Thus, the damage inflicted on EGF by HPLC treatment dramatically affected its cell binding potential. When we assayed p-ERK levels using the four targeting agents compared to unlabeled (not shown) or conjugated EGF from sources A and B (Figure 2B), the best performance of any HPLC-purified compound was 28% of unlabeled EGF stimulation. Reduced levels of ERK phosphorylation confirmed compromised function of the HPLC treated targeting agents.
Figure 2.
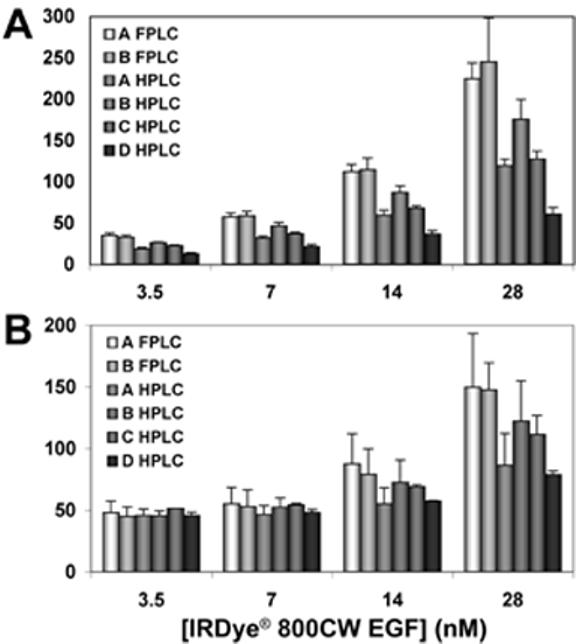
Biological activity elicited by labeled EGF. EGF from four sources (denoted as A,B,C,D) was conjugated to IRDye® 800CW and purified by HPLC. A431 cells at 90% confluence in 96-well plates were stimulated with the indicated concentrations of the respective agents or with the maximally active FPLC-purified conjugates corresponding to sources A and B. The plate was scanned to quantify binding of the IRDye® 800CW EGF targeting agents (A). The extent of ERK phosphorylation stimulated in each condition was assessed in the same plate by the in-cell western assay using antibodies to total ERK and p-ERK (B). Mean ± SD for three replicate wells is plotted.
Kinetics of IRDye® 800CW EGF clearance in vivo
Ultimately, our goal was to determine whether impaired function of the EGF receptor targeting agent in vitro would correlate with poor tumor targeting in mice. We first determined clearance kinetics of the targeting agent in normal and tumor-bearing mice. Clearance was monitored by injecting animals with 1 nmol of FPLC-purified IRDye® 800CW EGF via the tail vein and collecting NIR images over time from 15 minutes to 24 hours (Figure 3A). The pseudo-colored images depict the color-enhanced fluorescence intensity superimposed on white light images of a representative mouse. Fluorescence intensity in regions of interest encompassing the whole animal (large ROI) or limited to the abdominal area (small ROI) was quantified to evaluate clearance. We observed an initial increase in whole animal fluorescence intensity for the first 3-4 hours as the targeting agent circulated to the capillaries of the skin. Subsequently, the signal was seen to pass through the liver, intestines, kidneys and bladder. The analysis confirmed that 75% of the signal cleared after 8 hours and >90% cleared after 24 hours.
Figure 3.
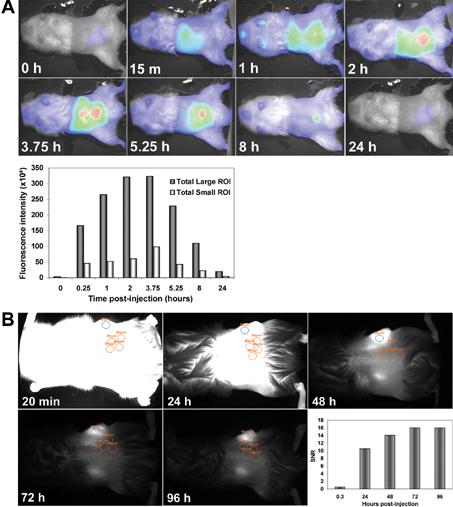
Clearance kinetics of IRDye ® 800CW EGF from normal and tumor-bearing mice.(A) IRDye® 800CW EGF (1 nmol) was injected IV into a normal NOD/SCID mouse. Images were collected prior to injection (0 h) and at the indicated times post-injection. Results are shown as color-enhanced fluorescence intensity superimposed on the white light images in the ventral view. Signal intensities (at 789 nm) were normalized to background fluorescence and plotted using the whole animal (large ROI, dark bars) or an arbitrary abdominal ellipse (small ROI, light bars) as regions of interest. (B) IRDye® 800CW EGF was injected IV into mice with subcutaneous PC3M-LN4 prostate tumor xenografts and images were collected at the indicated times post-injection. SNR was calculated using a single elliptical ROI denoted on the tumor in blue, and four background ellipses (orange) arbitrarily placed in irrelevant adjacent sites on the flank as shown. Fluorescence is also present in the kidneys in these dorsal views. Calculated SNR were plotted to obtain an optimal imaging time point for subsequent experiments.
We next monitored a similar time course for clearance of the targeting agent from tumor positive animals (Figure 3B). Mice bearing subcutaneous tumors of ≈0.5 cm in diameter (measured by digital calipers) were injected as above with IRDye® 800CW EGF, imaged immediately, and repeatedly imaged at 24-hour intervals. As observed for the tumor negative controls, whole animal and abdominal signal intensity peaked at 3-4 hours, but had diminished by >90% within 24 hours (data not shown). Analysis of signal-to-noise ratio (SNR) comparing mean fluorescence intensity in an arbitrarily chosen region of interest (ROI) within the tumor, designated as a blue ellipse, to the mean of four background areas of identical size, shown as orange ellipses, exhibited an initial time-dependent increase. Eventual saturation of SNR indicated that the time of maximal sensitivity for imaging was 72-96 hours, and subsequent images were collected at 72 hours (Figure 3B). In vivo, IRDye® 800CW EGF fluorescence following systemic clearance was amplified in the tumors relative to the whole animals, and was also specific as shown by antibody blocking. The sensitivity of this fluorescence signal lends support to the choice of EGF receptor as an epithelial or prostate tumor target and demonstrates IRDye® 800CW is an effective labeling fluorophore.
Subcutaneous tumor targeting by IRDye® 800CW EGF
To optimize targeting and tumor specific signal accumulation in noninvasive imaging, it is important to understand the functional parameters of the biomarker and its response to the target. Using the in-cell western, we detected impaired binding and response of the EGF receptor signaling cascade when the EGF conjugate was subjected to HPLC treatment. We performed an animal imaging study to determine if impaired binding function would translate to poor tumor signal. FPLC- or HPLC-purified IRDye® 800CW EGF from two sources (A and B) was injected via the tail vein into athymic mice bearing ≈ 0.5 cm subcutaneous A431 tumors. Mice were imaged three days post injection (Figure 4). Arrowheads indicate positions of detected tumor tissue in these representative fluorescence images, which have been corrected for background. Left panels (Figure 4, A and C) illustrate signal intensity in animals injected with the FPLC-purified targeting agent and panels on the right show the effect of the corresponding HPLC form (Figure 4, B and D). These images were analyzed to determine total fluorescence in the tumor (Table 1). Mice receiving HPLC-purified targeting agents from the two different preparations had ≈ 28% and ≈ 64% lower signal compared to the mice injected with minimally damaged agents (Table 1). Similar results were obtained when damaged agents were injected in mice bearing 22Rv1 prostate tumors, confirming that this phenomenon is not unique to the A431 cells. In images not shown, additional mice were injected with HPLC-purified probes from sources C and D, but the fluorescence signal from these animals was diminished 80-90% with respect to that calculated for the animals receiving optimized preparations (Table 1). Thus, the magnitude of the differences varied considerably, but a targeting agent exhibiting suboptimal signaling potential in vitro correlated strongly with poor signal strength in live animal imaging.
Figure 4.
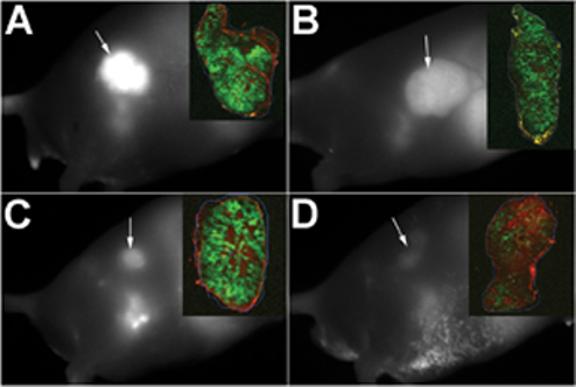
Targeting agent quality analysis in vivo. Male athymic Nu/Nu mice bearing A431 subcutaneous tumors (≈0.5 cm diameter) were injected IV with IRDye® 800CW EGF (1 nmol) purified by FPLC (A, C) or HPLC (B, D). Panels A and B illustrate use of the conjugate to EGF from source A in figure 3, and for panels C and D, source B was utilized. Images were collected on anesthetized animals at 72 hours post injection. Tumors were excised, fixed, paraffin embedded and sectioned for fluorescence signal determination by two-color scanning on the Odyssey® (inset panels).
Table 1.
Summary of tumor imaging results
| IRDye®800CW EGF | Normalized fluorescence | Signal loss | |||
|---|---|---|---|---|---|
| Preparation | Source | Intact animalsa | Tumor sectionsb | Intact animals | Tumor sections |
| FPLC | A | 1039.30 | 94.23 | ||
| FPLC | B | 512.77 | 113.39 | ||
| HPLC | A | 746.22 | 69.58 | 28%c | 26% |
| HPLC | B | 185.94 | 55.92 | (64%)d | (51%) |
| HPLC | C | 100.21 | 59.44 | 90% (80%) | 37% (48%) |
| HPLC | D | 85.63 | 55.75 | 92% (83%) | 41% (51%) |
Normalized fluorescence for intact animals: mean pixel fluorescence (tumor) minus mean pixel fluorescence (background).
800nm fluorescence per area (mm2).
Comparison to Source A FPLC to determine % Loss.
Comparison to Source B FPLC to determine % Loss (numbers in parentheses).
Finally, tumors were excised and sectioned for fluorescence scanning analysis to verify that signal intensities measured in the animal were attributable to fluorescence in the tumor tissue and that the intensity was consistent throughout the tumor. Sections were scanned on the Odyssey® in two channels and the corresponding merged images are shown as inset panels in Figure 4. Red represents tissue autofluorescence in the 700 nm channel and green denotes specific fluorescence in the 800 nm channel resulting from IRDye® 800CW EGF. Normalized fluorescence of the tissue sections is presented in Table 1. Signal intensity was evident throughout the tumors irrespective of targeting agent. Consistent with results from intact animal image analysis, sections exhibited signal reductions of ≈ 25-60%. Thus, again, performance of targeting agents damaged by HPLC purification, as identified in vitro by the assay for ERK phosphorylation, translated to poor outcome in whole animal imaging experiments.
In conclusion, we have characterized an effective targeting agent for noninvasive optical detection and tracking of tumors in mice. The fluorophore used, IRDye® 800CW, can be easily coupled to many ligand or targeting molecules of interest. We have found that an in vitro assay that detects binding and efficacy of EGF-conjugated products in stimulating downstream biological function is an accurate estimate of targeting efficacy in vivo. We expect this application to have further value in probe and drug development as a correlate assay for other biolabels to estimate competitive ligand and/or drug interactions in vivo.
Acknowledgements
The authors wish to acknowledge Rose Skopp for excellent technical assistant, and Drs. Joe Barycki and Mike Olive for critical evaluation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frangioni JV. Curr Opin Chem Biol. 2003;7:626–34. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Nat Biotechnol. 2005;23:313–20. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]
- 3.Choy G, Choyke P, Libutti SK. Mol Imaging. 2003;2:303–12. doi: 10.1162/15353500200303142. [DOI] [PubMed] [Google Scholar]
- 4.Troy T, Jekic-McMullen D, Sambucetti L, Rice B. Mol Imaging. 2004;3:9–23. doi: 10.1162/15353500200403196. [DOI] [PubMed] [Google Scholar]
- 5.Kang HW, Weissleder R, Bogdanov A., Jr. Amino Acids. 2002;23:301–8. doi: 10.1007/s00726-001-0142-2. [DOI] [PubMed] [Google Scholar]
- 6.Licha K, Debus N, Emig-Vollmer S, Hofmann B, Hasbach M, Stibenz D, Sydow S, Schirner M, Ebert B, Petzelt D, Buhrer C, Semmler W, Tauber R. J Biomed Opt. 2005;10:41205. doi: 10.1117/1.2007967. [DOI] [PubMed] [Google Scholar]
- 7.Carraway KL, 3rd, Cerione RA. Biochemistry. 1993;32:12039–45. doi: 10.1021/bi00096a014. [DOI] [PubMed] [Google Scholar]
- 8.Ke S, Wen X, Gurfinkel M, Charnsangavej C, Wallace S, Sevick-Muraca EM, Li C. Cancer Res. 2003;63:7870–5. [PubMed] [Google Scholar]
- 9.Citrin D, Lee AK, Scott T, Sproull M, Menard C, Tofilon PJ, Camphausen K. Mol Cancer Ther. 2004;3:481–8. [PubMed] [Google Scholar]
- 10.Becker A, Hessenius C, Licha K, Ebert B, Sukowski U, Semmler W, Wiedenmann B, Grotzinger C. Nat Biotechnol. 2001;19:327–31. doi: 10.1038/86707. [DOI] [PubMed] [Google Scholar]
- 11.Jobsis FF. Science. 1977;198:1264–7. doi: 10.1126/science.929199. [DOI] [PubMed] [Google Scholar]
- 12.Chance B. Annu Rev Biophys Biophys Chem. 1991;20:1–28. doi: 10.1146/annurev.bb.20.060191.000245. [DOI] [PubMed] [Google Scholar]
- 13.Aina OH, Marik J, Gandour-Edwards R, Lam KS. Mol Imaging. 2005;4:439–47. doi: 10.2310/7290.2005.05169. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Conti PS, Moats RA. Cancer Res. 2004;64:8009–14. doi: 10.1158/0008-5472.CAN-04-1956. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Z, Wu Y, Xiong Z, Gambhir SS, Chen X. Bioconjug Chem. 2005;16:1433–41. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Ke S, Wu Q, Charnsangavej C, Gurfinkel M, Gelovani JG, Abbruzzese JL, Sevick-Muraca EM, Li C. Mol Imaging. 2004;3:343–51. doi: 10.1162/15353500200404148. [DOI] [PubMed] [Google Scholar]
- 17.Kraus MH, Popescu NC, Amsbaugh SC, King CR. Embo J. 1987;6:605–10. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Lorenzo G, Tortora G, D’Armiento FP, De Rosa G, Staibano S, Autorino R, D’Armiento M, De Laurentiis M, De Placido S, Catalano G, Bianco AR, Ciardiello F. Clin Cancer Res. 2002;8:3438–44. [PubMed] [Google Scholar]
- 19.Levitzki A. Lung Cancer. 2003;41(Suppl 1):S9–14. doi: 10.1016/s0169-5002(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 20.Maihle NJ, Baron AT, Barrette BA, Boardman CH, Christensen TA, Cora EM, Faupel-Badger JM, Greenwood T, Juneja SC, Lafky JM, Lee H, Reiter JL, Podratz KC. Cancer Treat Res. 2002;107:247–58. doi: 10.1007/978-1-4757-3587-1_11. [DOI] [PubMed] [Google Scholar]
- 21.Kyritsis AP, Saya H. Curr Opin Oncol. 1993;5:474–80. doi: 10.1097/00001622-199305000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Sorkin A, Von Zastrow M. Nat Rev Mol Cell Biol. 2002;3:600–14. doi: 10.1038/nrm883. [DOI] [PubMed] [Google Scholar]
- 23.Kovar JL, Johnson MA, Volcheck WM, Chen J, Simpson MA. Am J Pathol. 2006;169:1415–26. doi: 10.2353/ajpath.2006.060324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Kovar J, Sissons S, Cox K, Matter W, Chadwell F, Luan P, Vlahos CJ, Schutz-Geschwender A, Olive DM. Anal Biochem. 2005;338:136–42. doi: 10.1016/j.ab.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 25.Heegaard NHH, Schou C. CRC Press; Boca Raton, FL: 2006. [Google Scholar]
- 26.Simpson MA, Wilson CM, McCarthy JB. Am J Pathol. 2002;161:849–57. doi: 10.1016/S0002-9440(10)64245-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harding J, Burtness B. Drugs Today (Barc) 2005;41:107–27. doi: 10.1358/dot.2005.41.2.882662. [DOI] [PubMed] [Google Scholar]
- 28.Lidke DS, Nagy P, Heintzmann R, Arndt-Jovin DJ, Post JN, Grecco HE, Jares-Erijman EA, Jovin TM. Nat Biotechnol. 2004;22:198–203. doi: 10.1038/nbt929. [DOI] [PubMed] [Google Scholar]