

Abstract
A single multi-domain viral protein, termed Gag, is sufficient for assembly of retrovirus-like particles in mammalian cells. We have purified the human immunodeficiency virus type 1 (HIV-1) Gag protein (lacking myristate at its N terminus and the p6 domain at its C terminus) from bacteria. This protein is capable of assembly into virus-like particles in a defined in vitro system. We have reported that it is in monomer–dimer equilibrium in solution, and have described a mutant Gag protein that remains monomeric at high concentrations in solution. We report that the mutant protein retains several properties of wild-type Gag. This mutant enabled us to analyze solutions of monomeric protein. Hydrodynamic studies on the mutant protein showed that it is highly asymmetric, with a frictional ratio of 1.66. Small-angle neutron scattering (SANS) experiments confirmed its asymmetry and yielded an Rg value of 34 Å. Atomic-level structures of individual domains within Gag have previously been determined, but these domains are connected in Gag by flexible linkers. We constructed a series of models of the mutant Gag protein based on these domain structures, and tested each model computationally for its agreement with the experimental hydrodynamic and SANS data. The only models consistent with the data were those in which Gag was folded over, with its N-terminal matrix domain near its C-terminal nucleocapsid domain in three-dimensional space. Since Gag is a rod-shaped molecule in the assembled immature virion, these findings imply that Gag undergoes a major conformational change upon virus assembly.
Keywords: HIV-1, virus assembly, molecular modeling, small angle neutron scattering, analytical ultracentrifugation
Introduction
Expression of a single protein, the human immunodeficiency virus type 1 (HIV-1) Gag polyprotein, is sufficient for efficient assembly of virus-like particles in mammalian cells.1 These particles are roughly spherical, with a diameter of approximately 100–150 nm, and contain several thousand Gag molecules. Individual Gag molecules are elongated rods in these particles, and are arranged radially, with their N termini in contact with the lipid bilayer that surrounds the particle and their C termini in the interior of the particle.
The molecular mechanisms underlying the assembly of the virus particle from the Gag protein are not well understood. One significant way of simplifying the analysis of these mechanisms is by studying assembly in a defined system in vitro. As first demonstrated by Campbell & Vogt,2 retroviral Gag proteins can be readily purified following expression in bacteria. When these proteins are incubated with nucleic acids in a buffer of moderate ionic strength, they can assemble into virus-like particles (VLPs) whose morphology closely resembles that of authentic immature retrovirus particles, except that they lack the lipid bilayer enclosing the authentic virion.2 This fundamental result shows that the ability to assemble into a rather regular, spherical structure is inherent in the Gag molecule, needing only nucleic acid as a cofactor. The role of the nucleic acid in the formation and maintenance of the structure is not understood, but almost any single-stranded nucleic acid, including short DNA homo-polymers, can perform this function in vitro.3,4 Interestingly although recombinant HIV-1 Gag protein forms spherical VLPs under these conditions,3 addition of a second cofactor, i.e. inositol pentakisphosphate (IP5) or a related molecule, is required for assembly of VLPs with the correct radius of curvature.5 (The reasons for this additional requirement, which appears to be unique to HIV-1, are not known and will not be addressed here. However, the interactions of HIV-1 Gag protein with inositol hexakisphosphate are analyzed in some detail in the accompanying paper6)
After a wild-type virus particle is released from the cell, the viral protease (PR) cleaves Gag into a series of smaller proteins; this event is termed virus maturation and is essential for the generation of an infectious virion. In the case of HIV-1, the cleavage products include (from N to C terminus) matrix (MA), capsid (CA), a short peptide termed p2, nucleocapsid (NC), the short peptide p1, and p6. Cleavage of Gag into these fragments leads to a global reorganization of the particle, including formation of a new structure, the mature viral core.
The ability to assemble into VLPs must ultimately derive from the three-dimensional structure of the Gag protein. Significant structural information has been obtained for several isolated domains of HIV-1 Gag, including MA; the N-terminal and C-terminal domains of CA (termed NTD and CTD, respectively); and NC in complexes with RNA stem-loops.7-15 However, except for one important study of a fragment comprising MA and the NTD of CA,16 no structures have been obtained for full-length Gag or even for proteins containing more than one of these segments. Further insight into the structure of the full-length protein would undoubtedly shed light on the molecular mechanisms underlying virus particle assembly. In the accompanying report,6 we have shown that our recombinant Gag protein (differing from authentic Gag in that it lacks myristate at its N terminus and the p6 domain at its C terminus3) is in monomer–dimer equilibrium in solution, using the dimer interface previously described 11,14 in the CTD of CA. We generated a double mutant which reduced the tendency of the protein to dimerize ∼100-fold (see the accompanying paper6). We now describe the use of this mutant protein (termed WM Gag), in which tryptophan 316 and methionine 317 (residues 184 and 185 of CA) have been replaced by alanine, for detailed studies on the monomer. Both small-angle neutron scattering (SANS) and hydrodynamic results indicate that the monomer is highly asymmetric in solution. Using the known structures of individual domains of Gag, we generated a large ensemble of structural models for the Gag protein. Only a small minority of these model structures were consistent with the experimental data; in all of these models, the protein was folded over, with its N terminus and C terminus relatively close together in three-dimensional space. The fact that Gag is apparently folded in solution implies that it must undergo a dramatic conformational change when it assembles into a virus particle.
Results
Effects of WM mutation on properties of Gag protein
The biophysical properties of the HIV-1 Gag protein are of considerable interest. The propensity of the wild-type protein to dimerize in solution is a serious complication for attempts to characterize the protein, since these solutions will contain mixtures of monomers and dimers, with the composition of the mixture determined by the Gag concentration. Therefore, the WM protein, which remains monomeric over a much wider concentration range, offers a crucial tool for biophysical studies. However, it is also possible that the mutation introduced global changes in Gag structure, as well as weakening the dimer interface. To test this possibility, we compared the mutant and wild-type proteins in several ways.
Our first comparisons were by circular dichroism (CD). As shown in Figure 1(a), the CD spectra of the two proteins are virtually superimposable. Thus, the mutation does not appear to have caused any major change in the secondary structure of Gag protein.
Figure 1.
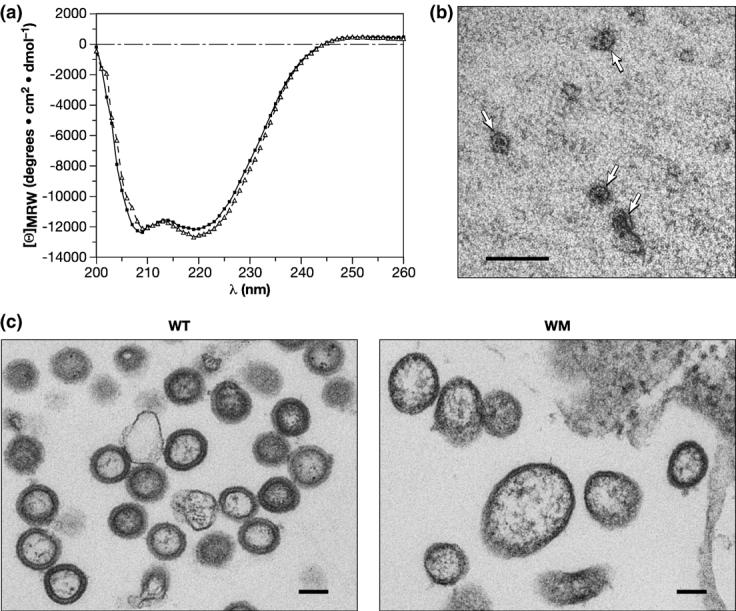
Properties of the WM Gag protein. (a) CD profile of wild-type (circles) and WM Gag (triangles). (b) VLPs assembled in vitro from WM Gag. WM Gag was mixed with tRNA, diluted, and analyzed by negative staining as described in Materials and Methods. VLPs are indicated by arrows. (c) Particles formed by wild-type and WM Gag proteins in mammalian cells. Particles associated with pelleted cells were analyzed by electron microscopy as described in Materials and Methods. The scale bars represent 100 nm.
As an additional test of the properties of the mutant protein, we measured its ability to assemble into small VLPs like its wild-type counterpart. WM Gag protein was diluted to a final concentration of 1 mg/ml in buffer containing 50 mM NaCl in the presence of yeast tRNA. A substantial fraction of the protein (50%) became pelletable under these conditions. Figure 1(b) shows an electron micrograph of negatively stained VLPs assembled from WM Gag; like those previously described for the wild-type protein,3 these VLPs are very small (∼25–30 nm in diameter). We found that these VLPs are more salt-labile than wild-type control VLPs (data not shown). We also tested the mutant protein for its ability to assemble, like wild-type Gag,5 into full-size VLPs in the presence of nucleic acid and IP5. We found no regular structures in these experiments (data not shown). Nevertheless, the ability of the mutant protein to assemble into the small VLPs is strong evidence that its overall structure has not been drastically changed by the replacement of CA residues tryptophan 184 and methionine 185 with alanine.
Finally, we expressed full-length wild-type and WM Gag proteins (both containing myristate at their N termini and p6 at their C termini) in human cells and tested their ability to assemble into virus particles under these conditions. We found (data not shown) that the level of WM Gag protein released from the cells in pelletable form was about fivefold lower than that seen with the wild-type control; this result is similar to findings of von Schwedler et al.17 on the individual mutants W184A and M185A. We also examined thin sections of the transfected cells by electron microscopy. As shown in Figure 1(c), the particles formed by the mutant protein tended to be somewhat irregular in size and shape, rather than roughly spherical with a nearly constant diameter, as seen in the wild-type control. Thus, mutation of the dimer interface does not prevent particle assembly in vivo, although it does cause more subtle quantitative and qualitative defects in the assembly process.
Hydrodynamic properties of WM Gag protein
The data presented above indicate that the WM mutant Gag protein is similar to wild-type Gag in its overall structure, yet remains monomeric over a broad concentration range (see the accompanying paper6). We therefore used the WM Gag protein in biophysical studies designed to characterize the solution properties of monomeric Gag.
The hydrodynamic radius (Rh) of WM Gag protein was determined by several independent methods. First, WM Gag protein was passed through a Superose 12 size-exclusion chromatography column. The elution profile is shown in Figure 2(a) (green profile), along with those of bovine serum albumin (BSA) (red profile) and of wild-type Gag (blue profile). It can be seen that the WM Gag eluted with a peak centered around ∼37.8 min. The column was also calibrated by chromatography of a series of other proteins of known Rh and the elution times were converted into the parameter (KD)1/3.18 As shown in Figure 2(b), the elution time of 37.8 min (indicated by the arrow) corresponds to an Rh value of ∼36 Å.
Figure 2.
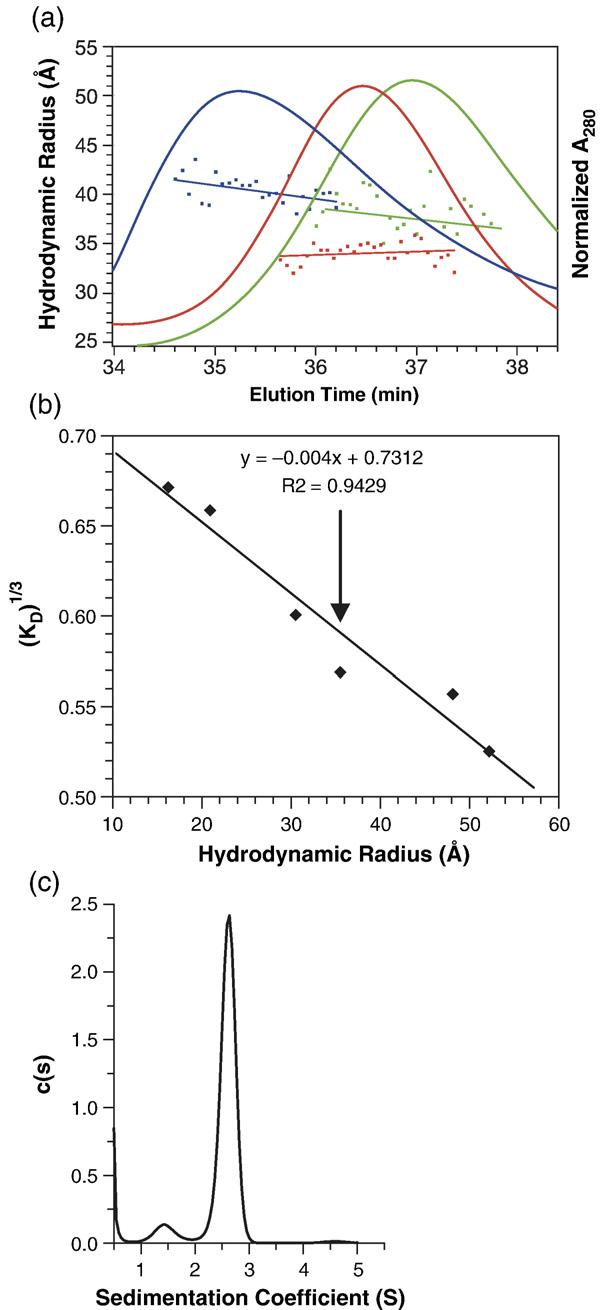
Hydrodynamic properties of the WM Gag protein. (a) WM Gag (green profile) was injected onto a Superose 12 column at 3 mg/ml, and the eluate was monitored by A280 (green elution profile) and simultaneously analyzed by QELS. The data points under the elution profiles show the Rh values obtained from QELS at each position in the elution and the lines are weighted fits through the points. Also shown are the elution profiles of wild-type Gag, loaded at 3 mg/ml (blue profile), and of BSA (5 mg/ml) (red profile). (b) The Rh value of WM Gag determined from its elution time in (a). The column was calibrated with a series of proteins of known Rh value, as described in Materials and Methods. The graph shows the cube-root of the column distribution coefficient KD18 plotted against the Rh value of the protein. The vertical arrow indicates the position of WM Gag on the curve. (c) Analysis of WM Gag by SV. WM Gag (0.64 mg/ml) was centrifuged at 58,000 rpm, and the boundary movement was monitored by A280. The graph shows the c(s) versus s plot from the Sednterp software program as described in Materials and Methods.
In addition, the Rh value of the protein in the column eluate was determined simultaneously by quasi-elastic light scattering (QELS). This technique yielded an Rh value of 38 Å (green data-points under WM Gag elution profile in Figure 2(a)) and a diffusion constant (D) of 6.36 × 10−7 cm2/s.
Finally, the properties of WM Gag were determined by sedimentation velocity (SV) analysis. Boundary SV measurements (not shown) were performed to determine the weight-average sedimentation coefficient of WM Gag and these data were analyzed by the Sedfit program. Figure 2(c) shows the c(s) versus s analysis of WM Gag resulting from this analysis. We obtained an s value of 2.62 S for WM Gag, corresponding to an s20,w value of 2.92 S. A spherical protein of molar mass 49,993 (the molecular mass of WM Gag) would have an s20,w value of 4.85 S. Therefore, WM Gag exhibits a frictional ratio (f/f0) of 1.66; this corresponds to an Rh value of 41 Å and a diffusion constant of 5.3 × 10−7 cm2/s. The hydrodynamic measurements are summarized in Table 1, showing good agreement between the different methods for the determinations of Rh and D.
Table 1.
Hydrodynamic parameters for WM Gag protein
| Method | Rh value (Å) | D (cm2/s)×107 |
|---|---|---|
| SEC | 36 | N.D. |
| QELS | 38 | 6.36 |
| SV | 41 | 5.3 |
N.D., not determined.
Analysis of WM Gag protein by small angle neutron scattering
We also explored the solution structure of WM Gag protein using SANS. This technique leads to a model-independent value for the radius of gyration, Rg, of the molecule under study. In addition, it results in a distance-distribution function, P(r). Both of these parameters provide information on the size and shape of the molecule in solution. Preliminary experiments showed very similar SANS profiles in H2O- and 2H2O-containing buffers, and in either 0.17 M or 0.5 M NaCl in 2H2O buffer. Moreover, sedimentation equilibrium studies (data not shown) showed that the Kd value for dimerization of wild-type Gag is ∼3.9 μM in 2H2O buffer, which is nearly the same as the value of 5.5 μM, obtained in the standard H2O buffer (Datta et al., accompanying paper6). Thus, the properties of Gag protein, and by extension WM Gag as well, are apparently affected little, if at all, by the difference between H2O and 2H2O. Taking advantage of the strength of SANS signals in 2H2O, we then analyzed WM Gag protein by SANS at concentrations of 0.5, 1.0, and 2.0 mg/ml in 2H2O buffer with 0.5 M NaCl. Guinier plots of the SANS data are shown in Figure 3(a), and the corresponding values for the radius of gyration, Rg, and the forward scattering, I(0) are shown in Table 2. The Rg values are consistent for the 0.5 and 1.0 mg/ml samples, with values of 33–35 Å. However, the data show an increase in Rg for the 2 mg/ml sample. It therefore appears that the WM mutant Gag protein is still capable of some level of oligomerization; this observation is qualitatively consistent with sedimentation-equilibrium data reported in the accompanying paper (Datta et al.6). We therefore used the SANS data obtained at 1.0 mg/ml in our efforts to model monomeric Gag protein, as described below. The best estimate of Rg obtained from the Guinier analysis of the SANS results is 34(±1) Å.
Figure 3.
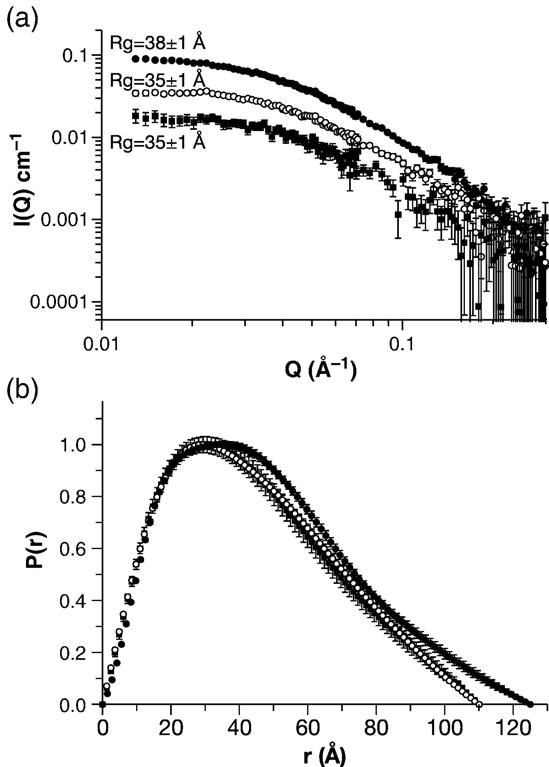
SANS analysis of the WM Gag protein. WM Gag solutions at 0.5, 1.0, and 2.0 mg/ml were analyzed by SANS as described in Materials and Methods. (a) Guinier plot. (b) Distance distribution function. Filled squares, 0.5 mg/ml; open circles, 1.0 mg/ml; filled circles, 2.0 mg/ml.
Table 2.
SANS results
| WM sample concentration (mg/ml) | Rg (Å) from Guinier | Rg (Å) from P(r) | Dmax (Å) | I(0) cm−1 from P(r) | MW (Da) |
|---|---|---|---|---|---|
| 2.0 | 37±1 | 38±1 | 125 | 0.10±0.01 | 67,000±5000 |
| 1.0 | 34±1 | 35±1 | 110 | 0.041±0.002 | 54,000±5000 |
| 0.5 | 33±2 | 35±1 | 110 | 0.019±0.001 | 51,000±4000 |
Errors on Rg and I(0) values are standard errors of the mean, taken from several fits to the data in the case of both Guinier and P(r) analyses. Mw values are obtained from equations (1) and (2) using the I(0) values from the P(r) analysis. Errors on the concentration values are assumed to be 5%. Dmax represents the optimal value obtained by testing several values of Dmax in the P(r) analysis and choosing the value that best fit the data and gave an Rg value in agreement with that from Guinier analysis.
The agreement between the Rg values at concentrations of 1 mg/ml and below suggests that WM Gag remains monomeric in this concentration range. As a further test of this hypothesis, we used equations (1) and (2) (see Materials and Methods) to calculate the molecular weight of the protein from the SANS data at each concentration. As shown in Table 2, the MW values are 51,000(±4000) Da for the 0.5 mg/ml sample and 54,000(±5000) Da for the 1.0 mg/ml sample, in good agreement with the expected MW value for monomeric WM Gag. As the Rg value of a sphere of 50,000 Da would be (3/51/2) times the radius of the sphere, or 18.9 Å, the value of 34 Å implies that WM Gag is quite asymmetric.
Another representation of the WM data is shown in Figure 3(b). The Figure shows the distance distribution function, i.e. the probability that two points in the molecule will be separated from each other by a distance r. This representation of the data gives a real-space view of the symmetry of the molecule. For example, a spherical molecule will have a symmetric P(r) distribution with a peak, representing the most probable distance in the molecule, at its radius. P(r) will be zero at r = 0 and at Dmax, the diameter of the sphere. On the other hand, if the molecule is asymmetric, the P(r) function will also be asymmetric. For example, a rod-shaped molecule will have a P(r) distribution with a peak at r = the radius and a Dmax equal to its length. Inspection of Figure 3(b) shows that, for 0.5 mg/ml and 1.0 mg/ml WM Gag, the peak is at r = 30 Å and Dmax = 110 Å. Thus, the distance distribution function, like the Guinier analysis, shows that the protein is notably asymmetric.
WM Gag modeling
The ultimate goal of our studies was to construct a model for WM Gag protein. Since the molecule is almost certainly flexible, it is unlikely that it can be adequately described by a single, rigid shape. We chose to model it using the following strategy. Gag contains the following domains, each connected to its neighbors by flexible linkers: MA; NTD of CA; CTD of CA; and NC. Detailed structural information is available for each of these domains, including multiple structures for NC. In addition a series of structures have been determined for a protein containing both MA and NTD. Accordingly, we attempted to join the known structures together in silico to generate a large ensemble of Gag structures. (Of course, it is possible that these domains have different structures when they are part of Gag from those determined for the free proteins. Indeed, it is clear that the N-terminal ∼50 residues of CA undergo a rearrangement following the release of CA from Gag.8,9,12,16,19 It has also been proposed that the structure of the C-terminal region of the CA domain of Gag is different, at least under some conditions, from the CTD of free CA protein.20) Because of the number of flexible sites in the protein, there exist a very large number of possible conformations of the protein. We searched through these models for arrangements/combinations that were consistent with the SANS and hydrodynamic data presented above.
One region of Gag for which there is very little direct structural information is the p2 linker peptide between CA and NC. This region plays a critical role in proper particle assembly.21,22 It has been suggested that p2 possesses a helical structure,21,23 but nuclear magnetic resonance (NMR) studies show that its helical content in solution is very limited.24,25 One step in our modeling was to evaluate a series of possible generic p2 structures, i.e. extended random coil, completely α–helical, and partially α–helical. In turn, the extended coil and fully helical p2 conformations were modeled to be on either the x, y, or z axis relative to the CTD of CA, while the partially helical conformation was simulated in two possible orientations. For each of these eight possibilities, we attached 20 MA-CA and 30 NC structures derived from NMR and crystallography (see Materials and Methods) to create a total of 4800 structural models for Gag.
The diversity of structures encompassed by these models is indicated pictorially in Supplementary Data, Figure S1, which depicts a composite of all 4800 structures. It is clear from the picture that the position of the MA domain (in blue) is not fixed relative to the NTD of CA (silver), but rather “sweeps out” a large volume of space around the latter. In addition, the diversity of NC structures (mauve), in combination with the eight hypothetical p2 structures (two of which are visible in purple), leads to wide variation in the overall shape and orientation of the C-terminal portions of the protein.
For each of the 4800 structures, the expected SANS spectrum and the corresponding Rg and Rh values were calculated from the atomic coordinates. These predicted results were then compared with the experimental SANS data in two steps. In Figure 4 we have divided the structures into three groups, representing the three trial p2 structures (linear, helical, and partially helical). Thus, there are 1800 structures each for the linear (Figure 4(a) and (d)) and helical (Figure 4(b) and (e)) p2 segments, along with 1200 structures for the partially helical structures shown in Figure 4(c) and (f). Figure 4(a)-(c) show the distribution of Rg values of the 4800 structures. It is obvious from Figure 4(a) that all of the structures with an extended, linear random coil for p2 are incompatible with the experimentally determined Rg value of 34 Å In contrast, the fully helical and the partially helical p2 sets (Figure 4(b) and (c)) both have a wide distribution of Rg values, including a minor fraction of structures with Rg values less than 40 Å.
Figure 4.

Evaluation of model structures for the WM Gag protein. (a)–(c) Rg frequency distributions for model structures with (a) extended random coil p2 structures; (b) helical p2 structures; and (c) partially helical p2 structures. (d)–(f) Detailed evaluation of the structures in each group exhibiting the best and worst fits to the experimental data. The main panel shows the calculated Guinier plot for the best (continuous line) and worst (broken line) in each group, compared with the Guinier plot obtained experimentally at 1 mg/ml. (d) Extended random coil p2 structures. (e) Helical p2 structures. (f) Partially helical p2 structures. Also shown in (d)–(f) are images of the best (black) and worst (colored) structures. The inset table shows the χ2 values for the Guinier plot and the calculated Rg and Rh values for the best and worst structures.
We also used χ2 analysis to quantitate the degree to which the computed SANS spectrum for each structure matched the experimental SANS results for WM Gag. Figure 4(d)-(f) shows, for each of the three groups, a comparison between the best and worst fits in the group with the data. The inset tables also show the χ2 values and predicted Rg and Rh values for the structures with the best and worst fits. Finally, the Figure also presents space-filling models of the best (black) and worst (grey) structures. In all cases the lower portion of the structure is the NC region of the full-length WM Gag molecule, and the structures are oriented so that one can follow the structure continuously to the MA or amino terminus of the protein. Since the CA structure was not varied in these models, the best and worst structures share their CA domain and only a single black CA region is shown. Again, it is obvious from the inset tables that structures with an extended linear p2 region (Figure 4(d)) provide very poor fits to the data, since the Rg value of the best structure (44 Å) is much higher than the experimental value (34 Å), and the χ2 value is 7.1. However, either of the helical p2 structures (Figure 4(e) and (f)) can lead to structures that are in fairly good agreement with the SANS spectra, with the best fit arising from the partially helical p2 structures (Figure 4(f)). Results from the Rh calculations follow the same trend, in that the predicted Rh value for the best structure in Figure 4(d) is considerably higher than the experimental value of 41 Å, while those in Figure 4(e) and (f) are reasonably close to this value.
Figure 5 displays the χ2 values for the entire ensemble of 4800 structures. In this analysis, the structures are not grouped by their p2 structures (as in Figure 4), but rather are divided into 20 groups (numbered 1–20) according to the structural orientation of theMAregion relative to the CA region of the protein. Since all of the structures in any given group have a common MA-CA orientation, all of the variation within a group arises from the differences in p2 and NC structures and orientations. Each group spans a wide range of χ2 values (Figure 5); thus the configuration of the C-terminal portion of the protein has a significant effect on the fit. (This is also clear in Figure 5 from the fact that structures denoted by circles and triangles, representing the helical p2 structures, tend to have lower χ2 values than those denoted by plus signs.) However, it is evident that there are four groups (i.e. groups 1, 6, 7, and 18) that give far better fits to the SANS data than the remaining 16 groups. The inset table in the left panel of the Figure shows the χ2 values and predicted Rg and Rh values for the best structure in each of these groups. Finally, space-filling models of these individual best structures are presented on the right side of the Figure. In all cases, regardless of the p2 structure used, the best fits are obtained with compact MA structures, and with MA in relatively close proximity to CA. It is also clear that the four structures shown are quite different from each other, but are nearly comparable in the degree to which they fit the data. Presumably, since Gag is a flexible molecule, there is no single correct structure; rather, this type of analysis helps eliminate improbable conformations and focus on families of favored structures.
Figure 5.
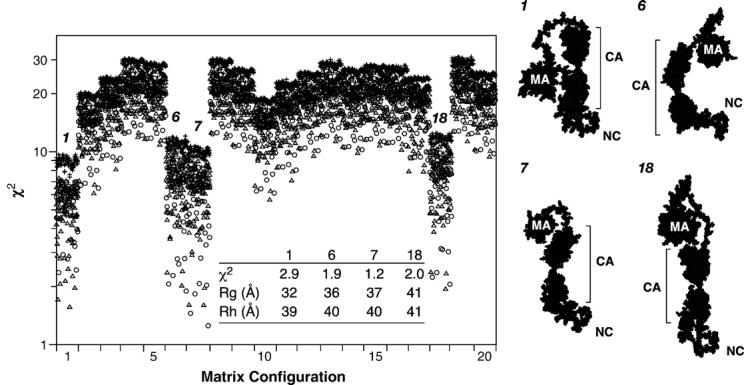
Evaluation of 4800 model structures of the WM Gag protein. For each model structure, the χ2 value for consistency with the SANS data was calculated. The model structures are divided into 20 groups according to the configuration of the MA domain. Structures with extended p2 regions are indicated by plus signs, those with helical p2 by triangles, and those with partially helical p2 by circles. Four groups, arbitrarily numbered 1, 6, 7, and 18, gave the lowest χ2 values. The inset table shows these values, along with the calculated Rg and Rh values, for the best structure in each of these four groups. These structures are depicted on the right side of the Figure. The approximate “vertical” dimensions of these structures are: group 1, 100 Å; groups 2 and 3, 110 Å; group 4, 130 Å.
Discussion
We have studied the solution properties of assembly-competent HIV-1 Gag protein, the polyprotein precursor which is responsible for formation of HIV-1 virus particles. To our knowledge, this represents the first analysis of this type for any retroviral Gag protein. As we report in the accompanying paper,6 the protein is in monomer–dimer equilibrium. Mutation of a pair of key residues at the dimer interface in the CTD of the CA domain greatly attenuates the tendency of the protein to dimerize; thus, it seems very likely that dimerization of Gag is mediated by this same interface. We now present biophysical and functional studies, which suggest that the mutant protein, which we refer to as WM Gag, is similar in overall structure to monomeric wild-type Gag (Figure 1).
The properties of WM Gag have been analyzed in some detail. The Rh and Rg values of the protein suggested that it is highly asymmetric, rather than globular (Figures 2 and 3). The large surface area of this multi-domain protein is presumably responsible for its high frictional ratio. Finally, modeling studies were performed to identify possible structures for WM Gag that would contain the known structures of the isolated MA, NTD, CTD, and NC domains and would be consistent with our hydrodynamic and SANS results. These studies suggested (Figures 4 and 5) that the most probable structures of WM Gag (and, presumably, of a monomer of wild-type Gag) are folded over, so that the N-terminal MA domain is relatively close to the C-terminal NC domain in three-dimensional space. This conclusion is entirely consistent with the results of inositol hexakisphosphate binding studies presented in the accompanying paper (Datta et al.6).
It is crucial to understand that there is probably no single, unique structure for the molecule, since it contains several flexible regions; rather, the modeling is an attempt to identify a population of possible structures that are consistent with the data. We also considered the possibility that the conformation of the protein is somewhat different at physiological ionic strength and in 0.5 M NaCl, where our studies were performed. In particular, the high ionic strength used here would be expected to decrease the repulsion between the MA and NC domains, which are both quite basic in character. However, additional SANS data showed that WM Gag protein gives virtually superimposable scattering spectra at 0.5 M and 0.17 M NaCl (data not shown); thus the properties of the protein under physiological conditions are probably very similar to what we observed at the higher salt concentration.
Three independent hydrodynamic tests, i.e. size-exclusion chromatography, QELS, and SV analysis, all gave frictional ratios for WM Gag protein of 1.59–1.67, suggesting that it is a highly asymmetric molecule. The hydrodynamic and SANS data showed that the protein has an Rg value of 34 Å and an Rh value of 41 Å. These values are clearly higher than those expected for a sphere of MW 5 × 104 Da (18.9 Å and 27.4 Å, respectively, assuming 0.3 g of H2O per g of protein in the Rh calculation). At the same time, they are far lower than those for a rod-shaped molecule of the same mass. For example, the Gag molecules in immature HIV-1 particles are approximately 250 Å long;26 the Rg value of these rods would be (1/12)1/2 times the length of the rod, or ∼72 Å. Since there are known to be several flexible regions in the molecule, it seems likely that the high frictional ratio does not reflect a rigid, extended structure like a prolate ellipsoid, but rather the multi-domain character of the protein.27 One would expect the peak value of P(r) for a protein composed of a series of globular domains to be the average of the peak P(r) values for the individual domains, while the Dmax value would be significantly larger than those for the domains. In accordance with these expectations, the peak P(r) for WM Gag, 30 Å (Figure 3(b)), is similar to the individual dimensions of the MA, NTD, and CTD domains, but the Dmax value for the protein is 110 Å (Figure 3(b)).
It seems possible that the folded conformation of Gag is caused by a specific interaction between its MA and NC domains. Alternatively, however, the conformation could be due to simple entropic effects: that is, in the absence of specific constraints, a fully extended arrangement of the domains in a multi-domain protein is relatively unlikely. While our data cannot exclude the possibility of a specific interaction between the terminal regions of the protein, we tend to favor the entropic explanation because of its simplicity. In addition, it is clear that the MA domain can adopt a wide variety of orientations relative to the NTD of CA (see Supplementary Data, Figure S1),16 and free NC is largely unstructured except for its two zinc fingers, as indicated by the diversity of structures in the 1MFS series. A specific interaction between the MA and NC domains also seems unlikely in view of these considerations. However, the data clearly indicate that MA and NC can be close together in three-dimensional space, and thus it is possible that both ends of the protein may interact with the same binding partner(s) under specific circumstances in vivo.
It is instructive to compare the evaluations of the 4800 models in Figures 4 and 5. In Figure 4, the models are divided into three groups according to their p2 structures, while in Figure 5 they are grouped according to their MA structures. Figure 4(a) and (d) shows that an extended p2 is incompatible with the data, since the best structure in this group has a relatively high χ2 value (7.1) when tested against the SANS data. However, the overall properties of the three groups are otherwise rather similar to each other: in each case, the Rg values span a range of ∼40 Å, and all three groups include structures with very poor fits (χ2 ≥ 25). In contrast, the disposition of the MA domain relative to CA appears to have a greater role in determining how well the model fits the SANS data. In this case (Figure 5), 16 of the 20 MA orientations give very poor fits to the data, with all of their χ2 values ranging between ∼10 and ∼30. In stark contrast, fits obtained with the remaining four orientations are far superior, giving χ2 values between <2 and ∼10. In all of the 960 structures in these four groups, the majority of MA is in close proximity to the majority of CA (see right side of Figure 5). In other words, it would appear that the most salient feature of a model that fits the data well is that MA is near CA.
Implications for HIV-1 particle assembly
The data presented here and in the accompanying paper strongly suggest that the dimer interface in the CTD of CA is exposed and active in the uncleaved Gag protein. Several lines of evidence indicate that this interface in CA is involved in the assembly of mature cores following maturation cleavage of Gag.17,28-30 However, it is not known whether the same interface in Gag is used in immature particle assembly. Our data show that the ability of the WM mutant Gag to dimerize is reduced by ∼ two orders of magnitude, but is not eliminated (Datta et al., accompanying paper6; see also Figure 3, above). We found that this protein can nevertheless assemble into small VLPs in vitro (Figure 1(b)) and into particles in mammalian cells (Figure 1(c)); these observations might tend to suggest that the interface is not essential for particle assembly. On the other hand, the mutation had definite effects on assembly: the small VLPs were more salt-labile than those assembled from wild-type Gag (data not shown); no large VLPs were observed in the presence of RNA and IP5 (data not shown); and the level of virus production in mammalian cells was modestly reduced by the mutation (data not shown).17 Most strikingly, the particles assembled in vivo from the mutant protein were considerably less regular and spherical than wild-type controls (Figure 1(c)).
Taken together, the data indicate that the dimer interface does participate in immature particle assembly. It is striking that a ∼100-fold change in the strength of dimeric association (accompanying paper) has such mild effects upon assembly. However, it is important to remember that a particle is formed by the association of hundreds or thousands of Gag molecules, and the sum of their interactions might well be sufficient to maintain particle structure even when each individual interaction is far weaker than in wild-type VLPs.31 The binding of Gag protein in the cell to the plasma membrane, to RNA, or to both, may also facilitate particle assembly in vivo by helping to concentrate Gag molecules at specific sites in the cell.
The foregoing discussion has dealt exclusively with assembly by the Gag protein, which forms the immature virion in vivo. After the immature particle is released from the cell, the cleavage of Gag by PR leads to a global structural reorganization of the particle. One of the defining features of the mature particle is the “mature core”, which is a densely staining region within the virion. The mature core is composed of CA molecules. It has recently been found that only ∼1/4–1/3 of the CA molecules in a mature HIV-1 particle are part of the mature core structure, while the remaining CA proteins are apparently free “in solution” within the particle.32-34 This observation suggests that the formation of the mature core represents a second, independent assembly event during the replication cycle of the virus, rather than a rearrangement or “adjustment” of the existing contacts between the CA domains of Gag proteins in the immature particle. The structure of the mature core is relatively well understood, and an extensive literature makes it clear that the dimer interface within the CTD of CA plays a critical role in this structure.11,17,28,30 Thus, one major conclusion that emerges from the present work is that the same interface probably participates in both of the assembly events, i.e. formation of the immature particle and the subsequent formation of the mature core. This dual functionality would appear to make this interface a particularly attractive target for therapeutic intervention.
As pointed out in the accompanying paper, the fact that Gag is evidently folded over in solution carries another significant implication for virus assembly. Since an immature retrovirus particle is composed of highly elongated, rod-shaped Gag molecules,26,35,36 a Gag molecule must undergo a drastic conformational change during the process of assembly. It would obviously be of great importance to learn what induces this change.
Materials and Methods
Protein expression and purification
Gag protein was expressed and purified as described in the accompanying paper (Datta et al.6). 2H2O was substituted for H2O in the buffer used for SANS experiments.
VLP production in mammalian cells
The full-length, myristylated HIV-1 Gag protein and the corresponding WM mutant protein were expressed in mammalian cells by transient transfection (using Transit 293 [Mirrus] according to the manufacturer's instructions) of the plasmid pCMV55M1-10 and the WM mutant of this plasmid into 293T cells. This plasmid encodes a Rev-independent HIV-1 gag gene, containing a number of silent mutations eliminating the Rev requirement, under the control of the cytomegalovirus major late promoter.37 Three days after transfection, the cells were fixed and processed for electron microscopy as described.38
Mutagenesis
Mutations were introduced into the mammalian Gag expression vector pCMV55M1-10 by QuikChange (Stratagene), according to the manufacturer's instructions.
In vitro assembly and analysis by EM
Proteins at 5–10 mg/ml in storage buffer were diluted to 1 mg/ml by dropwise addition of 20 mM Tris (pH 8.0), 10 mM DTT at room temperature. Yeast tRNA was added prior to dilution at a protein:RNA ratio of 4% (w/w). Assembly reactions were allowed to proceed for 2 h before examination by negative staining using 2% (w/v) uranyl acetate on Formvar-coated grids.
Size exclusion chromatography and light scattering
A Rainin HPXL solvent delivery system connected to a Rainin Dynamax UV-1 detector and a Wyatt systems Dawn EOS static and quasi-elastic light scattering detector was used to study the hydrodynamic properties of the proteins on a Superose 12 (GE Healthcare) column. Solvent viscosity of 0.00936 centipoise and a temperature dependence of (−1.95E-4 g/cm s K) were assumed. Data collected simultaneously from light detectors 5 through 18, with the exception of detector 13, was used for SLS, while detector 13 was modified for QELS measurements. Other details were as described in the accompanying paper (Datta et al.6). The column was calibrated with standards (GE Healthcare) including RNase A (Rh 16.4 Å); chymotrypsin (Rh 20.9 Å); ovalbumin (Rh 30.5 Å); BSA (Rh 35.5 Å); aldolase (Rh 48.1 Å); and catalase (Rh 52.2 Å).
Sedimentation velocity measurements
Boundary SV analysis was carried out in an Optima XLA analytical ultracentrifuge (Beckman-Coulter Instruments); 1 mM DTT was included in the buffer used in these experiments. SV analysis was performed at 20 °C. The centrifuge cell was filled with 400 μl of protein solution. Absorbance scans were obtained at either 280 nm or 250 nm. To deconvolute the boundary velocity data into sedimenting species, we analyzed the sedimentation coefficient distribution with previously described computational methodology.39 This approach is implemented using the public domain software Sedfit†. Briefly, the sedimentation boundary velocity data were subjected to maximum entropy regularization statistical analysis for the most parsimonious distribution of sedimenting species39 that best fit the data. The results of this computational analysis give c(s) versus s plots where c(s) is the concentration of protein divided by the sedimentation coefficient at the respective s position. Hydrodynamic analysis of the data was performed using the software Sednterp‡. Sednterp was also used to determine the partial specific volume of the proteins and the viscosity (η) and density (ρ) for the buffers used in the sedimentation analyses.
Circular dichroism
CD measurements were performed using an AVIV 202 spectropolarimeter. Proteins were analyzed at 10–15 μMin 10 mM sodium phosphate (pH 7.6),0.5 M NaCl,1 mM DTT. Far UV CD spectra were acquired in 0.1 cm path-length cells. The temperature was maintained at 25 °C using a Peltier temperature controller.
SANS measurements
SANS measurements were performed on the 30-meter SANS instruments at the NIST Center for Neutron Research in Gaithersburg, MD.40 The neutron wavelength, λ, was 5 Å, with a wavelength spread, Δλ/λ, of 0.15. Scattered neutrons were detected with a 64 cm × 64 cm two-dimensional position-sensitive detector with 128 × 128 pixels at a resolution of 0.5 cm/pixel. Raw counts were normalized to a common monitor count and corrected for empty cell counts, ambient room background counts and non-uniform detector response. Data were placed on an absolute scale by normalizing the scattered intensity to the incident beam flux. Finally, the data were radially-averaged to produce scattered intensity, I(Q), versus Q curves, where Q =4πsin(θ)/λ and 2θ is the scattering angle. Sample-to-detector distances of 5.0 m and 1.5 m were used in order to cover the range 0.009 Å−1 ≤ Q ≤ 0.25 Å−1. The scattered intensities from the samples were then further corrected for buffer scattering and incoherent scattering from hydrogen in the samples.
SANS data analysis
Initial data analysis was performed using the Guinier approximation, , on the low-Q portions of the data to obtain initial values for the radius of gyration, Rg, and the forward scattering intensity, I(0), of the samples. This analysis is valid only in the region where QRg ∼ 1. The GNOM program,41 which makes use of all of the data, rather than a limited data set at small Q values, was used to determine the distance distribution function, P(r); the radius of gyration, Rg;; and the forward scattering intensity, I(0). This analysis requires the stipulation of a maximum dimension, Dmax, beyond which P(r)= 0. Typically, several values of Dmax are explored in order to find the range over which the P(r) function is stable. Since all of the data are used, this approach typically leads to more accurate determinations of Rg and I(0).
The molecular weight, MW, of the Gag protein was calculated from the forward scattering, I(0), usng the equation:
| (1) |
where Δρ =(ρ−ρs) is the contrast, or the difference between the scattering length density of the molecule (ρ) and the solvent (ρs), n is the number density of molecules and V is the molecular volume. The number density can be written as n =cNA/Mw, where c is the concentration, and NA is Avogadro's number. The volume can be written as V = Mw/(NAd), where d is the mass density. Now, equation (1) can be rewritten as:
| (2) |
The only unknown parameter in equation (2) is the Mw, since all other parameters can be measured or calculated. The I(0) value is generally taken from the GNOM41 analysis of the data. The concentration can be directly measured during sample preparation and Δρ can be calculated from the chemical composition of the sample and solvent. The mass density, d, is taken as the inverse of the partial specific volume, which is assumed to be 0.73 cm3 /g. It is important to note that I(0) must be on an absolute scale, usually in cm−1 , in order to obtain accurate Mw values from equations (1) or (2).
Modeling the WM Gag protein
The basic strategy used to construct full-length models of the Gag protein was to connect the individual domains using atomic coordinates from previously determined experimental structures. In one case, the existing structures overlap in structurally rigid regions of the protein, making unambiguous constructions possible in this region. Additionally, we exploited the fact that a collection of structures has been determined for the MA (20 structures) and NC (30 structures) domains by NMR spectroscopy, thus allowing for a degree of experimentally grounded structural heterogeneity in our modeling effort.
For the purposes of this modeling, the protein was divided into four regions. Region A consists of residues 1–144 of Gag. This roughly corresponds to the MA domain (residues 1–132), but also contains 12 residues from the N terminus of CA. The coordinates for 20 versions of region A were taken from the 1L6N structure, which includes residues 1–278.16 They were joined to a single set of coordinates (structure 1E6J) for region B, which consisted of residues 145–352.42 This sequence constitutes nearly all of CA. Since the protein analyzed in the 1E6J structure has an N-terminal extension and lacks the authentic N-terminal proline of HIV-1 CA protein, it does not form the β-hairpin found at the N terminus of mature CA. The overlap between the 1L6N and 1E6J structures (i.e. residues 145–278) was aligned by the algorithm of Kabsch,43,44 using the backbone atoms of the connecting polypeptide fragments. Region C (resi-dues 353–377) contained the last 11 residues of CA together with the p2 “spacer” between CA and NC. There is no published structural information on this stretch of Gag, and eight hypothetical models of its structure were incorporated into the Gag models as described below. Finally, region D is identical to the 55 residues of mature NC protein (residues 378–432 of Gag). Thirty versions of region D were taken from 1MFS.45 Although the protein studied experimentally in this work also contains, at its extreme C terminus, the 16-residue “spacer” found between NC and p6 in authentic HIV-1 Gag protein, this spacer (approximately 3.5% of the protein) was not included in the models we constructed. Structures were minimized using the CHARMM-22 force field46 incorporated in the CHARMM molecular dynamics program.47 Use of the 1E6J coordinates for CA, containing a single structure comprising both the NTD and CTD of CA, may lead our models to slightly underestimate the flexibility of the Gag protein.
Since no definitive structural information exists for region C, we evaluated three possible models of this region of the protein with the goal of excluding certain structures or orientations by comparison of the theoretical SANS spectra with the experimental data. It was modeled as linear random coil (89 Å linear length) or completely helical (39 Å linear length) using CHARMM. In turn, each of these models was made parallel to one of the three principal axes of CA (see below). Two additional configurations of region C, termed “partially helical” structures (each 24 Å in length), were selected from a pair of simulated annealing runs. Briefly, a completely helical region C was attached to the coordinates of region B and solvated with TIP3P water48 for a total of ∼45,000 atoms. One hundred cycles of heating at 1400 K (100 ps), cooling to 300 K, and equilibration at 300 K (100 ps) were performed using periodic boundary conditions. The coordinates of the CA domain were held fixed. In both cases, the initial helical structure was largely lost after 20 cycles and only minor structural variations occurred after 50 cycles.
At present, there are no experimental coordinates at either the B:C or the C:D junction. Regions B and C were joined as follows. The backbone dihedral angles of the last four residues of region B were varied such that the first four residues of region C were aligned along one of the three principal axes of CA. After forming the peptide bond between regions B and C at each of the orientations, the atoms of the last four amino acids of B and the first four amino acids of C were energy-minimized while keeping all other atoms rigid. In contrast, regions C and D were joined simply by forming a peptide bond between the last amino acid of C and the first amino acid of D.
When assembled in all possible combinations, the 20 region A structures, single region B structure, eight region C structures, and 30 region D structures generated 4800 models of the WM Gag protein. The resulting models were then energy-minimized using CHARMM. Predicted SANS spectra were calculated for each of the models using the program Xtal2Sas.49,50 The calculated results were compared to the SANS intensity at 19 values of Q from 0.01 to 0.18 Å−1 sampled every 0.01 Å. I(0) was estimated from the Guinier extrapolation to Q = 0. The quality of the fit was determined by calculating:
| (3) |
value over the N = 19 grid points. Rh values for each structure were computed using HYDROPRO.51 For simplicity, region A is referred to as the MA domain, B the CA domain, C the p2 domain, and D the NC domain in the descriptions of the models elsewhere in this paper.
Supplementary Material
Acknowledgements
We thank Dr Barbara Felber for the kind gift of the pCMV551-10 plasmid. We also acknowledge the excellent technical assistance of Jane Mirro and Demetria Harvin, and electron microscopy by Ferri Soheilian and Kunio Nagashima. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations used
- HIV-1
human immunodeficiency virus type 1
- VLP
virus-like particle
- IP5
inositol pentakisphosphate
- PR
protease
- MA
matrix
- CA
capsid
- NC
nucleocapsid
- NTD
N-terminal domain of CA
- CTD
C-terminal domain of CA
- SANS
small angle neutron scattering
- SLS
static light scattering
- BSA
bovine serum albumin
- MW
molecular weight
- QELS
quasi-elastic light scattering
- SV
sedimentation velocity
- PMSF
phenylmethylsulfonyl fluoride
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2006.10.073
References
- 1.Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retro-viruses. Cold Spring Harbor Laboratory Press; Plainview, NY: 1997. pp. 263–334. [PubMed] [Google Scholar]
- 2.Campbell S, Vogt VM. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 1995;69:6487–6497. doi: 10.1128/jvi.69.10.6487-6497.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 1999;73:2270–2279. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu F, Joshi SM, Ma YM, Kingston RL, Simon MN, Vogt VM. Characterization of Rous sarcoma virus Gag particles assembled in vitro. J. Virol. 2001;75:2753–2764. doi: 10.1128/JVI.75.6.2753-2764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell S, Fisher RJ, Towler EM, Fox S, Issaq HJ, Wolfe T, et al. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl Acad. Sci. USA. 2001;98:10875–10879. doi: 10.1073/pnas.191224698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta SAK, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, et al. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J. Mol. Biol. 2006;365:799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional structure of the human immuno-deficiency virus type 1 matrix protein. J. Mol. Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 8.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 9.Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 10.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl Acad. Sci. USA. 1996;93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, et al. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 12.von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998;17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 14.Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 Å resolution. Acta Crystallog. sect D. 1999;55(Pt 1):85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- 15.Amarasinghe GK, De Guzman RN, Turner RB, Chancellor KJ, Wu ZR, Summers MF. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the psi-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 2000;301:491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 16.Tang C, Ndassa Y, Summers MF. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nature Struct. Bio. 2002;9:537–543. doi: 10.1038/nsb806. [DOI] [PubMed] [Google Scholar]
- 17.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 2003;77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel LM, Monty KJ. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- 19.Mortuza GB, Haire LF, Stevens A, Smerdon SJ, Stoye JP, Taylor IA. High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature. 2004;431:481–485. doi: 10.1038/nature02915. [DOI] [PubMed] [Google Scholar]
- 20.Ivanov D, Stone JR, Maki JL, Collins T, Wagner G. Mammalian SCAN domain dimer is a domain-swapped homolog of the HIV capsid C-terminal domain. Mol. Cell. 2005;17:137–143. doi: 10.1016/j.molcel.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Accola MA, Hoglund S, Gottlinger HG. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J. Virol. 1998;72:2072–2078. doi: 10.1128/jvi.72.3.2072-2078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krausslich HG, Facke M, Heuser AM, Konvalinka J, Zentgraf H. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 1995;69:3407–3419. doi: 10.1128/jvi.69.6.3407-3419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J. Virol. 2002;76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman JL, Butcher EW, Patel DT, Mikhaylenko Y, Summers MF. Flexibility in the P2 domain of the HIV-1 Gag polyprotein. Protein Sci. 2004;13:2101–2107. doi: 10.1110/ps.04614804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. Helical structure determined by NMR of the HIV-1 (345-392) Gag sequence, surrounding p2: implications for particle assembly and RNA packaging. Protein Sci. 2005;14:375–386. doi: 10.1110/ps.041087605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, et al. Organization of immature human immunodeficiency virus type 1. J. Virol. 2001;75:759–771. doi: 10.1128/JVI.75.2.759-771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia de la Torre J, Perez Sanchez HE, Ortega A, Hernandez JG, Fernandes MX, Diaz FG, Lopez Martinez MC. Calculation of the solution properties of flexible macromolecules: methods and applications. Eur. Biophys. J. 2003;32:477–486. doi: 10.1007/s00249-003-0292-0. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- 29.Ganser BK, Cheng A, Sundquist WI, Yeager M. Three-dimensional structure of the M-MuLV CA protein on a lipid monolayer: a general model for retroviral capsid assembly. EMBO J. 2003;22:2886–2892. doi: 10.1093/emboj/cdg276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 2004;78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zlotnick A. Are weak protein-protein interactions the general rule in capsid assembly? Virology. 2003;315:269–274. doi: 10.1016/s0042-6822(03)00586-5. [DOI] [PubMed] [Google Scholar]
- 32.Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nature Struct. Mol. Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- 33.Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nature Struct. Mol. Biol. 2004;11:676–677. doi: 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- 34.Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J. Mol. Biol. 2005;346:577–588. doi: 10.1016/j.jmb.2004.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuller SD, Wilk T, Gowen BE, Krausslich HG, Vogt VM. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr. Biol. 1997;7:729–738. doi: 10.1016/s0960-9822(06)00331-9. [DOI] [PubMed] [Google Scholar]
- 36.Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: implications for retroviral assembly mechanisms. Proc. Natl Acad. Sci. USA. 1998;95:7299–7304. doi: 10.1073/pnas.95.13.7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider R, Campbell M, Nasioulas G, Felber BK, Pavlakis GN. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J. Virol. 1997;71:4892–4903. doi: 10.1128/jvi.71.7.4892-4903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muriaux D, Costes S, Nagashima K, Mirro J, Cho E, Lockett S, Rein A. Role of murine leukemia virus nucleocapsid protein in virus assembly. J. Virol. 2004;78:12378–12385. doi: 10.1128/JVI.78.22.12378-12385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glinka CJ, Barker JG, Hammouda B, Krueger S, Moyer JJ, Orts WJ. The 30 m small-angle neutron scattering instruments at the National Institute of Standards and Technology. J. Appl. Crystallog. 1998;31:430–445. [Google Scholar]
- 41.Semenyuk AV, Svergun DI. GNOM–a program package for small-angle scattering data processing. J. Appl. Crystallog. 1991;24:537–540. [Google Scholar]
- 42.Berthet-Colominas C, Monaco S, Novelli A, Sibai G, Mallet F, Cusack S. Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 1999;18:1124–1136. doi: 10.1093/emboj/18.5.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallog. sect. A. 1976;32:922–923. [Google Scholar]
- 44.Kabsch W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallog. sect. A. 1978;34:827–828. [Google Scholar]
- 45.Lee BM, De Guzman RN, Turner BG, Tjandra N, Summers MF. Dynamical behavior of the HIV-1 nucleocapsid protein. J. Mol. Biol. 1998;279:633–649. doi: 10.1006/jmbi.1998.1766. [DOI] [PubMed] [Google Scholar]
- 46.MacKerell AD, Jr., Brooks B, Brooks CD, III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The energy function and its parametrization with an overview of the program. In: Schleyer P. v. R., Allinger NL, Clark T, Gasteiger J, Kollman PA, Schaefer HF, III, Schreiner PR., editors. Encyclopedia of Computational Chemistry. Vol. 1. John Wiley and Sons; Chichester: 1998. pp. 271–277. [Google Scholar]
- 47.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983;4:187–217. [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 49.Heidorn DB, Trewhella J. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry. 1988;27:909–915. doi: 10.1021/bi00403a011. [DOI] [PubMed] [Google Scholar]
- 50.Krueger S, Gorshkova I, Brown J, Hoskins J, McKenney KH, Schwarz FP. Determination of the conformations of cAMP receptor protein and its T127L,S128A mutant with and without cAMP from small angle neutron scattering measurements. J. Biol. Chem. 1998;273:20001–20006. doi: 10.1074/jbc.273.32.20001. [DOI] [PubMed] [Google Scholar]
- 51.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.