

Abstract
The microtubule-associated protein tau is important to normal neuronal function in the mammalian nervous system. Aggregated tau is the major component of neurofibrillary tangles (NFTs), present in several neurodegenerative diseases, including Alzheimer’s and frontotemporal dementia with Parkinsonism (FTDP). Splicing misregulation of adult-specific exon 10 results in expression of abnormal ratios of tau isoforms, leading to FTDP. Positions +3 to +16 of the intron downstream of exon 10 define a clustering region for point mutations that are found in FTDP. The serine/arginine-rich (SR) factor 9G8 strongly inhibits inclusion of tau exon 10. In this study, we established that 9G8 binds directly to this clustering region, requires a wild-type residue at position +14 to inhibit exon inclusion, and RNAi constructs against 9G8 increase exon 10 inclusion. These results indicate that 9G8 plays a key role in regulation of exon 10 splicing and imply a pathogenic role in neurodegenerative diseases.
Introduction
Alternative splicing is a versatile and widespread mechanism for generating multiple mRNAs from a single transcript (Grabowski, 1998; Graveley, 2001). Bioinformatics analysis of human genome indicates that more than 75% of human genes are alternatively spliced (Johnson et al., 2003; Kampa et al., 2004). Alternative splicing plays a critical role in controlling differentiation and development (Stamm et al., 2005), and misregulation of alternative splicing is the cause of many life-threatening human diseases (Faustino and Cooper, 2003; Stoilov et al., 2002).
Tau is a microtubule-associated protein enriched in axons of mature and growing neurons (reviewed in Andreadis, 2005). Hyperphosphorylated, microtubule-dissociated tau protein is the major component of neurofibrillary tangles, a hallmark of many neurodegenerative diseases (Goedert and Jakes, 2005).
The human tau gene contains sixteen exons, of which at least three (exons 2, 3, and 10) are alternatively spliced cassettes whose combinations produce six tau protein isoforms (Andreadis, 2005). Exons 2 and 3 modulate the N-terminus of tau protein that interacts with the plasma membrane (Brandt et al., 1995). Exon 10 modulates the C-terminus of the tau protein and encodes an additional microtubule binding domain (Lee et al., 1989). Exon 10 is adult-specific in rodents and humans (Goedert et al., 1989a, 1989b; Kosik et al., 1989) but with a crucial difference relevant to neurodegeneration: in adult rodents, exon 10 becomes constitutive (Kosik et al., 1989). In contrast, in adult humans exon 10 remains regulated in the central nervous system where the two possible isoforms (10+ and 10−) are present in a 1:1 ratio (Gao et al., 2000; Goedert et al., 1989b). The domain encoded by exon 10 increases the affinity of tau protein to microtubules and may be important in the transition from the more fluid fetal cytoskeleton to the more stable adult one (Himmler et al., 1989; Lee et al., 1989; Mandelkow et al., 1995). Misregulation of tau exon 10 splicing causes inherited frontotemporal dementia with Parkinsonism (FTDP-17; Goedert and Jakes, 2005). Tau exon 10 misregulation has also been observed in sporadic AD (Glatz et al., 2006).
Splicing is carried out by the spliceosome, a large and dynamic complex of proteins and small RNAs (Neubauer et al., 1998). The rules governing splice site selection are not fully understood; combinatorial control and “weighing” of splice element strength are used to enable precise recognition of the short and degenerate splice sites (Smith and Valcárcel, 2000). Despite the high fidelity of exon recognition in vivo, it is currently impossible to accurately predict alternative exons (Stoilov et al., 2002; Thanaraj et al., 2004). Exonic and intronic enhancers and silencers are involved in splicing regulation (Cartegni et al., 2003; Zheng, 2004). Mutations in these elements can result in human disease by causing aberrant splicing (Faustino and Cooper, 2003; Stoilov et al., 2002).
Exonic and intronic enhancers and silencers are regulated by splicing factors. Mammalian splicing regulators mostly belong to two superfamilies, the SR/SR-like and hnRNP proteins, neither of which is exclusively involved in alternative splicing (Dreyfuss et al., 2002; Graveley, 2000). The former are also components of the spliceosome, whereas the latter are also involved in pre-mRNA transport, RNA stability and translational regulation. Several mammalian splicing factors are enhanced in or restricted to neurons. Nevertheless, it appears that the exquisite calibration of mammalian alternative splicing is primarily achieved by SR and hnRNP proteins which show distinct ratios in tissues and during development, despite their ubiquitous distribution (Hanamura et al., 1998; Kamma et al., 1995).
Exon 10 splicing is affected by exonic and intronic enhancers and silencers (D'Souza et al., 1999; D'Souza and Schellenberg, 2000 D'Souza and Schellenberg, 2002; Gao et al., 2000; Grover et al., 1999; Hutton et al., 1998; Stanford et al., 2003; Wang et al., 2004), as well as by several trans factors and their phosphorylation (Broderick et al., 2004; Gao et al., 2000; Hartmann et al., 2001; Jiang et al., 2003; Kondo et al., 2004; Wang et al., 2004, 2005). Two regulatory elements and the factors that regulate them have been characterized within exon 10: an AT rich silencer that binds SRp30c and SRp55 which inhibit inclusion of exon 10 (Wang et al., 2005) and a purine-rich enhancer directly downstream of the silencer that binds htra2beta1 which activates inclusion of exon 10 (Jiang et al., 2003; Wang et al., 2005).
Investigations of dementia pedigrees have established that mutations in the downstream intron of exon 10 result in misregulation of tau exon 10 splicing and cause FTDP-17 (Goedert and Jakes, 2005; Hutton et al., 1998; Stanford et al., 2003). However, the regulators involved were unknown up to now. In this report we investigate the mechanism of action of SR protein 9G8, which inhibits inclusion of exon 10 (Wang et al., 2004;). Our results show that 9G8 directly interacts with the proximal downstream intron of exon 10, a clustering region for FTDP mutations (D'Souza et al., 1999; Hutton et al., 1998; Stanford et al., 2003).
Results
9G8 inhibits inclusion of tau exon 10 by interacting with the downstream intron of the exon
SR proteins most commonly act as splicing activators by binding to exonic splicing enhancers (ESEs; Graveley, 2000; Shen et al., 2004; Zheng, 2004). We found that SR proteins SRp30c, SRp55 and 9G8 strongly inhibit inclusion of exon 10 (Wang et al., 2004) and that SRp30c and SRp55 do so by binding to an AT-rich silencer near the 5’ end of exon 10 (Wang et al., 2005).
The inhibitory action of 9G8 on inclusion of exon 10 is shown in Fig. 1C. To dissect the mechanism of the 9G8 inhibitory action, we first co-transfected 9G8 expression constructs with miningene reporters containing exon 10 deletions that delimit exonic silencers whose interacting partners are still unknown (Δ8/9 and Δ15; Wang et al., 2005). Both deletions still show inhibition of exon 10 inclusion by 9G8 (Fig. 2B, lanes 3–6), indicating that neither is the site responding to this factor.
Fig. 1.
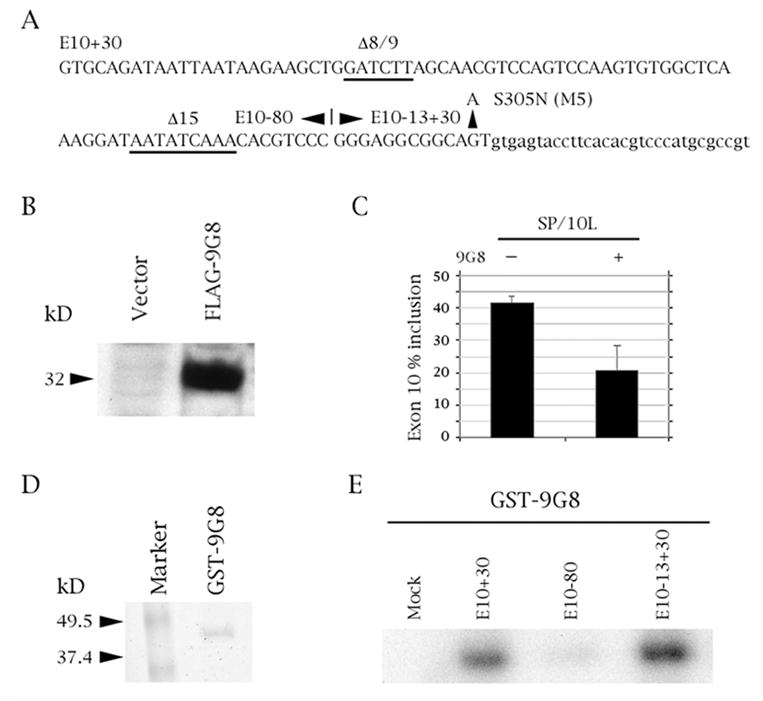
SR factor 9G8 inhibits splicing of tau exon 10 by binding to the proximal downstream intron. (A) Tau exon 10 (uppercase) and its downstream intron (lowercase). Relevant exonic deletions are underlined and point mutation S305N (M5) is shown. The boundaries of the three riboprobe constructs are indicated. E10+30 contains exon 10 plus 30 nucleotides of its downstream intron, E10-80 contains the 80 5’-most nucleotides of exon 10, E10-13+30 contains the 13 3’most nucleotides of exon 10 plus 30 nucleotides of its downstream intron. (B, D) Expression of FLAG-9G8 and GST-9G8 fusion proteins, respectively detected by (B) Anti-FLAG monoclonal antibody and (D) Coomassie blue staining. (C) RT-PCR of SP/10L in COS cells in the absence and presence of 9G8. The RT-PCR products come from 1:1 co-transfections of SP/10L and FLAG-9G8. The identities of the spliced species are indicated. Primer pair: SPL-LS/SPL-LN. % exon inclusion was calculated by scanning the bands from three independent transfections and measuring their areas using the OneDscan analysis program. (E) UV-crosslinking of GST-9G8 to the three riboprobes indicated in (A). Equal amounts of the fusion protein were present in each experiment.
Fig. 2.
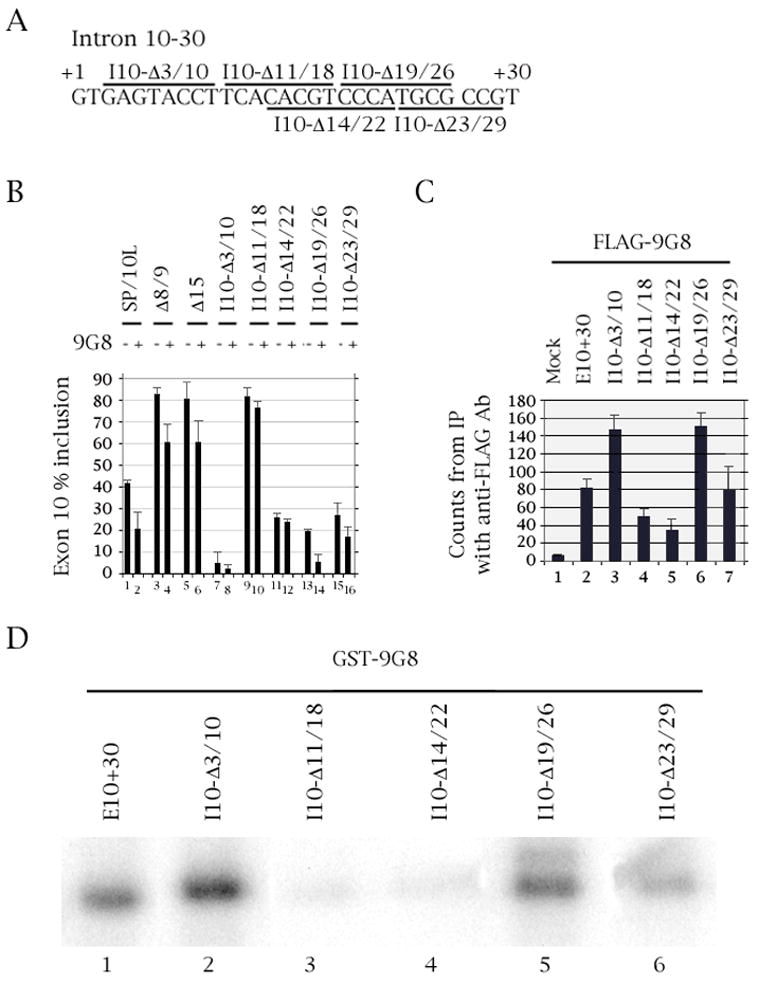
Deletion of residues 11-18 of the intron downstream of tau exon 10 abolishes 9G8 action and binding. (A) Nucleotides +1 to +30 of the intron downstream of tau exon 10. The deletions are demarcated by bars above or below the sequence. (B) RT-PCR of wild-type and deleted SP/10L in COS cells in the absence and presence of 9G8. The RT-PCR products come from 1:1 co-transfections of tau constructs and FLAG-9G8. Exon ratio calculations, primers and graph conventions are as in Fig.1C. (C) 32P-labeled riboprobes containing wild-type or deleted exon 10 were incubated with extracts from COS cells transfected with FLAG-9G8 and were immunoprecipitated by anti-FLAG monoclonal antibody. Amounts of riboprobe bound to 9G8 protein were calculated relative to the nonspecific binding of the mock vector transfection (means ± SD of three analyses). (D) UV-crosslinking of GST-9G8 to the deletion substrates indicated in (A). Equal amounts of the fusion protein were present in each experiment.
Next, we separated exon 10 and its proximal downstream intron into two “halves” (Fig. 1A). UV-crosslinking showed that 9G8 attaches to the 3’ “half”, which consists of the 13 3’most nucleotides of exon 10 and 30 bp of the intron (Fig. 1E). Based on this result, we concentrated our subsequent efforts on this region.
Identification of the intronic silencer downstream of exon 10 as the site of 9G8 influence
We created five overlapping deletions covering the 30 nucleotides directly downstream of exon 10 (Δ3/10, Δ11/18, Δ14/22, Δ19/26, Δ23/29; Fig. 2A). The splicing behavior of the deletions (Fig. 2B) is in agreement with the results from other laboratories which defined region 11–18 as an intronic splicing silencer (ISS) and 19–26 as an intronic splicing modifier (ISM; D’Souza and Schellenberg, 2002).
Δ3/10 almost entirely abolishes inclusion of exon 10, as it deletes the region required for interaction with the U1 snRNA (Fig. 2B, lane 7). Δ11/18 and Δ19/26 increase and decrease inclusion of exon 10, respectively (Fig. 2B, lanes 9 and 15), in agreement with the behavior of the FTDP point mutants in the same regions (discussed in the next section). Finally Δ14/22 and Δ23/29 also show decreased inclusion of exon 10 (Fig. 2B, lanes 11 and 13), confirming the conclusion that this element does not act as a splicing enhancer, but as a modifier of the silencer immediately upstream of it.
Two of the deletions (Δ11/18 and Δ14/22) no longer show inhibition of exon 10 inclusion by 9G8 in co-transfections (Fig. 2B, lanes 9–12). In RNA-protein pulldowns and UV-crosslinking experiments, the same two deletions show greatly decreased binding to 9G8 (Fig. 2C, lanes 4 and 5; Fig. 2D, lanes 3 and 4). Deletion Δ23/29 binds 9G8 as strongly as wild type exon 10 (Fig. 2C, lane 7; Fig. 2D, lane 6) in agreement with its behavior in co-transfections (Fig. 2B, lanes 15–16). Interestingly, deletions Δ3/10 and Δ19/26 bind 9G8 as strongly as wild type (Fig. 2C, lanes 3 and 6; Fig. 2D, lanes 2 and 5), though they show a decrease in inhibition of exon 10 inclusion by 9G8 in co-transfections (Fig. 2B, lanes 7–8 and 13–14).
The behavior of the exon 10 deletions in co-transfections, pulldowns and UV-crosslinking led us to the preliminary conclusion that 9G8 exerts its effect on a sequence that includes nucleotides 14–18 of the intron downstream of exon 10 within the ISS. To further pinpoint the region of influence, we created point mutations in the same region.
The FTDP mutation at position 14 downstream of exon 10 abolishes 9G8 action
We recreated six point mutations found in FTDP pedigrees (Fig. 3A). One (S305N, M5) is at position –2, at the 3’ end of exon 10. The others (M11, 12, 13, 14, 16) are in the corresponding positions of the downstream intron of exon 10 (D’Souza and Schellenberg, 2002; Gao et al., 2000; Wang et al., 2004). All point mutations significantly increase inclusion of exon 10 (Fig. 3B, odd-numbered lanes).
Fig. 3.
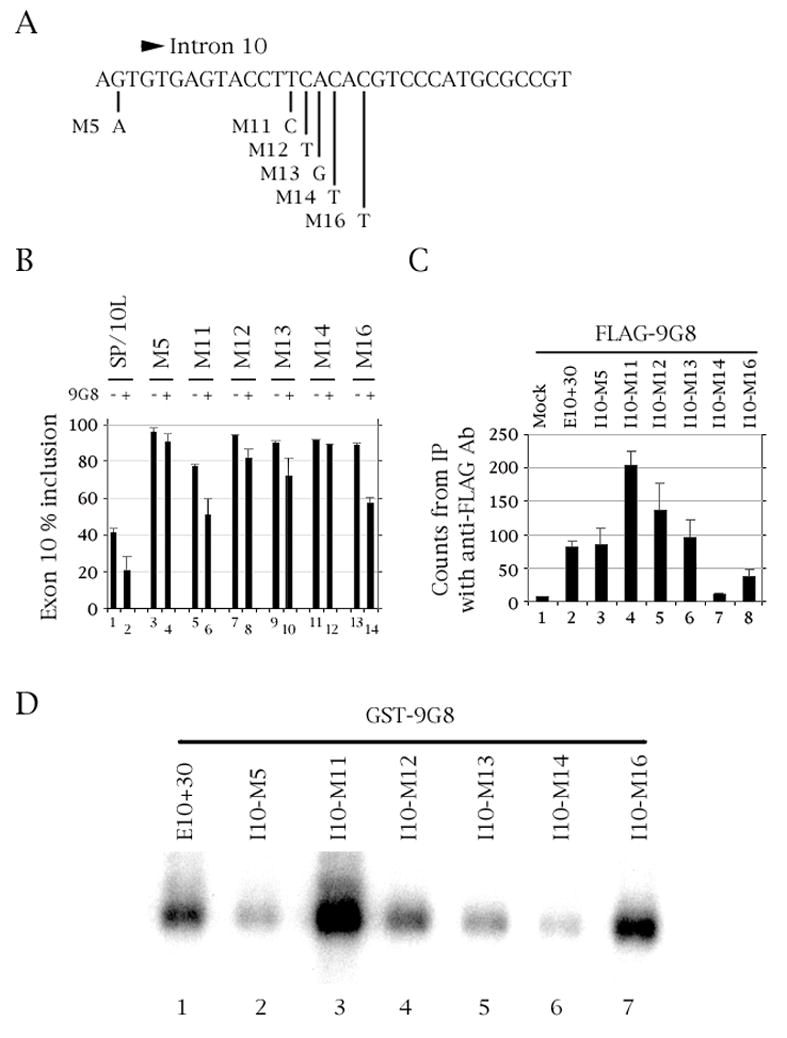
Mutation of residue +14 of the intron downstream of tau exon 10 abolishes 9G8 action and binding. (A) The last three nucleotides of exon 10 and nucleotides +1 to +30 of the intron downstream of tau exon 10. The point mutations are indicated. (B) RT-PCR of wild-type and point-mutated SP/10L in COS cells in the absence and presence of 9G8. The RT-PCR products come from 1:1 co-transfections of tau constructs and FLAG-9G8. Exon ratio calculations, primers and graph conventions are as in Fig. 1C. (C) 32P-labeled riboprobes containing wild-type or point-mutated exon 10 were incubated with extracts from COS cells transfected with FLAG-9G8 and were immunoprecipitated by anti-FLAG monoclonal antibody. Amounts of riboprobe bound to 9G8 protein were calculated relative to the nonspecific binding of the mock vector transfection (means ± SD of three analyses). (D) UV-crosslinking of GST-9G8 to the deletion substrates indicated in (A). Equal amounts of the fusion protein were present in each experiment.
In co-transfections, point mutations M11 and M16 show inhibition of exon 10 inclusion by 9G8 as much as wild type (~33% relative decrease in exon inclusion; Fig. 3B, lanes 5–6 and 13–14). Inclusion of exon 10 in mutants M5, M12 and M13 is partly inhibited by 9G8, the relative strength of the inhibition being M13~M12>M5 (Fig. 3B, lanes 3–4, 7–8 and 9–10). Finally, M14 no longer shows inhibition of exon 10 inclusion by 9G8 (Fig. 3B, lanes 11–12).
In RNA pulldowns with 9G8, mutants M5 and M13 interact with 9G8 as strongly as wild-type exon 10 (Fig. 3C, lanes 3 and 6). M11 and M12 show strong and moderate increase in interaction, respectively (Fig. 3C, lanes 4 and 5), whereas M16 shows interaction decreased by half but still ~3-fold above background (Fig. 3C, lane 8). However, M14 shows no interaction above background (Fig. 3C, lane 7).
In UV-crosslinking experiments, 9G8 binds to mutants M11 and M16 more strongly than to the wild-type probe (Fig. 3D, lanes 3 and 7). Binding to M12 is at wild-type level (Fig. 3D, lane 4) while M5 and M13 bind 9G8 roughly half as strongly as wild-type does (Fig. 3D, lanes 2 and 5). Last but not least, M14 shows extremely weak binding to 9G8 (Fig. 3D, lane 6).
Taken together, these results strongly suggest that the C residue in position +14 is critical for interaction of 9G8 with the tau exon 10 pre-mRNA.
The RNA binding domain of 9G8 is essential for regulation of exon 10 splicing
To establish which domains of 9G8 are required for regulation of exon 10, we created two 9G8 deletion variants as FLAG and GST fusions (Fig. 4A, 4C, 4E): 9G8N contains exons 1–5, which include the RNA recognition motif (RRM), a Zn knuckle domain (Zn) and the proximal half of the serine/arginine-rich (RS) region. 9G8C contains exons 3–8, which include the Zn knuckle and the entire RS domain (Cavaloc et al., 1994; Popielarz et al., 1995). Our attempts to create more extensive 9G8 deletion variants did not produce stable FLAG proteins.
Fig. 4.
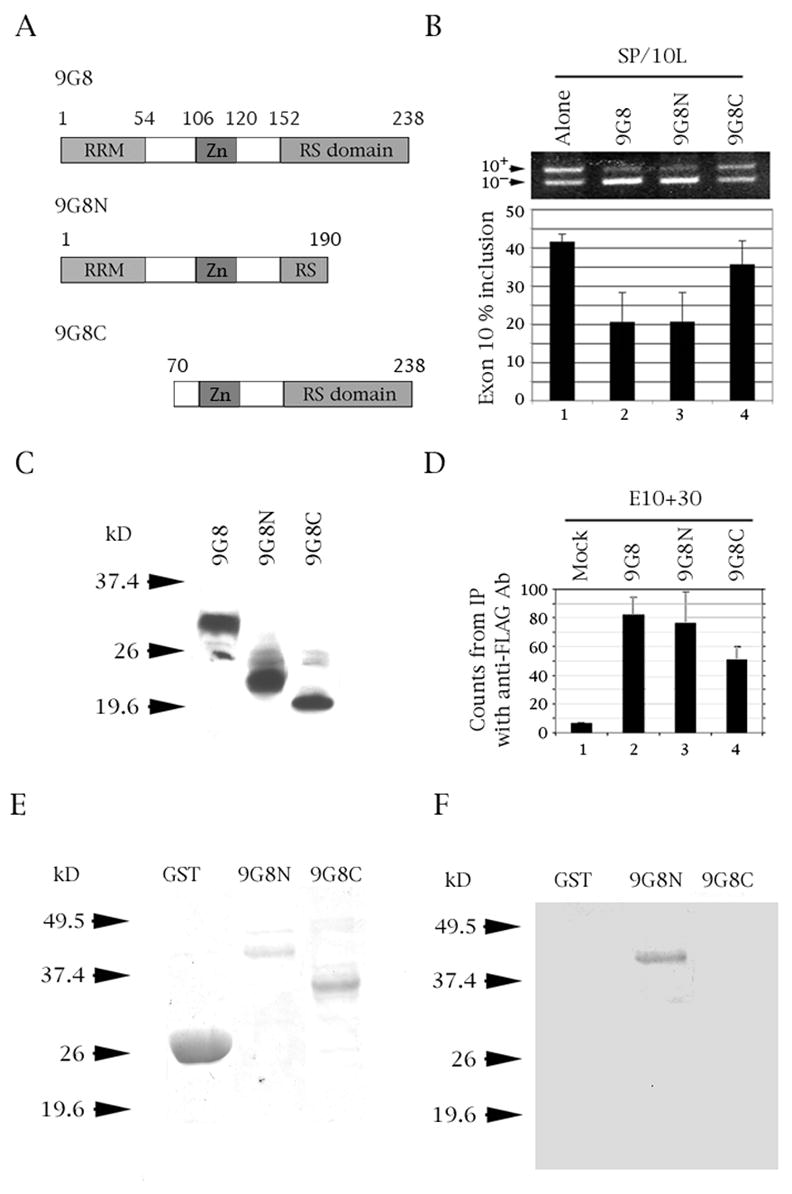
The RRM domain of 9G8 is required for inhibition of tau exon 10 splicing and binds to tau exon 10. (A) Diagram of 9G8 deletion variants. The amino acid number, RNA recognition motif (RRM), zinc knuckle (Zn) and serine/arginine-rich region (RS) are indicated. (B) RT-PCR of SP/10L in COS cells in the presence of full-length and deletion variants of 9G8. The RT-PCR products come from 1:1 co-transfections of SP/10L and FLAG-9G8 fusion constructs. Exon ratio calculations, primers and graph conventions are as in Fig. 1C. (C, E) Expression of FLAG-9G8 and GST-9G8 deleted fusion proteins, respectively detected by (C) Anti-FLAG monoclonal antibody and (E) Coomassie blue staining. (D) 32P-labeled riboprobes containing wild-type exon 10 were incubated with extracts from COS cells transfected with FLAG-9G8 variants and were immunoprecipitated by anti-FLAG monoclonal antibody. Amounts of riboprobe bound to 9G8 protein were calculated relative to the nonspecific binding of the mock vector transfection (means ± SD of three analyses). (F) UV-crosslinking of E10+30 to the 9G8 variants indicated in (A). Equal amounts of the fusion protein were present in each experiment.
In co-transfections, 9G8N is as effective as full-length 9G8 in decreasing exon 10 inclusion (Fig. 4B, lane 3), whereas 9G8C has essentially no effect (Fig. 4B, lane 4). In RNA-protein pull-downs, 9G8N associates with the exon 10 riboprobe as strongly as full-length 9G8 (Fig. 4D, lane 3), whereas 9G8C binds half as strongly (Fig. 4D, lane 4). Finally, in UV-crosslinking experiments, 9G8N binds the exon 10 riboprobe (Fig. 4F, lane 2), whereas 9GC does not (Fig. 4F, lane 3). The crosslinking results strongly indicate that the RRM domain of 9G8 is required for binding to the pre-mRNA template.
9G8 RNAi constructs reverse the inhibition of exon 10 inclusion
To establish that 9G8 is a native inhibitor of tau exon 10 inclusion, we first created and tested three RNAi constructs against the factor (9G8i-1, -2 and -3; shown in Table 1). We first tested the efficacy of these constructs on transfected tau. Upon addition of the same amounts of SP/10L and each 9G8i construct to COS cells, the three constructs but not the pFIV vector relieve the inhibition of exon 10 by 9G8 (Fig. 5A, left panel). The relative strength of the RNAi reversal effect is 1 ≈3>2 (Fig 5A, left panel, lanes 3–5).
Table 1.
Primers
| Name | Length | Strand | Location | Sequence |
|---|---|---|---|---|
| Tau intron 10 deletion constructs | ||||
|
| ||||
| I10-Δ 3/10S* | 39 | S | In tau intron 10 | GTCCCGGGAGGCGGCAGTGT^TCACACGTCCCATGCGCCG |
| I10-Δ 11/18S* | 44 | S | In tau intron 10 | GAGGCGGCAGTGTGAGTACCT^CCCATGCGCCGTGCTGTGGCTTG |
| I10-Δ 14/22S* | 40 | S | In tau intron 10 | GGCAGTGTGAGTACCTTCA^TGCGCCGTGCTGTGGCTTGAAT |
| I10-Δ 19/26S* | 40 | S | In tau intron 10 | GTGTGAGTACCTTCACACGT^CCGTGCTGTGGCTTGAATTA |
| I10-Δ 23/29S* | 39 | S | In tau intron 10 | AGTACCTTCACACGTCCCA^TGCTGTGGCTTGAATTATTA |
|
| ||||
| Tau exon/intron 10 point mutation constructs | ||||
|
| ||||
| E10-M5S* | 27 | S | End of tau exon 10 | CCGGGAGGCGGCA TGTGAGTACCTTC |
| I10-M11S* | 33 | S | In tau intron 10 | GGCAGTGTGAGTACCT CACACGTCCCATGCGC |
| I10-M12S* | 33 | S | In tau intron 10 | GCAGTGTGAGTACCTT ACACGTCCCATGCGCC |
| I10-M13S* | 33 | S | In tau intron 10 | CAGTGTGAGTACCTTC CACGTCCCATGCGCCG |
| I10-M14S* | 35 | S | In tau intron 10 | GGCAGTGTGAGTACCTTCA ACGTCCCATGCGCCG |
| I10-M16S* | 35 | S | In tau intron 10 | GGCAGTGTGAGTACCTTCACA GTCCCATGCGCCG |
|
| ||||
| PCR of endogenous and transfected tau 10 | ||||
|
| ||||
| HT7S3 | 24 | S | In tau exon 7 | CAACGCCACCAGGATTCCAGCAAA |
| HT11N | 24 | A | In tau exon 11 | ATGTTGCCTAATGAGCCACACTTG |
| SPL3-LS | 27 | S | In SPL3 vector | TCTGAGTCACCTGGACAACCTCAAAGG |
| SPL3-LN | 27 | A | In SPL3 vector | ATCTCAGTGGTATTTGTGAGCCAGGGC |
|
| ||||
| 9G8 deletion constructs | ||||
|
| ||||
| 9G8ex1S | 32 | S | Start of 9G8 exon 1 | CGGGATCCATGTCGCGTTACGGGCGGTACGGA |
| 9G8ex5N | 32 | A | End of 9G8 exon 5 | CCGCTCGAGTCATTGGAAATACCTCGATCCTT |
| 9G8ex3S | 32 | S | Start of 9G8 exon 3 | CGGGATCCGTGATTTGTGGCTCCCGAGTGAGT |
| 9G8ex8N | 32 | A | End of 9G8 exon 8 | CCGCTCGAGTCAGTCCATTCTTTCAGGACTTG |
|
| ||||
| 9G8 RNAi constructs | ||||
|
| ||||
| 9G8i-1S | 23 | S | In 9G8 exon 2 | AAAGAGGAGAGTTAGAAAGGGCT |
| 9G8i-1N | 23 | A | In 9G8 exon 2 | AAAAAGCCCTTTCTAACTCTCCT |
| 9G8i-2S | 23 | S | In 9G8 exon 3 | AAAGAGGGACATTATGCTTATGA |
| 9G8i-2N | 23 | A | In 9G8 exon 3 | AAAATCATAAGCATAATGTCCCT |
| 9G8i-3S | 23 | S | In 9G8 exon 6 | AAAGGATCAAGATCCAGGTCTAT |
| 9G8i-3N | 23 | A | In 9G8 exon 6 | AAAAATAGACCTGGATCTTGATC |
For creating deletions and point mutations, we used pairs of these primers and their reverse complements (not shown). S=sense, A=antisense. In the deletion primers, the caret indicates the position of the deletion. In the point mutation primers, the mutated nucleotide is boxed.
Fig. 5.
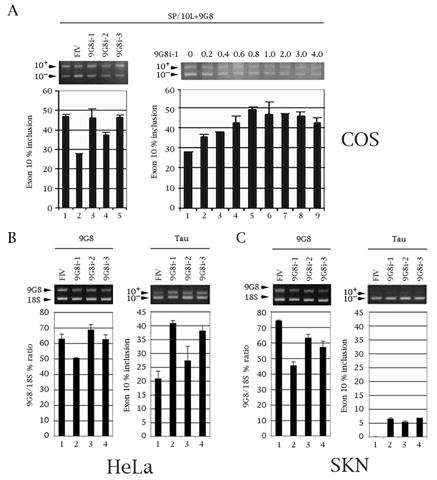
Three RNAi constructs against 9G8 decrease expression of endogenous 9G8 and counteract inhibition of endogenous tau exon 10 splicing in a sequence- and dosage-dependent manner. (A) Establishment of RNAi construct efficacy. RT-PCR of SP/10L and three 9G8 RNAi constructs in COS cells. Primer pair: SPL-LS/SPL-LN. (Left panel) 1 ug of SP/10L and 1 ug of FLAG-9G8 were co-transfected into the cells together with 1 ug of the RNAi constructs (9G8i-1, 2, 3) or the RNAi vector pFIV H1/U6 (FIV). (Right panel) RT-PCR of SP/10L with increasing amounts of construct 9G8i-1. 1 ug of SP/10L and 1ug of FLAG-9G8 were co-transfected in COS cells with the indicated amounts of RNAi-1 plasmid (in ug). (B, C) Effects of the three 9G8 RNAi constructs on endogenous 9G9 and tau in (B) HeLa and (C) SKN cells. (B, C, Left panels) Quantitative RT-PCR of endogenous 9G8. The % ratio of 9G8 to 18S is indicated. Primer pair: 9G8ex3S/9G8ex8N. (B, C, Right panels) Quantitative RT-PCR of tau exon 10. The % of tau exon 10 inclusion is indicated. Primer pair: HT7S3/HT11N. (A-C) Exon ratio calculations and graph conventions are as in Fig. 1C.
We then did a dosage curve with 9G8i-1, the construct most effective in relieving the inclusion inhibition, while keeping the amount of SP/10L and 9G8 constant at 1 ug (Fig. 5A, right panel). There is a linear response to increased amounts of the construct from 0.2 to 1 ug (Fig. 5A, right panel, lanes 2–5), after which it plateaus and even decreases slightly. This establishes that the effect is specific at low amounts of the RNAi.
We then introduced 1 ug of the three 9G8i constructs into HeLa and SKN human neuroblastoma cells and examined their effect on endogenous 9G8 and endogenous tau by quantitative RT-PCR. Both cell types express 9G8 and tau. Interestingly, the tau in HeLa is ~20% 10+ (Fig. 5B, right panel, lane 1), whereas the tau in SKN is almost exclusively 10− (Fig. 5C, right panel, lane 1). Consistent with their behavior in COS cells, the three 9G8 RNAi constructs decrease the total amount of 9G8 RNA (Fig. 5B and 5C, left panels, lanes 2–4). Concurrently, the presence of the three RNAi constructs results in a significant increase of exon 10 inclusion (Fig. 5B and 5C, right panels, lanes 2–4). Again, the relative order of efficacy of the 9G8 RNAi constructs is 1 ≈3>2.
These results unequivocally establish that 9G8 is a native inhibitor of tau exon 10 inclusion.
Discussion
9G8 inhibits inclusion of tau exon 10 by binding to the intronic silencer directly downstream of the exon
Previous work (D’Souza and Schellenberg, 2002; Gao et al., 2000; Wang et al., 2004, 2005) showed that tau exon 10 contains several silencers. Mutations in them appear in FTDP pedigrees (Goedert and Jakes, 2005) which can be separated into two groups: one is within exon 10 and includes N279K, L284L N296N (within deletions E10-Δ2/3/4, E10-Δ8/9 and E10-Δ15) and S305N (M5) (Fig. 1C); the other is in the proximal intron downstream of exon 10 and includes M3 (within Δ3/10) and M11, M12, M13, M14, M16 (within Δ11/18). Recently our laboratory extensively characterized E10-Δ2/3/4, an AT-rich silencer at the 5’ end of exon 10 which binds SRp55 and SRp30c (Wang et al., 2005).
The factors which act on E10-Δ8/9 and E10-Δ15 are not yet known, but our present work shows that 9G8 does not recognize any of the exonic silencers of exon 10. Instead, we discovered that 9G8 exerts its effect by binding to the intronic splicing silencer defined by deletion Δ11/18 and by a bevy of FTDP mutations (M11, 12, 13, 14, 16). This is unusual: in the traditional division of labor, SR proteins act on exonic elements whereas hnRNP and CELF proteins act on intronic elements (Sanford et al., 2005; Spellman et al., 2005). However, SR proteins also act as bridging elements for regulated exons (Sanford et al., 2005) and 9G8 may be fulfilling that role in this instance.
Our pulldown results (Fig. 3C) show that 9G8 specifically no longer influences or binds to mutant M14 and shows a weak effect on mutant M16 in cell lysates, whereas its interactions with other point mutants within the ISS are either essentially unchanged (M12, M13) or enhanced (M11). The in vitro UV-crosslinking experiments (Fig. 3D) show an identical qualitative profile. The slight qualitative differences may be due to the presence of additional factors in cell lysates and/or to the fact that the phosphorylation status of 9G8 is quite different in cell lysates and in bacterial extracts.
In co-transfections, pulldowns and crosslinking experiments the 9G8 interaction is consistently abolished in mutant M14 and strengthened in mutant M11. Additionally, mutant M5 is no longer inhibited by 9G8, although it still binds the factor. One possible explanation is that mutant M5 interacts with the U1 snRNA so strongly that it is no longer sensitive to the influence of 9G8 (the mutation changes a G to a T, creating an additional Crick/Watson base pair; Fig. 6B).
Fig. 6.
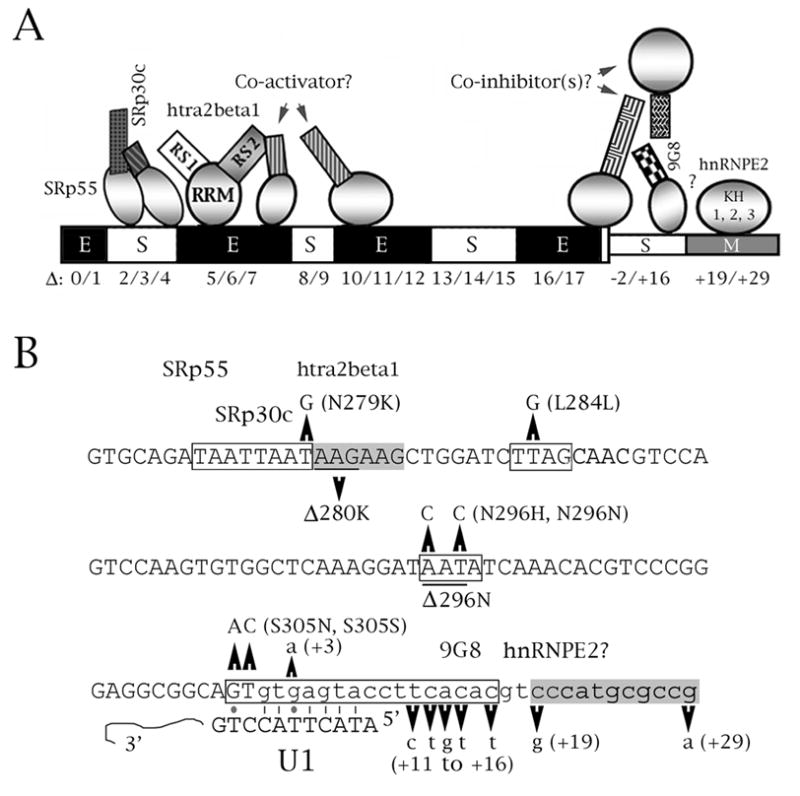
Speculative model of splicing regulation for two elements of tau exon 10. (A) Schematic representation of splicing factor interactions with exon 10. The exon is denoted by thick boxes, its downstream intron by thin boxes. The deletion nomenclature is according to D’Souza and Schellenberg (2000), Wang et al. (2005). The behavior of each region as delimited by the deletions is indicated by E (enhancer), S (silencer) or M (modifier). For the factors, circles represent RRM domains, squares represent RS domains. The model of the exonic silencer and enhancer at the 5’ end of the exon comes from Wang et al. (2005), with additional information from D’Souza and Schellenberg (2000), Jiang et al. (2003), Kondo et al. (2004). The model of the elements and factors in the proximal downstream intron come from this study and from Broderick et al. (2004). Question marks imply an unknown or hypothesized interaction. 5’ end: SRp30c and SRp55 bind to region E10-Δ2/3/4, whereas htra2beta1 binds to region E10-Δ5/6. The two inhibitors interact (or sterically interfere) with the RS1 domain of htra2beta1; its RS2 domain may interact with a putative co-activator which may bind to either E10-Δ6/7 or E10-Δ10/11/12. 3’ end: 9G8 binds to residues +11 to +18 but its influence is also abolished in mutant M5. 9G8 is probably part of a larger complex whose action may be modified or antagonized by hnRNPE2, which moderately activates splicing of exon 10. (B) Context of FTDP mutations that affect splicing within the cis regulatory elements depicted in (A). The sequence of exon 10 and its proximal downstream intron is shown. Exonic sequences are in uppercase, intronic in lowercase. FTDP point mutations and deletions are indicated. The boxed regions define enhancers (gray) or silencers (white). Also shown are the factors known or suspected to interact with regulatory cis elements, and the complementarity of the 5’ splice site with the U1 snRNA. Lines are regular Crick/Watson pairs, dots are G-T pairs.
Last, but most importantly, RNAi constructs against 9G8 decrease inclusion of exon 10 for both exogenous (Fig. 5A) and endogenous (Fig. 5B, 5C) tau. This definitively establishes 9G8 as a native regulator of tau exon 10. The behavior of 9G8 described in this study strongly suggests that it is part of a larger regulatory complex that interacts with the proximal downstream intron of exon 10.
9G8 as a regulator of other alternatively spliced exons
9G8 is a unique SR protein which contains a Zn knuckle that separates its RRM domain from its RS domain (Cavaloc et al., 1994; Shen et al., 2004). In other alternative splicing systems in which its action has been examined, 9G8 usually acts as a splicing activator (Cramer et al., 1999; Galiana-Arnoux et al., 2003; Li et al., 2000) and the sites it influences are also regulated by ASF/SF2 and/or SRp20. In our study, 9G8 acts as a strong splicing inhibitor and binds to an element which behaves as an intronic splicing silencer.
Splicing silencers are postulated to work by recruiting inhibitory factors or by blocking spliceosome assembly (Zheng, 2004). Our results suggest that 9G8 binds directly to the silencer defined by deletion Δ11/18 and that residue +14 is critical for its interaction with the exon 10 pre-mRNA. GAC/AAC/AC motifs have been defined by SELEX as binding sites for 9G8 (Cavaloc et al., 1999). Interestingly, deletion Δ11/18 overlaps an (AC)3 hexanucleotide (Fig. 2A), which conforms to the 9G8 binding site consensus.
Interaction with the U1 snRNA itself would encompass residues –2 to +7 (Fig. 4A). Thus, although 9G8 would not directly mask the site necessary for base pair formation with U1, it may sterically inhibit attachment of the U1 snRNP.
The FTDP-17 mutations initially clustered around the 5' splice site of exon 10, engendering the hypothesis that its splicing is partly modulated by a putative hairpin loop which hinders interaction with the U1 snRNP (Hutton et al., 1998; Grover et al., 1999, Jiang et al., 2000). However, all mutations which weaken the putative loop also increase complementarity to the 5' end of U1 and compensatory mutations for position +16 do not restore the wild type splicing ratio. Additionally, the proposed stem/loop structures have different exact configurations and free energies, all too low to be stable (Hutton et al., 1998; D’Souza et al., 2000). Finally compensatory mutations for position +16 of the downstream intron do not restore the wild type splicing ratio (Grover et al., 1999).
The alternative explanation was that this region may inhibit exon 10 inclusion by binding a factor (D’Souza and Schellenberg 2000, 2002). The behavior of 9G8 observed in our study argues in favor of the RNA being linear, rather than in the stem/loop structure.
In contrast to the human scenario, tau exon 10 becomes constitutive in adult rats and mice(Kosik et al., 1989). In this connection, it is interesting that the region around the 5' splice site of exon 10 is conserved only partially among human and mouse, diverging past position +8 (Grover et al., 1999). Given the strong effects of mutants in positions +14 to +29 (reviewed in Andreadis, 2005), this must be at least one region where regulation of this exon differs among species. This crucial difference in the behavior of exon 10 in rodents may well correlate with the repeated failure to find neurofibrillary tangles in aged rodents as well as the difficulty of inducing them artificially in transgenic models (Brandt et al., 2005).
Also interesting is the finding that the 9G8 variant that lacks the RRM domain does not activate inclusion of exon 10 (9G8C, Fig. 4B lane 4), as has been seen with equivalent mutants of other SR proteins, which are postulated to sequester other splicing factors (Cáceres et al., 1997; Wang et al., 2005). The different behavior of 9G8 underscores its uniqueness.
Regulation of tau exon splicing and frontotemporal dementia
Tau exon 10 codes for an additional microtubule binding domain, and its addition increases affinity of tau protein for microtubules (Andreadis, 2005; Himmler et al., 1989; Lee et al., 1989). The accumulation of abnormal tau filaments into tangles is a hallmark of many neurodegenerative diseases, including Alzheimer’s. Formation of NFTs is an early event in the dementia cascade, and the number of NFTs correlates with disease severity. In several neurodegenerative diseases, now collectively termed tauopathies, tau pathology is solely and directly responsible for neuronal death and development of the clinical dementia manifestations (Goedert and Jakes, 2005).
The tauopathy pedigrees analyzed thus far predominantly show mutations in tau exon 10, although several pedigrees carry mutations in tau exons 1, 9, 11, 12 and 13 which influence either microtubule binding or protein conformation (Goedert and Jakes, 2005). The exon 10 mutations fall in two categories -- those which influence microtubule binding and those which alter the ratio of exon 10 isoforms. Most mutations that affect exon 10 splicing increase exon inclusion whether they are in the exon or the downstream intron (the notable exception is D5; Andreadis, 2005; Goedert and Jakes, 2005).
The proximal downstream intron of exon 10 is a clustering region for FTDP mutations and contains two regulatory elements: an intronic splicing silencer (residues 11–18) and an intronic splicing modifier directly downstream of the ISS (residues 19–26). The present study is the first to discover the identity and mechanism of a splicing factor involved in the regulation of this region, which is critical to both the splicing regulation and the disease causing potential of the tau protein. 9G8 is an atypical SR protein, and its behavior in the tau system is novel – it acts as a splicing inhibitor and exerts its effect on an intronic element. Fig. 6A shows a model that summarizes our cumulative knowledge of the splicing regulation of tau exon 10.
Continued work on the basic molecular biology of the tau molecule may give us the tools to comprehend and combat not only FTDP, but also other types of dementia. These diseases vary widely both in clinical phenotype and brain pathology, but they share tangles as an invariable defining characteristic (Goedert and Jakes, 2005).
Experimental Methods
Plasmid construction and mutagenesis
The starting point for mutant constructs was SP/10L (Wang et al., 2004) which contains human tau exon 10 plus 471 bp of its upstream intron and 408 bp of its downstream intron inserted into the EcoRI site of vector pSPL3 (Invitrogen). Deletions within exon 10 (Δ8/9 and Δ15), or within the 30 bp downstream of exon 10 (I10-Δ3/10, I10-Δ11/18, I10-Δ14/22, I10-Δ19-26 and I10-Δ23/29) and point mutations reproducing several FTDP pedigree mutations (M5, I10-M11, I10-M12, I10-M13, I10-M14 and I10-M16) were created in SP/10L using the QuikChange Mutagenesis kit (Stratagene) as previously described (Wang et al., 2004, 2005). All mutants were verified by sequencing. The mutations are diagrammed in Fig. 2A (deletions) and 3A (point mutations) and the mutagenic primers are listed in Table 1.
To generate riboprobes, a human tau genomic fragment was inserted into vector pGEM-TE (Promega) to create construct E10+30. This contains human tau exon 10 plus 30 bp of its downstream intron. From E10+30, two constructs were subsequently created that separate the original insert in two at the SmaI site of tau exon 10 (Fig. 1A): E10-80 contains the 80 5’most bp of exon 10. E10-13+30 contains the 13 3’most bp of exon 10 plus 30 bp of its downstream intron. The deletions and point mutations created in SP/10L were also recreated in E10+30 as described in the previous paragraph. The mutagenic primers are listed in Table 1.
To express 9G8 in eukaryotic cells and bacteria, the 9G8 cDNA was cloned (respectively) into vectors pFLAG-CMV-6c (Sigma) and pGEX-4T-1 (Pharmacia). Domain variants of 9G8 region (diagrammed in Fig. 4A) were created by PCR and cloned into vector pCMV-Tag2B (Stratagene) and pGEX-4T-1 (Pharmacia). Figs. 4C and 4E show the expression of the respective fusion proteins. The primers used to create the variants are listed in Table 1. 9G8N contains the 5’ end of 9G8 (exons 1–5) and 9G8C contains the 3’ end of 9G8 (exons 3 to 8).
Three pairs of siRNA oligonucleotides for 9G8 were designed and synthesized (Table 1). Each pair of complementary oligonucleotides was annealed and cloned into the pFIV-H1/U6 vector (SBI).
Cell culture and transfection
Monkey kidney (COS), human epithelioma (HeLa) and human neuroblastoma SK-N-SH (SKN) cells were cultured as previously described (Gao et al., 2000). The cells were grown on 60 mm plates and transfected when they reached 30% confluence. All transfected plasmids were purified using Qiagen Tip-100s. Cells were transfected with plasmid DNA by the lipofectamine method (Mirus). For single transfections, 3 ug of plasmid were used. The exceptions were the RNAi experiments shown in Fig. 5B and 5C, in which 1 ug of each 9G8i plasmid were used. For double or triple co-transfections, 1 ug of each plasmid was used; again, the exception was the RNAi dosage experiment shown in the right panel of Fig. 5A. The medium was changed 16 hours after transfection. The cells were harvested 48 hours after transfection.
RNA preparation, reverse transcription and PCR reaction
Total RNA was isolated from transfected cells by the TRIzol method (Invitrogen). We performed reverse transcription and PCR as previously described (Wang et al., 2005). For analysis of endogenous 9G8 levels, we used the Ambion quantitative method as previously described (Gao et al., 2005), with a ratio of 3:7 18S primers to 18S competimers as the internal control. For PCR, we used the following primer pairs (shown in Table 1): SPL-LS/SPL-LN to trace exogenous (transfected) tau, HT7S3/HT11N for endogenous tau and 9G8ex3S/9G8ex8N for endogenous 9G8. The isoform ratio was calculated by scanning the bands from three independent transfections using the One-Dscan program and the Scanalytics IPLab software.
Cell lysates, riboprobe preparation, and RNA-protein immunoprecipitation
Expression of the FLAG-9G8 full-length and deletion constructs in COS cells was verified by using anti-FLAG M2 monoclonal antibody (Sigma) on Westerns of crude lysates from cells transfected with each construct (Fig, 1B, 4B).
Cell lysates were prepared from transfected cells using lysis buffer containing 50 mM Tris-HCl, 150mM NaCl, 1 mM EDTA and 1% Triton X-100. Radiolabeled riboprobes were transcribed from E10+30 and its variants using the riboprobe combination system (Promega), T7 RNA polymerase (Promega) and [32P]-CTP and [32P]-UTP (Amersham Pharmacia, specific activity 3000 Ci/mmol).
For RNA- protein immunoprecipitation, cell lysates were mixed with riboprobes in binding buffer (10 mM Hepes pH7.9, 1mM DTT, 50mM KCl and 5% glycerol)for 20 min at RT. Then anti-FLAG-antibody agarose beads (Sigma) were added and incubated at 4 C overnight. The beads were washed four times with wash buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl). After washing, the radioactivity retained by the beads was counted in a beta scintillation counter.
GST-fusion protein expression and UV-crosslinking
pGEX-4T-9G8 was transformed into E. coli strain BL21(DE3) (Stratagene). The strain was induced by IPTG and purified recombinant proteins were produced according to the Novagen instructions. Coomassie blue staining shows that the fusion protein is stably expressed (Fig. 1D).
For UV-crosslinking, riboprobes and purified GST-9G8 fusion proteins were incubated at RT for 20 min in binding buffer (10 mM Hepes pH7.9, 1 mM EDTA, 1 mM DTT, 5% glycerol, 50 mM KCl, 1 ug tRNA). Reaction mixtures were prechilled on ice and irradiated with UV light (254 nm) for 10 min. The samples were digested with RNase A at 37 °C for 1 h and run on 12% SDS-PAGE. The gels were fixed, dried and subjected to autoradiography.
Acknowledgments
This study was supported by NIA grant AG018486 to A. A. We would like to thank our colleagues Stefan Stamm and Gavin Screaton for their generous gift of the original 9G8 cDNA.
Abbreviations
- FTDP
frontotemporal dementia with Parkinsonism
- ESE
exonic splicing enhancer
- hnRNP
heterogeneous ribonuclear protein
- htra
human transformer
- ISM
intronic splicing modifier
- ISS
intronic splicing silencer
- NFT
neurofibrillary tangle
- RRM
RNA recognition motif
- RS
arginine/serine
- snRNA
small nuclear RNA
- SR protein
serine/arginine-rich protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochem Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochem Biophys Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Broderick JA, Wang J, Andreadis A. Heterogeneous nuclear ribonucleoprotein E2 binds to tau exon 10 and moderately activates its splicing. Gene. 2004;331:107–114. doi: 10.1016/j.gene.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Cáceres JF, Misteli T, Screaton GR, Spector DL, Krainer AR. Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J Cell Biol. 1997;138:225–238. doi: 10.1083/jcb.138.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaloc Y, Popielarz M, Fuchs JP, Gattoni R, Stevenin J. Characterization and cloning of the human splicing factor 9G8: a novel 35 kDa factor of the serine/arginine protein family. EMBO J. 1994;11:2639–2649. doi: 10.1002/j.1460-2075.1994.tb06554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaloc Y, Bourgeois CF, Kister L, Stevenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5:468–483. doi: 10.1017/s1355838299981967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Cáceres JF, Cazalla D, Kadener S, Muro AF, Baralle FE, Kornblihtt AR. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell. 1999;4:251–258. doi: 10.1016/s1097-2765(00)80372-x. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza I, Schellenberg GD. Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem. 2000;275:17700–17709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- D’Souza I, Schellenberg GD. Tau exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5’ and 3’ splicing site. J Biol Chem. 2002;277:26587–26599. doi: 10.1074/jbc.M203794200. [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Galiana-Arnoux D, Lejeune F, Gesnel MC, Stevenin J, Breathnach R, DelGatto-Konczak F. The CD44 alternative v9 exon contains a splicing enhancer responsive to the SR proteins 9G8, ASF/SF2, and SRp20. J Biol Chem. 2003;278:32943–32953. doi: 10.1074/jbc.M301090200. [DOI] [PubMed] [Google Scholar]
- Gao L, Tse SW, Conrad C, Andreadis A. Saitohin, which is nested in the tau locus and confers allele-specific susceptibility to several neurodegenerative diseases, inreracts with peroxiredoxin 6. J Biol Chem. 2005;280:39268–39272. doi: 10.1074/jbc.M506116200. [DOI] [PubMed] [Google Scholar]
- Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A. Complex regulation of tau exon 10, whose missplicing causes frontotemporal dementia. J Neurochem. 2000;74:490–500. doi: 10.1046/j.1471-4159.2000.740490.x. [DOI] [PubMed] [Google Scholar]
- Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, Riederer P, Moller HJ, Andreadis A, Henkel K, Stamm S. The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer's disease. J Neurochem. 2006;96:635–644. doi: 10.1111/j.1471-4159.2005.03552.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989a;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein isoforms in human brain. EMBO J. 1989b;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochem Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Grabowski PJ. Splicing regulation in neurons: tinkering with cell-specific control. Cell. 1998;92:709–712. doi: 10.1016/s0092-8674(00)81399-9. [DOI] [PubMed] [Google Scholar]
- Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- Grover A, Houlden H, Barker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. 5’ splicing site mutaions in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- Hanamura A, Cáceres JF, Mayeda A, Franza BR, Jr, Krainer AR. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- Hartmann AM, Rujescu D, Giannakouros T, Nikolakaki E, Goedert M, Mandelkow EM, Gao QS, Andreadis A, Stamm S. Regulation of alternative splicing of human tau exon 10 by phosphorylation of splicing factors. Mol Cell Neurosci. 2001;18:80–90. doi: 10.1006/mcne.2001.1000. [DOI] [PubMed] [Google Scholar]
- Himmler A, Drechsel D, Kirschner MW, Martin DW. Tau consists of a set of proteins with repreated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering- Brown S, Chakraverty S, Isaacs A, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Havlioglu N, Zhang X, Stamm S, Yan R, Wu JY. Mutations in tau gene exon 10 associated with FTDP-17 alter the activity of an exonic splicing enhancer to interact with Tra2beta1. J Biol Chem. 2003;278:18997–19007. doi: 10.1074/jbc.M301800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Kamma H, Portman DS, Dreyfuss G. Cell type-specific expression of hnRNP proteins. Exp Cell Res. 1995;221:187–196. doi: 10.1006/excr.1995.1366. [DOI] [PubMed] [Google Scholar]
- Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, Tammana H, Gingeras TR. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Yamamoto N, Murakami T, Okumura M, Mayeda A, Imaizumi K. Tra2 beta, SF2/ASF and SRp30c modulate the function of an exonic splicing enhancer in exon 10 of tau pre-mRNA. Genes Cells. 2004;9:121–130. doi: 10.1111/j.1356-9597.2004.00709.x. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- Lee G, Neve RL, Kosik KS. The microtubule binding domain of human tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- Lejeune F, Cavaloc Y, Stevenin J. Alternative splicing of intron 3 of the serine/arginine-rich protein 9G8 gene. J Biol Chem. 2001;276:7850–7858. doi: 10.1074/jbc.M009510200. [DOI] [PubMed] [Google Scholar]
- Li X, Shambaugh ME, Rottman FM, Bokar JA. SR proteins ASF/SF2 and 9G8 interact to activate enhancer-dependent intron D splicing of bovine growth hormone pre-mRNA in vitro. RNA. 2000;6:1847–1858. doi: 10.1017/s1355838200000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation and interaction with microtubles. Neurobiol Aging. 1995;16:3550362. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. Mass spectometry and EST-database searching allows characterization of the multiprotein spliceosome complex. Nat Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- Popielarz M, Cavaloc Y, Mattei MG, Gattoni R, Stevenin J. The gene encoding human splicing factor 9G8. Structure, chromosomal localization, and expression of alternatively processed transcripts. J Biol Chem. 1995;270:17830–17835. doi: 10.1074/jbc.270.30.17830. [DOI] [PubMed] [Google Scholar]
- Shen H, Kan JLC, Green MR. Arginine-serine-rich domains bound at splicing enhancers contact the branchpoint to promote prespliceosome assembly. Mol Cell. 2004;13:367–376. doi: 10.1016/s1097-2765(04)00025-5. [DOI] [PubMed] [Google Scholar]
- Sanford JR, Ellis J, Cáceres JF. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem Soc Trans. 2005;33:443–446. doi: 10.1042/BST0330443. [DOI] [PubMed] [Google Scholar]
- Smith CW, Valcárcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- Spellman R, Rideau A, Matlin A, Gooding C, Robinson F, McGlincy N, Grellscheid SN, Southby J, Wollerton M, Smith CW. Regulation of alternative splicing by PTB and associated factors. Biochem Soc Trans. 2005;33:457–460. doi: 10.1042/BST0330457. [DOI] [PubMed] [Google Scholar]
- Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Stoilov P, Meshorer E, Gencheva M, Glick D, Soreq H, Stamm S. Defects in pre-mRNA processing as causes of and predisposition to diseases DNA. Cell Biol. 2002;21:803–818. doi: 10.1089/104454902320908450. [DOI] [PubMed] [Google Scholar]
- Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, Martins RN, Kwork JBJ, Schofield PR. Mutations in the tau gene that cause an increase in three repeat tau and frontoteporal dementia. Brain. 2003;126:814–826. doi: 10.1093/brain/awg090. [DOI] [PubMed] [Google Scholar]
- Thanaraj TA, Stamm S, Clark F, Riethoven JJ, Le Texier V, Muilu J. ASD: the Alternative Splicing Database. Nucleic Acids Res. 2004;32:D64–69. doi: 10.1093/nar/gkh030. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J Neurochem. 2004;88:1078–1090. doi: 10.1046/j.1471-4159.2003.02232.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang J, Gao L, Lafyatis R, Stamm S, Andreadis A. Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c / SRp55 complex that either recruits or antagonizes htra2beta1. J Biol Chem. 2005;280:14230–14239. doi: 10.1074/jbc.M413846200. [DOI] [PubMed] [Google Scholar]
- Zheng ZM. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and mammalian gene expression. J Biomed Sci. 2004;11:278–294. doi: 10.1159/000077096. [DOI] [PMC free article] [PubMed] [Google Scholar]