

Abstract
Increasing evidence supports an association between inflammation and plaque rupture. Macrophages and vascular smooth muscle cells are a source of cytokines and growth factors, which contribute to ongoing inflammation during atherogenesis. In a rabbit model of atherosclerosis, we evaluated the effect of the ACE inhibitor quinapril on different parameters implicated in the pathogenesis of the plaque, such as the presence of chemokines (interleukin-8, monocyte chemoattractant protein-1), collagen I, and vascular smooth muscle cell proliferation (PDGF-B). Since nuclear factor κB (NF-κB) has been implicated in the control of chemokine transcription and cell proliferation, we also investigated its activation and localization in the lesion. Quinapril administration for 28 days caused a down-regulation in arterial expression of interleukin-8 and monocyte chemoattractant protein-1 (mRNA and protein). However, collagen I expression (mRNA and protein) was not modified. PDGF-B expression was reduced in both the intima and the media. Active NF-κB, found in both macrophages and vascular smooth muscle cells, was also reduced by quinapril. Nevertheless, no significant changes were noted in the mild neointima formation, although a certain trend toward normalization was found in the quinapril-treated group. In conclusion, our results show that quinapril treatment attenuates several parameters associated with inflammation within the atherosclerotic lesions that are controlled by NF-κB, although it has no effect on collagen I expression. Both effects could contribute to the stabilization of the atherosclerotic plaque.
Cardiovascular diseases are the main cause of death in the western world. There is increasing evidence that the rupture of atherosclerotic plaques followed by thrombus formation is the phenomenon responsible for most fatal clinical events. The breakdown of the plaque occurs more frequently at points where the fibrous cap is thinner and there is a great infiltration of inflammatory cells such as macrophages and T lymphocytes. 1 Accumulation of these cells contributes to the inflammatory reaction that takes place inside the plaques and is favored by the presence of chemotactic factors such as interleukin 8 (IL-8) and monocyte chemoattractant protein-1 (MCP-1). 2 MCP-1 is involved in the migration of monocytes into the subendothelial space. It is not detectable in normal rabbit aortas but is overexpressed after vascular damage 3 or hypercholesterolemia, 4 and in human lesions. 3 This chemokine is also chemotactic for T lymphocytes 5 that are also present in human lesions, 6 although the main chemoattractant for these cells is IL-8, which is present in macrophage-rich areas of human atherosclerotic plaques, 7,8 and mitogenic and chemotactic for vascular smooth muscle cells (VSMC). 9 Its expression can be induced in these cells in response to interleukin-1 10 and produced by macrophage foam cells in human atheroma. 11
Accumulation of VSMC in the neointima is another feature of atherosclerosis and a consequence of their migration from the media and their proliferation within the lesion. Platelet derived growth factor B (PDGF-B) is a potent inductor of VSMC migration and proliferation. It can be produced by VSMC and macrophages and is overexpressed during experimental vascular damage 12,13 and in human lesions. 14
Nuclear factor κB (NF-κB) is a common transcription factor involved in the regulation of many proinflammatory genes 15 including MCP-1, 16 IL-8, 17 and PDGF-B, 18 as well as in the control of VSMC proliferation. 19 Its activation (the release of the inhibitory IκB subunit and the translocation of the active p50-p65 heterodimer to the nucleus) can be induced by different stimuli such as cytokines and oxidative stress. 15 The relevance of this nuclear factor in the development of inflammatory processes, including atherosclerosis, is becoming evident. 20,21
A high level of activity of the angiotensin converting enzyme (ACE) has been found in the neointima following experimental vascular injury 22,23 and in humans up-regulation of ACE expression is associated with enhanced risk of myocardial infarction. 24 In animal models, ACE inhibitors diminished neointima formation 25 and in humans they reduce the incidence of reinfarction. 26 Although several potential mechanisms of these effects have been proposed, including plaque stabilization 27 and improvement of endothelial function, 28 there is not yet any clear explanation.
In this work we have used an experimental atherosclerosis model in rabbits to study the effect of the ACE inhibitor quinapril on several parameters of plaque development and stability such as the presence of chemokines involved in the recruitment of inflammatory cells (IL-8, MCP-1), the VSMC mitogen and chemoattractant PDGF-B, and the amount of collagen I. We also studied the activation and localization of NF-κB, a common modulator of MCP-1, IL-8, and PDGF-B gene expression.
Methods
Experimental Model
Atherosclerosis was induced according to a previously described technique with minimal modifications. 29 Thirty-three white New Zealand male rabbits weighing 3.5–4 kg were anesthetized by intramuscular injection of 5 mg/kg xylazine (Rompun, Bayer AG, Leverkusen, Germany) and 35 mg/kg ketamine (Ketolar, Parke-Davis, Ann Arbor, MI). Local relief from pain was achieved by subcutaneous injection of 2% Lidocaine (Braun, Barcelona, Spain). A prophylactic intramuscular injection of 125 mg/kg of cefazoline (Llorente Laboratories, Madrid, Spain) was administered 30 minutes before the surgical procedure and all techniques were done in sterile conditions. Ten mL of blood were drawn from the ear vein for lipid determination by standard methods and serum ACE activity measurement. Arterial pressure was estimated by a sphygmomanometer in the femoral artery. Proximal bilateral femoral arteriotomies were performed and proximal and distal ligatures were placed to isolate a segment of approximately 2 cm, which was cannulated with a 27-gauge needle. A vent was made by needle puncture and blood was removed by a flush of saline. Endothelial damage was induced by the passage of industrial nitrogen at a rate of 80 mL/minute for 8 minutes. The isolated segments were then flushed again with saline and the ligatures were removed. Hemostasis was achieved by local pressure and the wound was closed with a 4.0 vicryl subcuticular suture. After surgery the animals were placed on a 2% cholesterol, 6% peanut oil diet (Letica, Barcelona, Spain) and maintained for 28 days in individual cages until killed. Four control animals were included in the study for comparison and five animals were killed after 2 days of quinapril consumption to quantify vascular ACE activity.
Two days before the surgical procedure the animals were randomized to quinapril (n = 9) (Accupril, Parke-Davis) or no treatment (n = 15). Quinapril was freshly dissolved every other day in drinking water and given to the rabbits at 1 mg/kg/day. The consumption of water was measured every other day and the weight of the animals was recorded weekly during the experiment to confirm the appropriate administration of the drug. Control animals were fed standard chow with no experimental intervention.
Sample Collection and Measurement of ACE Activity
At the time of sacrifice animals were anesthetized with ketamine/xylazine and 10 mL of blood were drawn from the ear vein for biochemical determinations. Both femoral arteries were exposed and ligatures were placed to isolate the damaged segments and flushed with saline. One of the arteries was removed, the adventitial layer was carefully peeled, and the artery was immediately snap-frozen in liquid nitrogen. The aorta was ligated and flushed with saline and a piece was removed and snap-frozen in liquid nitrogen. The animal was euthanized with an overdose of pentobarbital (Abbot, Madrid, Spain) and the other femoral artery was cannulated and fixed in situ with 100 mL of 4% buffered formaldehyde at a pressure of 100 mm Hg. It was then removed and kept for 24 hours in 4% buffered formaldehyde and afterwards in 70% ethanol until embedded in paraffin.
ACE activity determinations were done in serum and in samples of frozen uninjured artery (aorta) by a spectrophotometric method (Sigma Chemicals, St. Louis, MO). The activity is expressed as relative units per milligram protein of tissue as determined by the BCA method (Pierce, Rockford, IL) or as units per mL in serum.
Immunohistochemistry
Paraffin-embedded arteries were cross-sectioned in 4 μm-thick pieces at 5-mm intervals from the proximal to the distal end, dewaxed, and rehydrated. Identification of macrophages was performed using a monoclonal antibody for rabbit macrophages (RAM11, Dako, Carpinteria, CA). 30 MCP-1 and IL-8 were detected with a polyclonal goat anti-human MCP-1 antibody and a monoclonal anti-human IL-8 antibody, respectively (Immugenex Corp., Los Angeles, CA). For collagen I, a polyclonal anti-human antibody (Southern Biotechnology, Birmingham, AL) was used, and for VSMC a monoclonal anti-rabbit α-actin (HHF35; ENZO Diagnostic, New York, NY). Endogenous peroxidase activity was quenched by incubating the sections in 3% hydrogen peroxide:methanol (1:1) for 30 minutes. Nonspecific antibody binding was blocked by incubation of the tissue section for 1 hour in suppressor serum consisting of 6% goat serum and 4% bovine serum albumin (BSA) in phosphate buffered saline (PBS), pH 7.0, for HHF35 and anti-IL-8 antibodies, and 6% horse serum and 4% BSA in PBS, pH 7.0, for anti-MCP-1 and RAM11 antibodies. For collagen I, the suppressor serum was 2% horse serum and 1% BSA in PBS. HHF35 was applied directly for 1 hour. Anti-MCP-1 and anti-collagen I antibodies were applied overnight and RAM11 for 1 hour in 1% horse serum and 4% BSA in PBS and anti-IL-8 was applied overnight in 1% goat serum and 4% BSA. As secondary antibody, a goat anti-mouse IgG HRPO-conjugated (Seralab, Sussex, UK) was used for RAM11 and HHF35. For MCP-1 and collagen I a donkey anti-goat IgG HRPO-conjugated (The Binding Site, Birmingham, UK) was used and for IL-8 a goat anti-mouse IgG biotin labeled. Secondary antibodies were applied for 30 minutes to 1 hour and then sections were stained for 10 minutes at room temperature with 0.05% 3,3′-diaminobenzidine tetrahydrochloride (Dako) and 0.01% hydrogen peroxide in PBS (RAM11, HHF35, collagen I and MCP-1) or streptavidin-biotin/alkaline phosphatase (Dako) for 30 minutes plus fast red substrate for 10 minutes (IL-8). Finally, sections were counterstained with hematoxylin and mounted in Pertex (Medite, Burgdorf, Germany). In each experiment, negative controls without the primary antibody or using an unrelated antibody were included to check for nonspecific staining.
Quantification was assessed by two independent observers and the sections with the maximal lesion in each animal were chosen. The MCP-1 and macrophages-occupied area was quantified automatically as described below, but IL-8- and collagen I-stained areas were assessed with a semiquantitative score due to the diffuse pattern of staining. Computer-assisted morphometric analysis was performed with the Cue-2 semiautomatic image analysis system (Olympus, Hamburg, Germany). The arterial cross-sections stained with antibodies were digitized by an Olympus microscope (BH-2) connected to a CCD video camera. The labeled areas in the intima and the media were delimited and after image enhancement and segmentation (transformation to a binary image) a grey value ranging from 0 to 255 was assigned to each pixel and automatic analysis was performed. Results were expressed as immunostained area and fractional area of intima and media.
The lesion size was measured on orcein-stained arterial sections with the NewSketch 1212 graphic tablet (Genius, Ontario, CA) linked to a microcomputer using the autoCADD 10.0 software (Autodesk AG) as previously described. 20
RNA Extraction, Reverse Transcriptase Polymerase Chain Reaction, and Northern Blot
Frozen femoral arteries were pulverized in a metallic chamber. Total RNA was obtained by the acid guanidine-thiocyanate-phenol-chloroform method 31 and quantified by absorption at 260 nm in duplicate. Equal amounts of RNA (5 μg) were denatured and electrophoresed in a 1% agarose-formaldehyde gel to check the absence of degradation of RNA. Equal amounts of RNA from each animal of every group (control, untreated, and quinapril-treated) were pooled. For RT-PCR, 1 μg of RNA from every pool was applied and primers based on the published sequences for IL-8, MCP-1, 32 and glyceraldehyde-3′-phosphate dehydrogenase (G3PDH) 33 were used.
For Northern blot, 30 μg of RNA were denatured and electrophoresed in a 1% agarose-formaldehyde gel, transferred to nylon membranes (Genescreen, New England Nuclear, Boston, MA) and carried out as previously described. 31 The probes for collagen I 34 and G3PDH 33 were obtained from PCR products and labeled with α-[32P]-dCTP (Amersham, Buckinghamshire, UK) by the random priming method (Promega, Madison, WI). G3PDH was used as internal control to calculate the relative expression of each mRNA. Films were scanned using the Image Quant densitometer (Molecular Dynamics, Sunnyvale, CA).
In Situ Hybridization
Digoxigenin-labeled single-stranded RNA probe of PDGF-B was prepared using a nonradioactive RNA labeling kit (Boehringer Mannheim, Mannheim, Germany) according to the manufacturer’s instructions. Sense and antisense PDGF-B riboprobes were synthesized from the linearized plasmid PCR 3 (Invitrogen, San Diego, CA) containing a PCR fragment (462 bp) of rat PDGF-B 35 as run-off transcripts with T3 and T7 RNA polymerases in the presence of digoxigenin-11-UTP.
Paraffin-embedded tissue sections were cut 4 μm thick and floated onto APES (Sigma) coated slides. The tissue sections were heated at 65°C overnight, then fixed with 1.5% paraformaldehyde-1.5% glutaraldehyde. After dewaxing, tissues were incubated in 5 mmol/L levamisole to inhibit endogenous phosphatase. Deproteinization was carried out in 0.2 mol/L HCl followed by digestion with proteinase K and washed twice in 0.2% glycine for 5 minutes. After digestion, all sections were postfixed as above, dehydrated through a graded ethanol series, and air dried at room temperature. The slides were hybridized with 10 ng/mL denatured digoxigenin-11-UTP-labeled riboprobes in hybridization buffer (2× SSC, 1× Denhardt’s solution, 0.1 mol/L sodium phosphate, pH 6.5, 10% dextran sulfate in formamide). Sealed cover slips were placed over the tissue sections and hybridization was allowed to occur overnight at 42°C in a moisturized chamber. The cover slips were removed and the slices were washed in 2× SSC for 1 hour at room temperature and 0.2× SSC for 30 minutes at 37°C. The sections were incubated with an antidigoxigenin antibody conjugated with alkaline phosphatase (Boehringer Mannheim) for 30 minutes at 37°C. Colorimetric detection of RNA-RNA hybrids was performed with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolylphosphate (X-phosphate) in the dark for 1–4 hours. The color reaction was stopped with 10 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0, and cover slips applied with 60% glycerol before microscopic examination. The negative controls included hybridization with the sense probe, RNase treatment before hybridization, and omission of the RNA probe. Semiquantitative scores were assigned.
NF-κB Determination by Electrophoretic Mobility Shift Assay
For protein extraction from tissue samples the method of Negoro et al 36 was used with modifications. Briefly, frozen arterial pieces were pulverized in a metallic chamber and resuspended in 1 mL cold extraction buffer containing 20 mmol/L HEPES-NaOH, pH 7.6, 20% (vol-vol) glycerol, 0.35 mol/L NaCl, 5 mmol/L MgCl2, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol (DTT), 0.5 mmol/L phenylmethyl sulfonyl fluoride (PMSF), and 1 μg/mL pepstatin A. The homogenate was vigorously shaken for 30 minutes, insoluble materials were precipitated by centrifugation at 40,000 × g for 30 minutes at 4°C, and the supernatant was dialyzed overnight against a binding buffer containing 20 mmol/L HEPES-NaOH, pH 7.6, 20% (vol/vol) glycerol, 0.1 mol/L NaCl, 5 mmol/L MgCl2, 0.1 mmol/L EDTA, 1 mmol/L DTT and 0.5 mmol/L PMSF. The dialysate was cleared by centrifugation at 10,000 × g for 15 minutes at 4°C, pooled according to the treatment groups, and frozen at −80°C in aliquots until use. Protein concentration was quantified by the BCA method (Pierce).
Gel shift assays were performed with a commercial kit according to the instructions of the manufacturer (Promega). Briefly, NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) consensus oligonucleotide was [32P]-end-labeled by incubation for 10 minutes at 37°C with 10 U T4 polynucleotide kinase (Promega) in a reaction containing 10 μCi [γ-32P]ATP (3000 Ci/mmol) (Amersham), 70 mmol/L Tris-HCl, 10 mmol/L MgCl2, and 5 mmol/L DTT. The reaction was stopped by adding EDTA to a final concentration of 0.05 mol/L. Cellular protein (10 μg) was equilibrated for 10 minutes in a binding buffer containing 4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 50 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 7.5, and 50 mg/mL of poly(dI-dC) (Pharmacia LKB, Uppsala, Sweden). Labeled probe (0.35 pmols) was added to the reaction and incubated for 20 minutes at room temperature. When competition assays were performed, a 100-fold excess of the cold probe was added to this buffer 10 minutes prior to the addition of the labeled probe. For supershift assays, 1 μg of anti-p50 and anti-p65 antibodies (Chemikon, Temecula, CA and Santa Cruz Biotechnology, Santa Cruz, CA, respectively) were added and incubated for 1 hour. Positive controls were done using Hela cell nuclear extracts and negative controls were done setting the reaction without nuclear extract. The reactions were stopped by addition of gel loading buffer (250 mmol/L Tris-HCL, 0.2% bromophenol blue, 0.2% xylene cyanol and 40% glycerol) and run on a nondenaturing, 4% acrylamide gel at 100 V at room temperature in TBE. The gel was dried and exposed to X-ray film.
NF-κB Determination by Southwestern Histochemistry
This method was developed to detect the distribution and DNA-binding activity of NF-κB in situ using a digoxigenin-labeled double-stranded DNA probe with a specific NF-κB consensus sequence according to a previously described technique with modifications. 37 The synthetic sense (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and antisense (5′-GCCTGGGAAAGTCCCCTCAACT-3′) probes (Genosys Biotechnology, London) were annealed by heating at 80°C for 2 minutes. The probes were labeled with digoxigenin (DIG oligonucleotide 3′-end labeling, Boehringer Mannheim). Arterial sections were dewaxed, rehydrated, incubated with 5 mmol/L levamisole for 30 minutes, and fixed with 0.2% paraformaldehyde. They were then digested with 0.5% pepsin in 1 N HCl for 30 minutes and washed with HEPES buffer (10 mmol/L HEPES, 40 mmol/L NaCl, 10 mmol/L MgCl2, 1 mmol/L DTT, 1 mmol/L EDTA, 0.25% BSA, pH 7.4). Sections were incubated with 0.1 mg/mL DNAse I during 30 minutes and washed once with HEPES buffer with 10 mmol/mL EDTA instead of MgCl2. The DNA binding reaction was performed by incubation with 100 pmol/mL of the labeled DNA probe overnight at 37°C. The sections were washed with HEPES buffer, washing buffer (0.3% Tween 20 in 0.1 mol/L maleic acid, 0.15 mol/L NaCl, pH 7.5) and blocking solution (0.1× SSC, 0.1% SDS diluted 1:10 in washing buffer) for 1 hour. Preparations were incubated with an anti-digoxigenin antibody conjugated with alkaline phosphatase (Boehringer) overnight at 4°C and then washed with 0.1 mol/L Tris-HCl, 0.1 mol/L NaCl, 50 mmol/L MgCl2, pH 9.5, and with washing buffer. Afterwards, preparations were incubated with 0.4% NBT and 0.32% X-Phosphate for 1 hour. Reaction was stopped by washing with 10 mmol/L Tris, pH 8.0, and 1 mmol/L EDTA. Preparations were mounted with glycerol. Preparations without probe were used as negative controls and a mutant NF-κB probe was used to test the specificity of the technique (sense: 5′-AGTTGAGGCTCCTTTCCCAGGC-3′ and antisense: 5′-GCCTGGGAAAGGAGCCTCAACT-3′). For the identification of the cell types showing NF-κB activity in the lesions, simultaneous detection of NF-κB and macrophages or VSMC was done. Sections were washed in PBS before mounting and immunohistochemistry was done according to the above described technique.
A semiquantitative score ranging from 0 to 5 was assigned to each preparation.
Statistical Analysis
Results are expressed as the mean ± SD. Significance was established using GraphPAD InStat (GraphPAD Software). Student’s t-test and Wilcoxon nonparametric test were used to compare the data. Differences were considered significant when P < 0.05.
Results
Plasma Cholesterol Levels, ACE Activity, and Morphometric Analysis of the Lesions
Total serum cholesterol increased about 30 times over the basal values after 4 weeks of 2% cholesterol diet (1703 ± 536 versus 53 ± 37 mg/dl). Quinapril treatment did not induce any significant change compared to untreated animals.
At the moment of injury, after two days of quinapril consumption, there was a significant diminution in serum ACE activity (2.5 ± 1 × 10−2 versus 8 ± 3.6 × 10−2 U/mL; P < 0.03) and in arterial blood pressure (56 ± 16 versus 83 ± 13 mmHg; P < 0.0001) in the animals receiving quinapril. However, there was no significant difference between vascular ACE activity in quinapril-treated and untreated rabbits (1.2 ± 0.2 × 10−2, n = 5, versus 1.1 ± 0.4 × 10−2 U/mg; n = 4). At the time of death (28 days), serum ACE activity could not be quantified due to the turbidity caused by the high amount of lipids. Vascular ACE activity (determined in the aorta) was increased about fourfold in the arteries of the untreated rabbits compared to the initial values (4.8 ± 0.2 × 10−2 U/mg, n = 9), but it was significantly lower in quinapril-treated rabbits (2.14 ± 0.2 × 10−2 U/mg, n = 7; P = 0.023).
Maximal lesion size was reduced about 60% in the quinapril group compared to the untreated animals determined as intima/media ratio (1.01 ± 0.8 versus 0.39 ± 0.4) and as maximal stenosis (328,691 ± 263,104 versus 101,547 ± 109,977 μm2). However, the reduction did not reach statistical significance.
Effect of Quinapril Treatment on Expression of Chemokines and Presence of Macrophages in the Lesions
Healthy control animals did not show any stain for MCP-1 and IL-8 (Figure 1A) ▶ . In contrast, untreated animals presented a marked neointimal stain for MCP-1 and IL-8 that was significantly reduced in the group treated with quinapril (185,642 ± 169,784 versus 45,582 ± 68,963 μm2; P < 0.05 and 13.7 ± 1.2 versus 8.6 ± 3.8 semiquantitative score; P < 0.04 respectively) (Figure 1A) ▶ . The medial layers were also stained for MCP-1 and IL-8; although there was a lower presence of both chemokines in the animals treated with quinapril compared to the untreated (20% and 25% respectively), the reduction was not statistically significant.
Figure 1.


MCP-1, IL-8, and macrophage detection by immunohistochemistry in arterial sections. Arterial sections were stained with anti-MCP-1, anti-IL-8, or RAM11 antibodies. A: In the upper part is shown MCP-1 immunostaining of a control (1), an untreated (2), and a quinapril-treated (3) animal. In the lower part is shown the staining for IL-8 of a control (2), an untreated (5), and a treated (6) animal. B: Staining for macrophages corresponding to a control (1), an untreated (2), and a quinapril-treated (3). Magnification, ×400. In the fourth panel of fig 1B ▶ is shown the bar graph corresponding to the mean neointimal area occupied by the macrophages (closed bars), the MCP-1 (open bars) and the semiquantitative score for IL-8 (hatched bars). The figure illustrates the association between the three parameters. *, P < 0.04; **, P < 0.05; ***, P < 0.04.
We next studied whether the diminution in the expression of chemokines in the neointima was associated with a reduction in the number of infiltrating macrophages. As can be seen in Figure 1B ▶ , there were no macrophages in the control arteries, while a certain number were present in the neointima of the untreated rabbits. The treatment with quinapril reduced by 80% the neointimal area occupied by these cells (105,729 ± 196,696 μm 2 versus 20,213 ± 52,957 μm2; P < 0.04). Furthermore, in the quinapril-treated animals there was also a 50% reduction in the medial area occupied by macrophages, although it did not reach statistical significance. In Figure 1B ▶ a bar graph depicts the association between the neointimal areas occupied by MCP-1 and macrophages and the semiquantitative score for IL-8. No staining was observed in the negative controls included in each experiment (data not shown).
MCP-1 and IL-8 mRNA expression was quantified in the femoral arteries by semiquantitative RT-PCR (30 cycles of amplification) in a pool of total RNA from each group. Control animals showed no detectable mRNA expression of MCP-1 or IL-8 (Figure 2) ▶ , whereas untreated rabbits presented a marked arterial expression of IL-8 (9.9 arbitrary units) and MCP-1 (4.8 arbitrary units). Quinapril-treated rabbits showed a diminution in arterial IL-8 expression (2.1 versus 9.9 a.u.) and MCP-1 expression (2.4 versus 4.8 a.u.) compared to the untreated animals.
Figure 2.
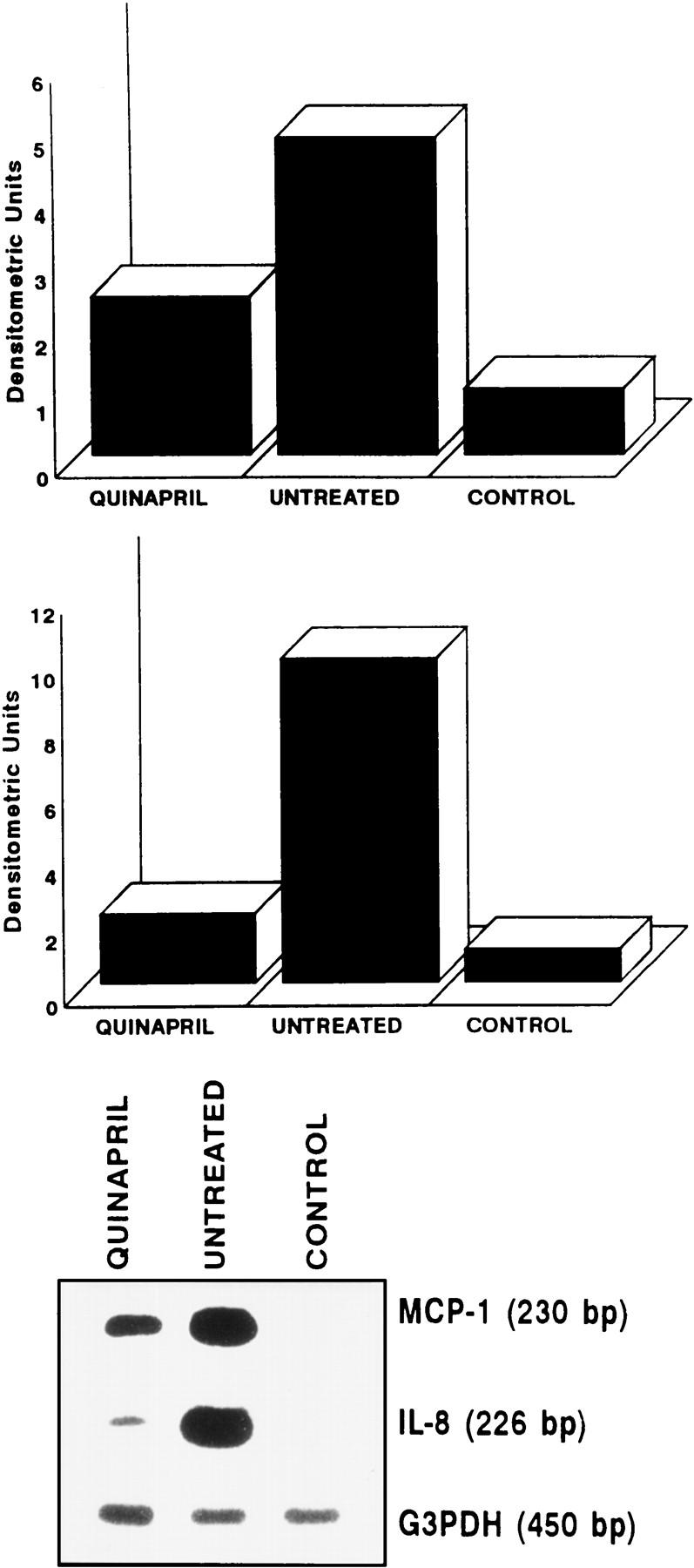
Arterial MCP-1 and IL-8 mRNA expression determined by RT-PCR. MCP-1 and IL-8 mRNA expression was quantified in a pool of total RNA of each group by a semiquantitative RT-PCR method consisting of amplifying a fragment of both cDNAs in the same conditions of a fragment of the housekeeping gene G3PDH. Amplification was carried out for 30 cycles after checking the linearity of the reaction up to 35 cycles. Densitometric analysis of the RT-PCR products is shown. Arbitrary units were calculated in relation to the expression of the G3PDH expression. Autoradiography of the dried gels showing the products of the PCR and the G3PDH control is shown in the lower part of the figure (representative PCR of two different done).
Effect of Quinapril Treatment on Collagen I Expression in the Lesions
As depicted in Figure 3A ▶ , healthy animals showed stain for collagen I in the medial layer. Collagen I staining was found increased in the media of the injured arteries compared to the healthy ones, but no difference was noted between the untreated and treated groups (8.2 ± 4.8 versus 10 ± 7; NS, semiquantitative score) (Figure 3, B and C) ▶ . Collagen I was also present in the neointima of both groups in similar amounts (10.5 ± 5 versus 11.8 ± 7; NS) (Figure 3, B and C) ▶ .
Figure 3.

Collagen I detection by immunohistochemistry in arterial sections. Photomicrographs showing immunostaining of cross sections from femoral arteries stained with anti-collagen I antibody: (A) control, (B) untreated, and (C) quinapril-treated animals. Both the intima and the media were stained for collagen I in the injured arteries and also the media in the control. Magnification, ×400.
A similar result was found in the arterial collagen I mRNA expression, quantified by Northern blot in a pool of RNA of the different groups. Control animals showed a basal expression that was increased in untreated rabbits (3.7-fold) but not modified by quinapril treatment (3.5-fold) (Figure 4) ▶ .
Figure 4.

Arterial collagen I mRNA expression determined by Northern blot. The left panel shows the densitometric analysis of the Northern blot obtained from the mRNA of the pool of control, quinapril-treated, and untreated rabbits is shown. The right panel shows the bands obtained in the hybridization.
Effect of Quinapril Treatment on PDGF-B Expression in the Lesions
Because PDGF-B is one of the main mitogens involved in VSMC proliferation and migration, we studied the effect of quinapril treatment on PDGF-B mRNA expression by in situ hybridization. Control animals showed no stain for PDGF-B although there was a great expression of this factor in both the intima and media of the untreated animals (10.2 ± 4 and 10.6 ± 3.3, respectively) (Figure 5) ▶ , coinciding with the areas stained for macrophages. Quinapril-treated animals presented a marked reduction in the expression of this cytokine in the intima (5.5 ± 5.2, not significant) and in the media (4.8 ± 5, P < 0.03). As negative controls we used the labeled sense probe, the treatment with RNAse, and the omission of the probe. As expected, no staining was seen in these controls (not shown).
Figure 5.

Arterial PDGF-B mRNA expression determined by in situ hybridization. Arterial sections from a control (A), an untreated (B), and a quinopril-treated (C) animal were hybridized with a digoxigenin-labeled oligonucleotide complementary to a fragment of the PDGF-B mRNA. The control animal did not show any stain for PDGF-B, whereas in the untreated animal a great stain appeared in the intima as well as in the media. In the quinapril-treated animal a marked reduction in the expression of this growth factor can be observed in both the neointima and the media, reaching statistical significance in the latter. Magnification, ×400. D: Bar graph showing semiquantitative quantification.
Localization of NF-κB Activity in the Lesions
The presence of NF-κB in the lesions was evaluated by Southwestern histochemistry, a technique that allows determination of its in situ activation. We studied the activation of this factor in a representative number of animals of each group. We found NF-κB activity in the neointima and media of four out of five animals in the untreated group but in only two out of five in the quinapril-treated group (3 ± 1 versus 1.2 ± 0.8 and 3.4 ± 1 versus 0.8 ± 0.5 respectively, semiquantitative score).
Figure 6A ▶ presents an example of a control rabbit artery where no staining was detected (Figure 6A ▶ 1), an untreated (Figure 6A ▶ 2), and a quinapril-treated rabbit (Figure 6A ▶ 3). A graph indicating semiquantitative scores in the neointima and the media for both groups is also shown. We have previously seen that cultured monocytes and VSMC possess NF-κB activity that increases in response to inflammatory stimuli commonly overexpressed in the atherosclerotic lesion. 21 However, it is not known which of these cells is responsible for the increased activity of this nuclear factor in vivo. To identify which cellular types possessed NF-κB activity in the lesion, we performed Southwestern technique and immunohistochemistry using specific antibodies for rabbit macrophages and VSMC. Figure 6B ▶ 1 shows a double immunohistochemistry of an untreated animal where the brown area refers to VSMC and the pink to macrophages. This section corresponds to the lesion of the untreated rabbit of Figure 5A ▶ 2 and it can be observed that NF-κB activity is present in the area occupied by both cell types. Figure 6B ▶ 2 shows a double staining of the cap zone of the lesion for NF-κB and VSMC. The arrow points to a cell with the NF-κB-activated nuclei in dark blue and the cytoplasm in brown. Figure 6B ▶ 3 is a detail of the macrophage-occupied zone also showing activated nucleus in blue (arrow) and the cytoplasm of the macrophages in brown. We found no difference between the two groups in the cellular distribution of activity.
Figure 6.


Arterial localization of the NF-κB activity determined by Southwestern histochemistry. Arterial sections were hybridized with an oligonucleotide containing the consensus sequence of the NF-κB recognition site. A: Control animals (1) did not present activated nuclei, whereas untreated animals (2) showed extensive staining both in the intima and in the media (arrow). Quinapril-treated rabbits (3) showed a marked decrease in the presence of activated nuclei in both the intima and the media. Incubation of arterial sections from untreated animals with a mutant NF-κB oligonucleotide showed no staining (4). Magnification, ×400. B: In panel B 1, it can be seen a detail of a lesion showing the presence of VSMC in brown and macrophages in light pink, (this section corresponds to the untreated one shown in panel A2). Magnification, ×400. A detail of the localization of the NF-κB activity by double staining with anti-α actin antibody is shown in B2 and with -macrophage in B3 (Magnification, ×1000). The arrows indicate the presence of a nuclei stained for NF-κB and surrounded by a cytoplasm stained with the antibody specific for VSMC (B2) and macrophages (B3). C: The graph shows the semiquantitative score of the NF-κB activity in the intima and media of the untreated (NT) and quinapril-treated (Q) animals.
NF-κB Activity in the Uninjured Vessels
Because short-term hyperlipidemia alone does not cause the formation of atherosclerotic plaques in rabbits but increases ACE activity in the vascular wall (as shown in our model), we also studied the activation of NF-κB by gel shift assay in a pool of proteins obtained from the uninjured arteries of the animals on high cholesterol diet for 4 weeks. As can be seen in Figure 7 ▶ , NF-κB activity was increased in the arteries of the untreated group (lane 1) compared to the quinapril-treated animals (lane 2), whereas no NF-κB activity was present in the control group that did not receive the atherogenic diet (lane 3). We then characterized the activity induced by the diet in the aortas of rabbits by supershift assay with the antibodies for the most common subunits of NF-κB, p50 and p65. Both subunits were present in the activated NF-κB of the untreated animals (lane 4) because the anti-p50 antibody caused a diminution of the main band and the appearance of a super-retarded one (arrow, lane 5) whereas the anti-p65 antibody induced almost the disappearance of the main complex (lane 6). A densitometric analysis of the heterodimer band appears at the bottom of the figure.
Figure 7.
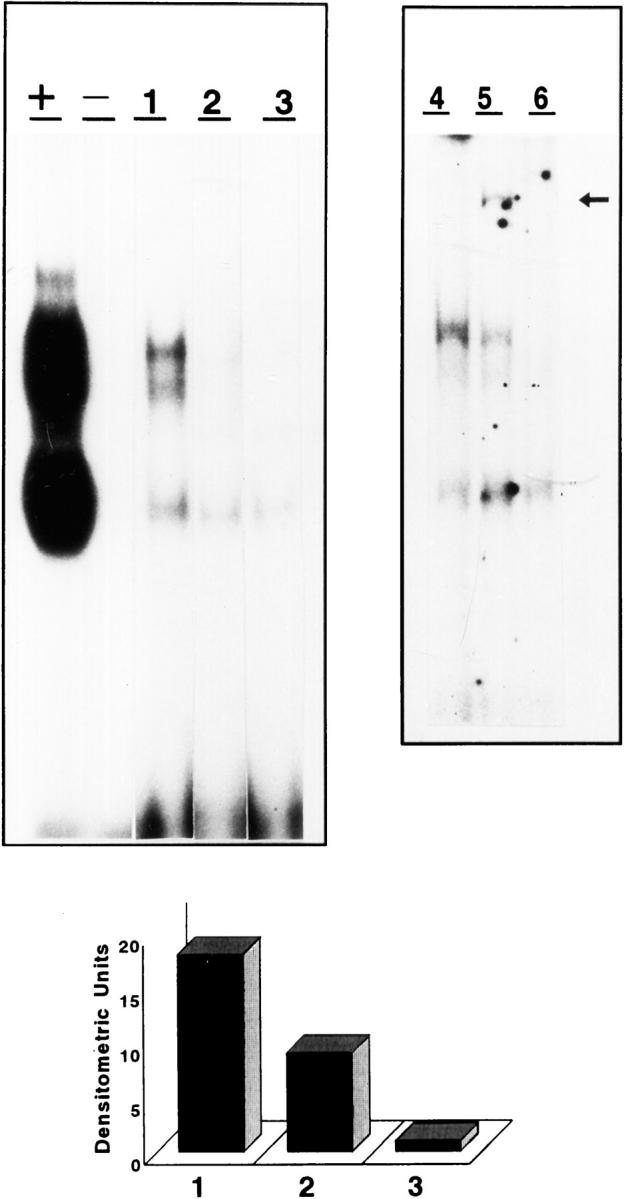
Arterial activation of NF-κB determined by gel shift assay. Aortic protein extracts from animals of the same group were pooled and incubated with a consensus NF-κB oligonucleotide labeled with 32P. A representative assay of two done is shown for untreated (1), quinapril-treated, (2) and control (3) animals. Characterization of the activity was done in the untreated pool (4) by supershift assay with anti-p50 (5) and anti-p65 (6) antibodies. As can be seen in the autoradiography of the gel, incubation with anti-p50 antibody induced reduction in the intensity of the band and the appearance of a super-retarded one (arrow), whereas the anti-p65 antibody induced almost the disappearance of the band. The graph shows a densitometric analysis of the main band corresponding to the p50-p65 heterodimer for untreated (1), quinapril-treated (2), and control (3) animals. +, positive control of the experiment with Hela cells nuclear extract; −, negative control without nuclear extract.
Discussion
The pathogenesis of atherosclerosis involves inflammatory infiltration of the vessel wall, cellular proliferation, fibrous plaque formation, and, finally, plaque rupture and occlusive thrombosis. Rupture of the fibrous plaque is associated with increased numbers of macrophages and T lymphocytes that may release cytokines and metalloproteinases, which in turn may be responsible for the loss of the fibrous cap. The main finding of this work is that arterial ACE inhibition by quinapril reduced several factors implicated in the recruitment of inflammatory cells (MCP-1 and IL-8) and VSMC migration and proliferation (PDGF-B) in the atherosclerotic lesion. Consequences were a lower macrophage infiltration rate and smaller lesions. A common characteristic of these cytokines is that they have a NF-κB binding site in their promoters. Moreover, ACE inhibition reduced arterial NF-κB activation in macrophages and VSMC, two key cells in the pathogenesis of atherosclerosis.
Although the role of vascular renin-angiotensin system activation in the development of atherosclerotic lesions is rather well-established, 38 it is not clear whether the tissular RAS may also participate in the inflammatory process leading to plaque rupture and thrombosis. However, some data suggest a relationship. For example, it has been shown that the main sources of tissue ACE in human atherosclerotic plaques are areas with inflammatory cells, especially those with clustered macrophages. 39 In addition, ACE inhibitors have been found to reduce the incidence of acute ischemic syndromes. 26
The diminution of chemokine expression in the neointima of quinapril-treated rabbits is probably due to the significant reduction in the number of macrophages invading that area; these cells are a source of and a target for PDGF-B, a potent monocyte chemotactic factor that can also elicit the expression of tissue factor. 40 VSMC are also targets for PDGF-B which can cause their proliferation through the activation of NF-κB 41 and the expression of MCP-1. 42 In humans, PDGF-B seems to play a role in the development of atherosclerotic plaques because in coronary arteries after angioplasty it is found colocalized with the PDGF-β receptor. 43 In several experimental models 44 the administration of neutralizing anti-PDGF-B antibodies reduced the intimal thickening. In addition, in VSMC this growth factor induces a stable increase in the activity of low-density lipoprotein receptor-related protein. 45
Several studies have demonstrated the effectiveness of ACE inhibitors in preventing the neointima formation in vessel wall. 46,47 In our model, we have noted a diminution in the neointima formation in quinapril-treated animals, though the reduction did not reach statistical significance. Higher doses of ACE inhibitors for the inhibition of neointima formation have been used in the above referenced studies, suggesting that the need for a higher dose may be due to the relative difficulty of inhibiting ACE tissue activity. Rakugi et al 22 have inhibited vascular ACE approximately 60%, similar to the result found with our model and reduced neointima formation in an experimental model administering 50 mg/Kg/day to rats. However, we have shown an attenuation in the size of the plaque employing a more therapeutic dose of quinapril (1 mg/Kg/day). It is possible that higher doses of quinapril could have a major effect on the lesion size but, as is well known, rabbits are very sensitive to ACE inhibition and we had high mortality rates with doses higher than 1 mg/Kg/day (not shown).
The presence of NF-κB in human atherosclerotic lesions in the nuclei of macrophages, VSMC, and endothelial cells 48 has been recently demonstrated. We have seen that in our model both macrophages and VSMC express activated NF-κB and that quinapril treatment reduces the activity equally in both cell types. We have previously shown that Ang II elicits NF-κB activation and MCP-1 expression in VSMC and monocytic cells. 20 Therefore, inhibition of Ang II generation due to quinapril treatment could account for the reduction of NF-κB activation and of IL-8, MCP-1, and PDGF-B expression in the atherosclerotic lesions. Because it has been shown that ACE inhibition promotes nitric oxide production by inhibiting degradation of bradykinin 49 and PDGF-B seems to inhibit the induction of nitric oxide synthase activity, 50 one may speculate that an increment in NO generation may play a role in the overall reduction of inflammatory cells because NO seems to be involved in the down-regulation of IL-8 and MCP-1 expression in endothelial cells 51,52 and adhesion molecules ICAM and VCAM-1 in VSMC 53 through the blockade of NF-κB generation. It is also noteworthy that NF-κB is necessary for VSMC proliferation 19 and diminution of its activation could therefore contribute to reduce the amount of VSMC in the lesion. This effect can be potentiated by a lower migration of VSMC into the neointima, because there are less chemokines and angiotensin II that can induce them to migrate. 54 As an overall consequence, there is a diminution in the number of VSMC in the treated animals that, in addition to a lower macrophage presence, may account for the reduction in lesion size observed after treatment with ACE inhibitors. 25
Correlation between the susceptibility to development of fatty streaks and the activation of vascular 55 and hepatic NF-κB in response to an atherogenic diet 56 has been demonstrated. In our model we observed an increase in arterial NF-κB activation in response to the atherogenic diet and a reduction of the activation in response to the treatment. Because we also observed that after 4 weeks of high-cholesterol diet there was a four-fold increase in vascular ACE activity, one may also speculate that the lower NF-κB activity observed in the uninjured aortas of the quinapril-treated group could be due to reduction of Angiotensin II generated by the hyperlipidemia. 57
Collagen I is the main extracellular matrix protein in the fibrous cap that, together with collagen III, accounts for around 60% of the total protein and at least 90% of the total collagen of the lesion. 58 The integrity of the fibrous cap and thus its resistance to rupture depends critically on the collagenous extracellular matrix of the plaque’s fibrous cap. Collagen I is produced mainly by VSMC and a diminution in collagen synthesis due to a decreased number of VSMC, as well as an increment in its degradation due to metalloproteinases secreted by the invading macrophages, have been associated with the weakening of the fibrous cap making it prone to rupture. 59 Angiotensin II may increase plaque stability by favoring collagen I expression, an effect that could theoretically be reversed by ACE inhibition. Paradoxically, the treatment with quinapril did not modify the expression of collagen I. The reasons for this apparent discrepancy are unknown but one possible explanation could be that whereas MCP-1, IL-8, and PDGF-B are NF-κB-dependent genes, the expression of collagen I is regulated by the transcription factor AP-1. 60 This suggests that the blockade of NF-κB activation as a potential control step of certain genes may be achieved with quinapril treatment, although angiotensin II can also regulate gene expression via AP-1 activation. 61 Further studies are needed to clarify this issue. Consequently, untreated and quinapril-treated animals showed plaques with fibrotic morphology, but only those treated with quinapril presented a diminution of the inflammatory components IL-8, MCP-1, PDGF-B, and macrophages.
However, it is also possible that some of the effects observed with the ACE inhibitor quinapril could be due in part to the accumulation of bradykinins or the generation of NO and prostaglandins. In fact, although previous authors have shown that other ACE inhibitors or AT1 antagonists could cause diminution of the atherosclerosis lesion, 62 the potential mechanisms of this beneficial effect are mostly unknown. To establish the relative roles of Angiotensin II and bradykinin in the actions of quinapril, similar studies with bradykinin or AT1 antagonist may be necessary; the aim of our study was not to compare the effect of these drugs.
On the whole, our results support the conclusion that in a model of atherosclerosis in rabbits, ACE inhibition improves some inflammatory and proliferative parameters of the lesion without affecting the fibrotic component and could therefore contribute to stabilization of the plaque.
Acknowledgments
The authors thank Natalia Duque for helpful assistance in development of the PDGF-B in situ hybridization and Dr. Carmen Gómez for helpful assistance in the development of Southwestern histochemistry.
Footnotes
Address reprint requests to Jesús Egido, M.D., Research Laboratories, Fundación Jiménez Díaz, Avda Reyes Católicos 2, 28040 Madrid, Spain. E-mail: evasc@uni.fjd.es.
Supported by grants from Ministerio de Educación y Ciencia (MEC) (PB 94/0211; SAF 97-55 PM97/85), Fundación Mapfre Medicina and Parke-Davis, Spain.
References
- 1.van der Wal AC, Becker AE, van der Loos, Das PK: Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89:36-44 [DOI] [PubMed] [Google Scholar]
- 2.Wysocki SJ, Zheng MH, Smith A, Lamawansa MD, Iacopetta BJ, Robertson TA, Papadimitriou JM, House AK, Norman PE: Monocyte chemoattractant protein-1 gene expression in injured pig artery coincides with early appearance of infiltrating monocyte/macrophages. J Cell Biochem 1996, 62:303-313 [DOI] [PubMed] [Google Scholar]
- 3.Ylä-Herttuala S, Lipton BA, Rosenfeld ME, Särkioja T, Yoshimura T, Leonard EJ, Witztum JL, Steinberg D: Expression of monocyte chemoattractant protein 1 in macrophage rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci USA 1991, 88:5252-5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X, Dluz S, Graves DT, Zhang L, Antoniades HN, Hollander W, Prusty S, Valente AJ, Schwartz CJ, Sonenshein GE: Elevated expression of monocyte chemoattractant protein 1 by vascular smooth muscle cells in hypercholesterolemic primates. Proc Natl Acad Sci USA 1992, 89:6953-6957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA: Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci USA 1994, 91:3652-3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonasson L, Skalli O, Bondjers O, Hansson GK: Regional accumulation of T cells, macrophages and smooth muscle cells in the human atherosclerotic plaque. Arterioscler Thromb 1986, 6:131-138 [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Hulten LM, Wiklund O: Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arterioscler Thromb Vasc Biol 1997, 17:317-323 [DOI] [PubMed] [Google Scholar]
- 8.Wang N, Tabas I, Winchester R, Ravalli S, Rabbani LE, Tall A: Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J Biol Chem 1996, 271:8837-8842 [DOI] [PubMed] [Google Scholar]
- 9.Yue TL, McKenna PJ, Gu JL, Feuerstein GZ: Interleukin-8 is chemotactic for vascular smooth muscle cells. Eur J Pharmacol 1993, 240:81-84 [DOI] [PubMed] [Google Scholar]
- 10.Wang JM, Sica A, Peri G, Walter S, Padura IM, Libby P, Ceska M, Lindley I, Colotta F, Mantovani A: Expression of monocyte chemotactic protein 1 and interleukin-8 by cytokine activated human vascular smooth muscle cells. Arterioscler Thromb 1991, 11:1166-1174 [DOI] [PubMed] [Google Scholar]
- 11.Apostolopoulos J, Davenport P, Tipping PG: Interleukin-8 production by macrophages from atheromatous plaques. Arterioscler Thromb Vasc Biol 1996, 16:1007-1012 [DOI] [PubMed] [Google Scholar]
- 12.Uchida K, Sasahara M, Morigami N, Hazama F, Kinoshita M: Expression of platelet-derived growth factor B-chain in neointimal smooth muscle cells of balloon injured rabbit femoral. Atherosclerosis 1996, 124:9-23 [DOI] [PubMed] [Google Scholar]
- 13.Hart CE, Clowes AW: Platelet-derived growth factor and arterial response to injury. Circulation 1997, 95:555-556 [DOI] [PubMed] [Google Scholar]
- 14.Wilcox JN, Smith KM, Williams LT, Schwartz SM, Gordon D: Platelet-derived growth factor mRNA detection in human atherosclerotic lesions by in situ hybridization. J Clin Invest 1988, 82:1134-1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes PJ, Karin M: Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 1997, 336:1066-1071 [DOI] [PubMed] [Google Scholar]
- 16.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T: NF-κB and SP1 regulate transcription of the human monocyte chemoattractant protein 1 gene. J Immunol 1994, 153:2052-2063 [PubMed] [Google Scholar]
- 17.Mukaida N, Okamoto S, Ishikawa Y, Matsushima K: Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol 1994, 56:554-558 [PubMed] [Google Scholar]
- 18.Kachigian LM, Resnick N, Gimbrone NA, Collins T: Nuclear factor κB interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J Clin Invest 1995, 96:1169-1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellas RE, Lee JS, Sonenshein GE: Expression of a constitutive NF-κB-like activity is essential for proliferation of cultured bovine vascular smooth muscle cells. J Clin Invest 1995, 96:2521-2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernández-Presa M, Bustos C, Ortego M, Tuñón J, Renedo G, Ruiz-Ortega M, Egido J: Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-κB activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 95:1532-1541 [DOI] [PubMed] [Google Scholar]
- 21.Bourcier T, Sukhova G, Libby: The nuclear factor kappa-B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J Biol Chem 1997, 272:15817-15824 [DOI] [PubMed] [Google Scholar]
- 22.Rakugi H, Kim DK, Krieger JE, Wang DS, Dzau VJ, Pratt RE: Induction of angiotensin converting enzyme in the neointima after vascular injury. Possible role in restenosis. J Clin Invest 1994, 93:339-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jandeleit-Dahm K, Burrell LM, Johnston CL, Koch KM: Elevated vascular angiotensin converting enzyme mediates increased neointima formation after balloon injury in spontaneously hypertensive rats. J Hypertens 1997, 15:643-650 [DOI] [PubMed] [Google Scholar]
- 24.Pitt B: Angiotensin-converting enzyme inhibitors in patients with coronary atherosclerosis. Am Heart J 1994, 128:1328-1332 [DOI] [PubMed] [Google Scholar]
- 25.Powel JS, Glozel JP, Muller RK, Kuhn H, Hefti F, Hosang M, Baumgartner: Inhibitors of angiotensin-converting enzyme prevent myointimal proliferation after vascular injury. Science 1989, 245:186-188 [DOI] [PubMed] [Google Scholar]
- 26.Yusuf S, Pepine CJ, Garces C, Pouleur H, Salem D, Kostis J, Benedict C, Rousseau M, Bourassa M, Pitt B: Effect of Enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet 1992, 240:1173-1178 [DOI] [PubMed] [Google Scholar]
- 27.Aberg G, Ferrer P: Effects of captopril on atherosclerosis in cynomolgus monkeys. J Cardiovascular Pharmacol 1990, 15(suppl 5):S65-S72 [PubMed] [Google Scholar]
- 28.Pepine CJ: Improved endothelial function with angiotensin-converting enzyme inhibitors. Am J Cardiol 1997, 79:29-32 [DOI] [PubMed] [Google Scholar]
- 29.Le Veen RF, Wolf GL, Villanueva TG: New rabbit atherosclerosis model for the investigation of transluminal angioplasty. Invest Radiol 1982, 17:470-475 [DOI] [PubMed] [Google Scholar]
- 30.Tsukada T, Rosenfeld ME, Ross R, Gown AM: Immunocytochemical analysis of cellular components in atherosclerotic lesions: use of monoclonal antibodies with the Watanabe and fat-fed rabbit. Arteriosclerosis 1986, 6:601-613 [DOI] [PubMed] [Google Scholar]
- 31.Chomczynski P, Sacchi N: Single-step of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 32.Yoshimura T, Yuhki N: Neutrophil attractant/activation protein-1 and monocyte chemoattractant protein-1 in rabbit. J Immunol 1991, 146:3483-3488 [PubMed] [Google Scholar]
- 33.Kaneto H, Morrissey J, Klahr S: Increased expression of TGF-β1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int 1993, 44:313-321 [DOI] [PubMed] [Google Scholar]
- 34.Wiggins R, Goyal M, Merrit S, Killen PD: Vascular adventitial cell expression of collagen I messenger ribonucleic acid in antiglomerular basement membrane antibody induced crescentic nephritis in the rabbit. Lab Invest 1993, 68:557-565 [PubMed] [Google Scholar]
- 35.Herren B, Weyer KA, Rouge M, Lotscher P, Pech M: Conservation in sequence and affinity of human and rodent PDGF ligands and receptors. Biochim Biophys Acta 1993, 1173:294-302 [DOI] [PubMed] [Google Scholar]
- 36.Negoro N, Kanayama Y, Haraguchi M, Umetani N, Nishimura M, Konishi Y, Iwai J, Okamura M, Inoue T, Takeda T: Blood pressure regulates platelet-derived growth factor A-chain gene expression in vascular smooth muscle cells in vivo. J Clin Invest 1995, 95:1140-1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang XB, Watanabe Y, Osugi T, Ikemoto M, Hirata M, Miki N: In situ DNA-protein binding: a novel method for detecting DNA-binding activity of transcription factor in brain. Neurosci Lett 1992, 146:25-28 [DOI] [PubMed] [Google Scholar]
- 38.Cody RJ: The integrated effects of angiotensin II. Am J Cardiol 1997, 79:9-11 [DOI] [PubMed] [Google Scholar]
- 39.Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ: Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation 1996, 94:2756-2767 [DOI] [PubMed] [Google Scholar]
- 40.Ernofsson M, Siegbahn A: Platelet-derived growth factor-BB and monocytic chemotactic protein-1 induce human peripheral blood monocytes to express tissue factor. Thromb Res 1996, 83:307-320 [DOI] [PubMed] [Google Scholar]
- 41.Obata H, Biro S, Arima N, Kaieda H, Kihara T, Eto H, Miyata M, Tanaka H: NF-kappa B is induced in the nuclei of cultured rat aortic smooth muscle cells by stimulation of various growth factors. Biochem Biophys Res Commun 1996, 224:27-32 [DOI] [PubMed] [Google Scholar]
- 42.Marmur JD, Poon M, Rossikhina M, Taubman MD: Induction of PDGF-responsive genes in vascular smooth muscle. Implications for the early response to vessel injury. Circulation 1992, 86:53-60 [PubMed] [Google Scholar]
- 43.Tanizawa S, Ueda M, van der Loos CM, van der Wal AC, Beck AE: Expression of platelet derived growth factor B chain and beta receptor in human coronary arteries after percutaneous transluminal coronary angioplasty: an immunohistochemical study. Heart 1996, 75:549-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutherford C, Martin W, Salame M, Carrier M, Anggard E, Ferns G: Substantial inhibition of neo-intimal response to balloon injury in the rat carotic artery using a combination of antibodies to platelet-derived growth factor-BB and basic fibroblast growth factor. Arteriosclerosis 1997, 130:45-51 [DOI] [PubMed] [Google Scholar]
- 45.Weaver AM, McCabe M, Kim I, Allietta MM, Gonias SL: Epidermal growth factor and platelet-derived growth factor-BB induce a stable increase in the activity of low density lipoprotein receptor-related protein in vascular smooth muscle cells by altering receptor distribution and recycling. J Biol Chem 1996, 271:24894-24900 [DOI] [PubMed] [Google Scholar]
- 46.Jandeleit-Dahm K, Burrell LM, Jhonston CL, Koch KM: Elevated vascular angiotensin converting enzyme mediates increased neointima formation after balloon injury in spontaneously hypertensive rats. J Hypertens 1997, 15:643-650 [DOI] [PubMed] [Google Scholar]
- 47.Rakugi H, Wang DS, Dzau V, Pratt RE: Potential importance of tissue angiotensin-converting enzyme inhibition in preventing neointima formation. Circulation 1994, 90:449-455 [DOI] [PubMed] [Google Scholar]
- 48.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D: Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest 1996, 97:1715-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kichuk MR, Zhang X, Oz M, Michler R, Kaley G, Nasjletti A, Hintze TH: Angiotensin-converting enzyme inhibitors promote nitric oxide production in coronary microvessels from failing explanted human hearts. Am J Cardiol 1997, 80:137A-142A [DOI] [PubMed] [Google Scholar]
- 50.Schini VB, Durante W, Elizondo E, Scott-Borden T, Junquero DC, Schafer AL, Vanhoutte PM: The induction of nitric oxide synthase activity is inhibit by TGF-β, PDGFAB and PDGFBB in vascular smooth muscle cells. Eur J Pharmacol 1992, 216:379-383 [DOI] [PubMed] [Google Scholar]
- 51.Andrew PJ, Harant H, Lindley IJ: Nitric oxide regulates IL-8 expression in melanoma cells at the transcriptional level. Biochem Biophys Res Commun 1995, 214:949-956 [DOI] [PubMed] [Google Scholar]
- 52.Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R: Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res 1995, 76:980-986 [DOI] [PubMed] [Google Scholar]
- 53.Shin WS, Hong YH, Peng HB, De Caterina R, Libby P, Liao JK: Nitric oxide attenuates vascular smooth muscle cell activation by interferon-γ. The role of constitutive NF-κB activity. J Biol Chem 1996, 271:11317-11324 [DOI] [PubMed] [Google Scholar]
- 54.Liu G, Spinosa E, Oemar BS, Lüscher T: Bimodal effects of angiotensin II on migration of human and rat smooth muscle cells: direct stimulation and indirect inhibition via transforming growth factor-β1. Arterioscler Throm Vasc Biol 1997, 17:1251-1257 [DOI] [PubMed] [Google Scholar]
- 55.Liao F, Andalibi A, Qiao JH, Allayee H, Fogelman AM, Lusis AJ: Genetic evidence for a common pathway mediating oxidative stress, inflammatory gene induction, and aortic fatty streak formation in mice. J Clin Invest 1994, 94:877-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao F, Andalibi A, deBeer FC, Fogelman AM, Lusis AJ: Genetic control of inflammatory gene induction and NF-kappa B-like transcription factor activation in response to an atherogenic diet in mice. J Clin Invest 1993, 91:2572-2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitani H, Bandoh T, Kimura M, Totsuka T, Hayashi S: Increased activity of vascular ACE related to atherosclerotic lesions in hyperlipidemic rabbits. Am J Physiol 1996, 271:H1065-H1071 [DOI] [PubMed] [Google Scholar]
- 58.Barnes MJ: Collagens in atherosclerosis. Coll Relat Res 1985, 5:65-97 [DOI] [PubMed] [Google Scholar]
- 59.Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT: Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol 1996, 7:330-335 [DOI] [PubMed] [Google Scholar]
- 60.Chung KY, Agarwal A, Uitto J, Mauviel A: An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem 1996, 271:3272-3278 [DOI] [PubMed] [Google Scholar]
- 61.Takeuchi K, Nakamura N, Cook NS, Pratt RE, Dzau VJ: Angiotensin II can regulate gene expression by the AP-1 binding sequence via a protein kinase C-dependent pathway. Biochem Biophys Res Commun 1990, 172:1189-1194 [DOI] [PubMed] [Google Scholar]
- 62.Keidar S, Attias J, Smith J, Breslow JL, Hayek T: The angiotensin-II receptor antagonist, losartan, inhibits LDL lipid peroxidation and atherosclerosis in apolipoprotein E-deficient mice. Biochem Biophys Res Commun 1997, 236:622-625 [DOI] [PubMed] [Google Scholar]