

Abstract
We studied acute responses of rat lungs to inhalation of urban particulate matter and ozone. Exposure to particles (40 mg/m3 for 4 hours; mass median aerodynamic diameter, 4 to 5 μm; Ottawa urban dust, EHC-93), followed by 20 hours in clean air, did not result in acute lung injury. Nevertheless, inhalation of particles resulted in decreased production of nitric oxide (nitrite) and elevated secretion of macrophage inflammatory protein-2 from lung lavage cells. Inhalation of ozone (0.8 parts per million for 4 hours) resulted in increased neutrophils and protein in lung lavage fluid. Ozone alone also decreased phagocytosis and nitric oxide production and stimulated endothelin-1 secretion by lung lavage cells but did not modify secretion of macrophage inflammatory protein-2. Co-exposure to particles potentiated the ozone-induced septal cellularity in the central acinus but without measurable exacerbation of the ozone-related alveolar neutrophilia and permeability to protein detected by lung lavage. The enhanced septal thickening was associated with elevated production of both macrophage inflammatory protein-2 and endothelin-1 by lung lavage cells. Interestingly, inhalation of urban particulate matter increased the plasma levels of endothelin-1, but this response was not influenced by the synergistic effects of ozone and particles on centriacinar septal tissue changes. This suggests an impact of the distally distributed particulate dose on capillary endothelial production or filtration of the vasoconstrictor. Overall, equivalent patterns of effects were observed after a single exposure or three consecutive daily exposures to the pollutants. The experimental data are consistent with epidemiological evidence for acute pulmonary effects of ozone and respirable particulate matter and suggest a possible mechanism whereby cardiovascular effects may be induced by particle exposure. In a broad sense, acute biological effects of respirable particulate matter from ambient air appear related to paracrine/endocrine disruption mechanisms.
Human health impacts of air pollution have been recognized for over a century. In recent years, episodic variations of respirable particles of less than a hundred micrograms per cubic meter have been associated with increases, on following days, of respiratory and cardiovascular morbidity and mortality. 1-4 The consistency between epidemiological studies argues for a causal association, but the attribution of acute health effects to such low concentration levels appears to defy current toxicological and pathophysiological paradigms. Acceptance of a biological plausibility for a causal association will require the identification of direct biological effects of respirable particles and their mechanisms of interference with the course of diseases. Surprisingly, there is a paucity of data on the direct in vivo biological effects of particulate matter from ambient air, especially for experimental inhalation of environmental materials.
Although inhalation exposure of rats to urban particulate matter may not cause significant acute injury in the lung parenchyma, particles can enhance centriacinar lesions induced by ozone. 5 It is conceivable that phagocytes activated by particles could amplify the complex cellular dynamics at sites of a primary lesion, through elaboration of inflammatory, chemotactic, and mitogenic cytokines. 6,7 This refers to the classical concept of macrophage activation followed by outpouring of free radicals, eicosanoids, and cytokines and a cascade of cellular responses, leading to adverse effects. Such mechanisms may well be relevant to enhancement of inflammatory reactions by particles in sensitive subjects, eg, asthmatics. In cell culture studies, urban particles are not markedly cytotoxic but can nevertheless alter cell physiology, as revealed by inhibition of the oxidative burst of macrophages 8 or by the induction of a heat-shock response in epithelial cells. 9 This suggests a second aspect to biological effects of complex particles, which could be metabolic or physiological blocks in target cells. 10,11 Such responses might be relevant to potential alterations of coordinated immunological defense in individuals with lung infections. For example, enhancement of bacterial infectivity in mice by intratracheal injection of urban dust suspensions has been reported more than a decade ago. 12
It is noteworthy that low concentrations of respirable particles are associated epidemiologically with acute cardiac outcomes, in addition to being associated with pulmonary outcomes. 4 Ozone has been associated with pulmonary effects but generally not with cardiac morbidity and mortality. Although this distinction may not be definitive, it can be rationalized on the basis of expected differences in toxicodynamics between particles and reactive gases. 4 Ozone is consumed in the respiratory tract and the proximal segment of alveolar ducts, sparing the distal parenchyma. Respirable particles, however, should access the distal alveolar surface area, where bioavailable components leached from particles could affect the capillary bed in a more diffuse fashion. The lung capillaries regulate the levels of a number of circulating vasoactive substances that affect cardiovascular homeostasis. Endothelin (ET)-1 is a potent vasoconstrictor produced not only by endothelial cells of the lung capillaries but also by bronchial epithelial cells, macrophages, and pulmonary neuroendocrine cells. Elevated levels of ET-1 have been reported in a number of pathologies and acute lung injury models. 13 ET-1 is thought to play an active role in the maintenance of basal systemic vascular tone 14-17 and, interestingly, may create a hypercoagulable state and act as an antifibrinolytic factor. 18,19
The first objective of our study was to substantiate previous observations of focal histological changes in the lungs of animals exposed to ambient air particulate matter and ozone 5 by an assessment of cytological and biochemical changes in bronchoalveolar lavage fluids. In particular, we sought to extend analyses to patterns of functional changes in lung macrophages, such as altered production of cytokines and nitric oxide or reduced phagocytic activity, that would be consistent with impacts of particles on lung health and that may help to clarify the synergistic interaction of particles and ozone in centriacinar lesions. 5 The second objective of our study was to investigate whether circulating levels of ET-1, a potent vasoconstrictor that can affect cardiovascular homeostasis, could be modulated by inhalation of urban dust, ozone, or dust plus ozone, and to determine whether such changes of ET-1 would correlate with or would be dissociated from the intensity of the centriacinar tissue lesions.
Materials and Methods
Animals
Specific-pathogen-free Fischer 344 rats (males, 200 to 250 g) were purchased from Charles River (St. Constant, Québec, Canada) and received in filter cages. The animals were inspected, randomized, and housed in shoe-box plexiglass cages on wood chip bedding, under a constant charcoal-filtered and 0.2-μm-filtered air stream, and in a 12-hour dark/light cycle. Food and water were provided ad libitum. Animals were kept in accordance with the standard operating procedures of the Animal Resources Division and as set forth in the Guidelines of the Canadian Council on Animal Care. All experimental protocols were reviewed by the Animal Care Committee of the Health Protection Branch, Health Canada.
Experimental Atmospheres
The urban dust EHC-93 was recovered by vacuuming of bag-house filters (100% ambient air) of the Environmental Health Center in Ottawa and cleaned through a 36-μm sieve. Comparative data on chemical composition and biological reactivity were presented before. 9 In brief, EHC-93 contains profiles and amounts of polycyclic aromatic hydrocarbons, ions, and metals comparable to those of the standard reference materials 1648 (urban particulate matter) and 1649 (organics) of the National Institute of Standards and Technology (Gaithersburg, MD), and to a range of fine particulate materials recovered by field sampling. The dust was dispersed in a venturi (powder disperser model 3433, TSI, St. Paul, MN) and directed to a flow-past, nose-only-exposure, 12-port inhalation manifold (CH Technologies, Westwood, NJ). 5 One or two stacks of inhalation manifolds were used, for a flow rate of 12 to 24 L/minute (1 L/minute per port). The concentration of EHC-93 was evaluated at the inhalation ports by isokinetic sampling on 0.2-μm Teflon filters (TF-200, 47 mm; Gelman Sciences, Ann Arbor, MI). Real-time particle count and size distribution measurements were obtained by isokinetic sampling at the inhalation ports (Lasair model 301, Particle Measuring Systems, Boulder, CO). Count median diameter (CMD) determined by laser-optic counting was 0.5 to 0.6 μm, with a geometric SD of 1.9. The mass median aerodynamic diameter (MMAD) verified by cascade impactor analyses was 3 to 4 μm (seven-stage Mercer type, Intox, Albuquerque, NM). Ozone was produced from pure oxygen in a silent arc generator (model 200, Sanders, Uetze, Germany), introduced in the experimental atmosphere, and continuously monitored by ultraviolet spectrometry (model 1003-AH, Dasibi Environmental, Glendale, CA) at the inhalation ports by sampling through a Teflon filter. 5 Ozone concentrations were stabilized by feedback control and varied ±5% from the 0.8 parts per million (ppm) O3 target concentration. Environmental factors, mechanical factors related to the dust-atmosphere generation system, and sampling and analytical variabilities, can all affect the variance of particle concentrations over time. For the series of exposures presented here, the target EHC-93 dust time-weighted average concentration was 50 mg/m 3 over 6 hours. The average measured concentration from a series of 2-hour filter samples (two or three sequential filters per exposure run) was 40 ± 2 mg/m 3 (mean ± SE; n = 51 filters). This value is not significantly different from that in our previous report (48 mg/m3). 5 The test particle concentration to which we will uniformly refer for lung lavage and cellular endpoints is 40 mg/m 3 EHC-93. Morphometry data were extracted from animals exposed to 48 mg/m 3 EHC-93.
Inhalation Exposures
The animals were progressively trained in the nose-only exposure tubes over 5 days. Animals were then exposed for 4 hours to clean air, to 40 mg/m 3 EHC-93, to 0.8 ppm O3, or simultaneously to 40 mg/m 3 EHC-93 plus 0.8 ppm O3, during a single session (1-day exposure) or on three consecutive days (3-day exposure). The animals were returned to normal housing conditions immediately after exposure and euthanized 20 hours after the last exposure. For cytological and biochemical endpoints, five separate experiments were performed: three experiments consisting of 1-day exposures and two experiments consisting of 3-day repeated exposures. All four exposure conditions (clean air, particles alone, ozone alone, and particles plus ozone) were blocked within each experiment, with six animals per treatment group. A sixth 4-hour single-exposure experiment was conducted for qualitative histological examination of lungs; group size was four animals, and recovery in clean air was 20 hours. Finally, additional morphometric analyses were conducted on lung tissues of animals exposed under equivalent conditions (48 mg/m 3 EHC-93); group size was six animals, and recovery in clean air was 32 hours. 5
Lung Histology
Animals were anesthetized by intraperitoneal injection of pentobarbital sodium (65 mg/kg). Lungs were fixed by two different procedures. For qualitative examination of intra-alveolar inflammatory cells and protein, and to avoid displacement of these changes, the lungs were collapsed and fixed by vascular perfusion. The thoracic cavity was exposed, and the lungs were perfused via the pulmonary artery with 0.2 mol/L sodium cacodylate, followed by a 2% glutaraldehyde solution containing 0.085 mol/L sodium cacodylate, 0.05% (w/v) calcium chloride, pH 7.4. For morphometry, the lungs were inflated via the trachea with the glutaraldehyde solution at a pressure of 25 cm H2O above the chest for 10 to 15 minutes. The lungs were excised and immersed in the glutaraldehyde solution. For light microscopy, three lung tissue blocks sampled below the hilum of the left lobe were post-fixed in formalin for 24 hours, dehydrated in ethanol, and embedded in glycol methacrylate. Sections (0.75 to 1 μm) were stained with toluidine blue. For electron microscopy examination, lung samples were post-fixed in osmium tetroxide, stained en bloc with uranyl acetate, and embedded in Spurr resin. Thin sections were mounted directly on copper grids and stained with lead citrate.
Morphometry
Light microscopy morphometric analyses were performed according to established procedures. 20 Briefly, low-magnification digitized images of alveolar ducts were captured (10× objective) using Image (U.S. National Institutes of Health, public domain software) and printed to generate maps. The septal edges along alveolar duct paths were identified on the maps and coded, and their individual distance from the terminal bronchiole-alveolar duct junction was recorded. Each septal edge was digitized at high magnification (100× objective) and retrieved in Stereology Toolbox (Morphometrix, Davis, CA). A counting overlay was applied (staggered cycloid test lattice, 135 points/45 lines per picture), and points on septum (excluding capillary lumen) and points on type II epithelial cell profiles as well as intercepts with type I and type II cells apical surfaces were tallied. Within each animal, points and intercepts for images of septal edges within the distance intervals of 0 to 500 μm (proximal region) and 500 to 1000 μm (distal region), relative to the terminal bronchiole, were added. Three lung sections were analyzed, for a total of four to five alveolar ducts and 45 to 50 pictures per animal. The estimates of normalized volume density (μm) of septum and type II cells were calculated from the ratio of volume density (μm3/μm3) over the epithelial surface density (μm2/μm3). 20 The normalized volume density is analogous to an arithmetic mean thickness. An upward shift in the value of the normalized volume density of the septum can be due to a number of factors, such as interstitial edema and inflammatory cell infiltration and epithelial hyperplasia/hypertrophy. A thickening of the type II cell compartment is more specific for hyperplasia/hypertrophy of this progenitor cell population and is typically interpreted as a repair proliferation.
Bronchoalveolar Lavage
The animals were anesthetized by intraperitoneal injection of pentobarbital sodium (65 mg/kg). Blood was collected into heparinized or EDTA-treated syringes from the dorsal aorta. The animals were then exsanguinated, and the trachea was exposed and cannulated. The diaphragm was punctured to collapse the lungs, and the lungs were filled by intratracheal instillation of warm (37°C) calcium- and magnesium-free Dulbecco’s phosphate buffered saline (Sigma Chemical Co., St. Louis, MO) at a ratio of 35 ml/kg body weight. The saline was aspirated and re-injected twice more, and the bronchoalveolar lavage fluid was recovered in cold centrifuge tubes. This first lavage fluid was used for protein and fibronectin assays. Cell recovery was optimized by following the first lavage with sequential single instillation of saline, for a total lavage volume of 40 ml. The free cells were sedimented by centrifugation at 350 × g for 15 minutes at 4°C. Aliquots of supernatants for protein and fibronectin analyses were snap-frozen in liquid nitrogen and stored at −80°C.
Biochemical and Cytological Analyses
Protein
Protein was determined by the Coomassie blue dye-binding assay using Pierce reagent (Pierce, Rockford, IL) essentially as described before. 21
Fibronectin
Lavage fluid supernatant was mixed with loading buffer (1:1) and applied to 6% SDS-polyacrylamide minigels at 25 μg of total protein per well. Electrophoresis was performed at 100 V for 2 hours. The proteins were electroeluted to a nitrocellulose membrane, which was incubated overnight with a polyclonal rabbit anti-rat fibronectin antibody (Calbiochem, San Diego, CA), followed by a 2-hour incubation with a goat anti-rabbit alkaline phosphatase-conjugated secondary antibody (Bio-Rad, Hercules, CA). The fibronectin was revealed, and the dried membranes were analyzed by imaging densitometry. The cumulative area under the curve for the A and B chains of fibronectin was computed as relative optical density units.
Cell Counting
Free lavage cells were resuspended in saline and counted in a Coulter Multisizer II (Coulter Electronics, Burlington, Ontario, Canada). Average yield for air control animals was 6.8 ± 0.4 × 10 6 cells (mean ± SE; n = 30 animals).
Differential Cytology
Lavage cells (1 × 104) were sedimented on glass slides using a cytocentrifuge (Cytospin-3, Shandon, Pittsburgh, PA) at 350 × g for 8 minutes. The slides were stained with DiffQuick (Baxter Diagnostics, Montréal, Québec, Canada). Differential counts for macrophages and neutrophils were determined by microscopic examination. Lavage cells from control animals were >99% macrophages and 0.02% neutrophils. In all treatment groups, the frequency of neutrophils was always less than 2%, and so individual cell types were not separated.
Macrophage Functional Assays
The cells were suspended in M199 culture medium (Sigma) supplemented with penicillin (50 U/ml), streptomycin (50 μg/ml), l-glutamine (2 mmol/L) and seeded in 96-well plates (6 × 10 4 cells/well; 2 × 10 5 cells per cm2). The culture medium contained 5% (v/v) fetal bovine serum for the viability, phagocytosis, and nitric oxide production assays and 2 mg/ml bovine serum albumin for the cytokine assays (tumor necrosis factor (TNF)-α, macrophage inflammatory protein (MIP)-2, and ET-1). Measurements in the cell culture medium for each rat were made in triplicate. Attachment of cells was verified routinely by NaOH solubilization and assay of protein with the Coomassie blue dye-binding assay, and all cell culture endpoints were normalized to cell protein for statistical analyses. Control values are provided below per 10 6 cells/24-hour incubation for reference.
Cell Viability
Viability was determined by the alamarBlue assay (Alamar Biosciences, Sacramento, CA) essentially as described before. 22
Phagocytosis
Cells were incubated with fluorescein-labeled Escherichia coli (Molecular Probes, Eugene, OR) for 2 hours. The extracellular fluorescence was quenched with trypan blue, and phagocytosis by the cell monolayers was determined by fluorescence measurement (relative units) at 485-nm excitation and 530-nm emission (Cytofluor 2350, Millipore, Bedford, MA).
Nitrite
Nitric oxide production was determined by the assay of nitrite with the Griess reagent 23 in 24-hour culture supernatants of macrophages stimulated with 5 μg/ml Salmonella thyphimurium lipopolysaccharide (LPS; Sigma). Nitrite production was suppressed by >90% with the addition of 100 μmol/L nitric oxide synthase inhibitor NG-methyl-l-arginine (data not shown). Basal production of nitrite was highly variable in cell cultures not stimulated with LPS and was not considered in our analyses. Nitrite values were reproducible in cells induced with LPS. Corresponding nitrite values for cells from clean air control animals were 126 ± 9 nmol/10 6 cells/24 hours (mean ± SE; n = 28 animals).
TNF-α
TNF-α was measured in 24-hour supernatants by ELISA (Biosource, Camarillo, CA). Basal levels of TNF-α secretion by macrophage cultures were 26 ± 5 ng/10 6 cells/24 hours (mean ± SE; n = 18 animals).
MIP-2
MIP-2 was measured in 24-hour culture supernatants by ELISA (Biosource). Secretion of MIP-2 by air control cells was 31 ± 4 ng/10 6 cells/24 hours (mean ± SE; n = 20 animals).
ET-1
ET-1 was measured in 24-hour cell culture supernatants. Proteins were precipitated with 1.5 vol of acid/acetone (acetone/1 N HCl/H2O, 40:1:5) and sedimented at 3000 × g for 20 minutes. The supernatant was evaporated by vacuum centrifugation at 37°C for 6 to 10 hours, the pellet was resuspended for a four- to sixfold concentration, and ET-1 was measured by ELISA (R&D Systems, Minneapolis, MN). Secretion of ET-1 by clean air control cells was 7.3 ± 1.3 pg/10 6 cells/24 hours (mean ± SE; n = 16 animals).
Plasma ET-1
Blood (3 to 5 ml) recovered from the dorsal aorta into heparin-treated or EDTA-treated syringes was centrifuged at 350 × g for 8 minutes. Plasma aliquots were processed immediately as indicated below or stored at −40°C until analysis. Plasma proteins were precipitated with 1.5 vol of acid/acetone (acetone/1 N HCl/H2O, 40:1:5) and sedimented at 3000 × g for 20 minutes. The supernatant was evaporated by vacuum centrifugation at 37°C for 6 to 10 hours, and the resulting pellet was resuspended in ELISA buffer in one-sixth of the initial sample volume (ie, for a sixfold concentration). ET-1 was measured directly by ELISA (R&D Systems) with human ET-1 as standard. Plasma levels of immunoreactive ET-1 in clean air control animals were 0.25 ± 0.03 pg/ml (mean ± SE; n = 24 animals). The cross-reactivities of ET-1-related proteins in the ELISA are reported by the manufacturer as follows: big-endothelin, <1%; ET-2, 45%; and ET-3, 14%. The normal ET-1 value in human plasma with this ELISA is reported as 0.6 pg/ml, with a range of 0.3 to 0.9 pg/ml. Our measurements in rat plasma were thus conducted within the performance specifications of the assay.
Data Analyses and Statistics
Data for lavage protein and cell numbers were computed as total yield per animal. Lavage fibronectin data were calculated as total yield (relative fluorescence units) per animal, and each value was then computed as percentage of the air control mean, matched by date of assay. Cell culture data (macrophage viability, phagocytosis, nitric oxide production, and cytokine secretion) were normalized to cellular protein and then computed as percentage of the air control mean, matched by date of assay. Plasma ET-1 data were computed as concentration (pg/ml). Absolute values for all endpoints are provided in relevant sections of Materials and Methods for reference. Data for the entire panel of endpoints were not available for every animal studied. To identify consistent patterns of effects, lung lavage endpoints (protein, fibronectin, neutrophil numbers, and macrophage numbers) and macrophage function endpoints (cell viability, phagocytosis, nitric oxide, TNF-α, MIP-2, and ET-1) were first analyzed, for those animals with complete data sets, by multivariate analysis of variance (MANOVA) for the groups of dependent variables indicated in parentheses. Factors used in the MANOVAs were EHC (0 and 40 mg/m 3 EHC-93), OZONE (0 and 0.8 ppm O3), and DAYS (1-day and 3-day). Treatment effects were observed for all variables except for macrophage viability. The entire database for each dependent variable was then analyzed by univariate multiway ANOVA with EHC (0 and 40 mg/m 3 EHC-93), OZONE (0 and 0.8 ppm O3), and DAYS (1-day and 3-day) as factors, followed by Tukey’s multiple comparison procedure (α = 0.05). The raw data for plasma ET-1 (pg/ml) were analyzed by a general linear model procedure with EHC (0 and 40 mg/m 3 EHC-93), OZONE (0 and 0.8 ppm O3), DAYS (1-day and 3-day), and DATE (dates of the ELISAs) as factors. The raw data for the normalized volume densities of septum and type II cells were analyzed by multiway repeated measures analysis of variance with EHC (0 and 48 mg/m 3 EHC-93), OZONE (0 and 0.8 ppm O3), and DISTANCE (0 to 500 and 500 to 1000 μm) as factors. Statistical software used was SAS (SAS Institute, Cary, NC) and SigmaStat (Jandel Co., San Rafael, CA).
Results
Histology
To minimize washing and displacement of cells and protein in the lumen of bronchioles and alveolar ducts, a series of lungs were collapsed and fixed by vascular perfusion. Lungs of air control rats were normal with only occasional intravascular neutrophils being seen where capillaries were not completely flushed by fixative (not shown). Histological features after a single exposure to the urban dust alone were unremarkable. Occasional neutrophils were observed in the central acinus, but there was no evidence of primary lung injury such as epithelial damage (Figure 1A) ▶ . Exposure of the animals to ozone for 4 hours resulted in a centriacinar injury, observed after a 20-hour recovery in clean air, with some edema and fibrin deposition in the alveolar duct lumen, as well as a limited intra-alveolar and interstitial infiltration by neutrophils (Figure 1B) ▶ . In contrast, simultaneous exposure to EHC-93 plus ozone produced profound histological changes in the central acinus. By qualitative examination, there was a mild amplification of the intra-alveolar inflammation but no clear increase of the extent of plasma protein transudation after exposure to particles in combination with ozone. However, the interstitial septal cellularity was markedly increased in animals exposed simultaneously to ozone and the urban dust, and many interstitial neutrophils were observed (Figure 1C) ▶ . Electron microscopy examination of the lungs of animals exposed to the dust plus ozone combination confirmed the epithelial injury and the neutrophilic infiltration to the lung interstitium (Figure 1D) ▶ .
Figure 1.

Lung histology assessed after a single, 4-hour inhalation exposure to the pollutants, followed by 20-hour recovery in clean air. The lungs were collapsed and fixed by vascular perfusion to minimize displacement of focal intra-alveolar changes. A: Alveolar duct region after exposure to EHC-93. A few neutrophils (arrows) can be seen in tissue closest to the duct, but the lung structure is otherwise normal. GMA section; toluidine blue; magnification, ×540. B: Alveolar duct region after ozone exposure. There is some increase in cellularity, and several neutrophils are found in the lung tissue. GMA section; toluidine blue; magnification, ×560. C: Alveolar duct region after combined dust plus ozone exposure. Septal cellularity is much increased, and a few neutrophils and macrophages are seen with edema fluid in the alveolar space. Several neutrophils are seen in the septum. GMA section; toluidine blue; magnification, ×560. D: Electron micrograph of alveolar duct wall after combined dust plus ozone exposure. Cell debris and edema are seen in the lumen at an area of epithelial cell injury. In the interstitium, neutrophils are observed. Magnification, ×5400. AD, alveolar duct; E, edema; EP, epithelial cell; P, neutrophil.
Morphometry
Morphometric measurements of septal changes were obtained from lungs prepared by intratracheal inflation with fixative. The thickening of septal edges in alveolar ducts (Figure 2A) ▶ and the alveolar type II cell hyperplasia/hypertrophy (Figure 2B) ▶ were quantitated for two anatomical regions assimilated to the proximal segment of alveolar ducts (0 to 500 μm), and the distal segments and lung parenchyma (500 to 1000 μm). Changes were measured in animals exposed to the pollutants for 4 hours, followed by 32 hours in clean air. The significant OZONE × EHC × DISTANCE factor interaction by repeated measures analysis of variance, for both septum thickness (P < 0.008) and type II cell thickness (P < 0.002), confirmed a synergistic toxicological interaction of ozone and particles in the central acinus, with no effect in the distal parenchyma. Inhalation of dust alone had no measurable effects on septal or type II cell thickness. The injury induced by ozone in the central acinus, observed histologically, did not translate into significant shifts of the morphometric estimates. However, inhalation of dust plus ozone resulted in a thickening of the septum and the type II cell compartment in the proximal path of the alveolar ducts but not distally (0 mg/m3 versus 48 mg/m 3 EHC-93 within 0.8 ppm O3 and 0 ppm versus 0.8 ppm O3 within 48 mg/m 3 EHC-93 in the 0- to 500-μm alveolar duct segment; Tukey, α = 0.05).
Figure 2.
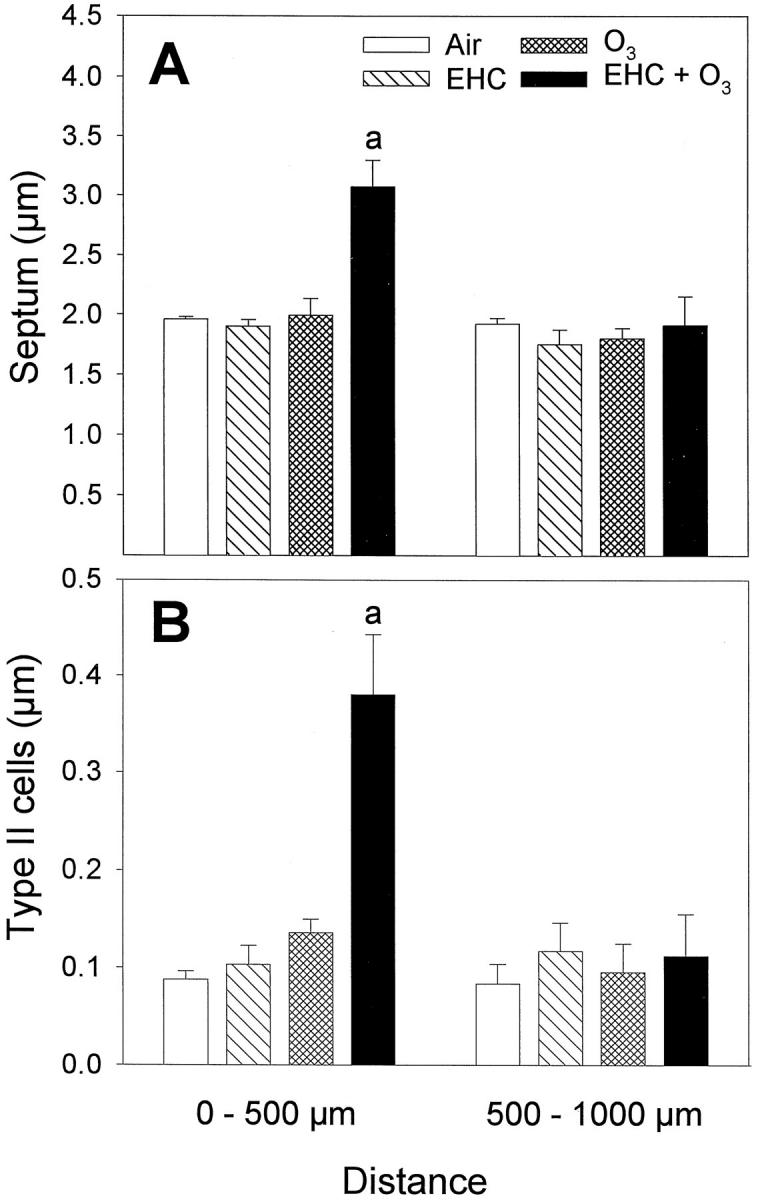
Lung morphometry after a single, 4-hour inhalation exposure to the pollutants, followed by 32-hour recovery in clean air. Lungs were fixed by intratracheal inflation. Results are shown as normalized volume density (volume to surface ratio, in microns) of septum and type II cells of septal edges in alveolar duct paths 0 to 500 μm (proximal) and 500 to 1000 μm (distal), relative to terminal bronchiole. Bars are mean ± SE; n = 6 animals. Repeated measures analyses of variance indicated significant EHC × OZONE × DISTANCE factor interactions for septum (P = 0.008) and type II cells (P = 0.002). By Student t-test, proximal overall was different from distal (α = 0.05). Two-way analyses of variance and multiple comparisons for the proximal (0 to 500 μm) measurements are summarized below. A: Septum thickness, excluding capillary lumen. aEHC × OZONE factor interaction, P < 0.001. Tukey, in the 0- to 500-μm alveolar duct segment, 0 mg/m3 versus 48 mg/m 3 EHC-93 within 0.8 ppm O3 and 0 ppm versus 0.8 ppm O3 within 48 mg/m 3 EHC-93 (α = 0.05). B: Type II alveolar epithelial cell hyperplasia/hypertrophy. aEHC × OZONE factor interaction, P < 0.001. Tukey, in the 0- to 500-μm alveolar duct segment, 0 mg/m3versus 48 mg/m 3 EHC-93 within 0.8 ppm O3 and 0 ppm versus 0.8 ppm O3 within 48 mg/m 3 EHC-93 (α = 0.05).
Lung Lavage Cytology and Biochemistry
Exposure of the animals to ozone for 4 hours followed by 20 hours of recovery in air resulted in higher recoveries of protein (Figure 3A) ▶ , fibronectin (Figure 3B) ▶ , and neutrophils (Figure 3C) ▶ in lavage fluids for both the 1-day and 3-day exposure groups (factor OZONE, P < 0.001). In contrast, these endpoints were not increased after inhalation of the urban dust EHC-93. Furthermore, co-exposure to EHC-93 plus ozone did not amplify those biochemical and cytological changes measured in lung lavage fluids after exposure to ozone alone. Lung lavage observations were generally consistent with the intra-alveolar changes examined histologically in the collapsed lungs (Figure 1A ▶ -D). It should be noted that the suggestion of lower lavage protein values in the ozone-plus-dust exposure group, when compared numerically to ozone alone, was not statistically significant (EHC × OZONE factor interaction, P > 0.05) in the multiway ANOVA (Figure 3A) ▶ . Macrophages remained the dominant cell population recovered by bronchoalveolar lavage of pollutant-exposed animals (Figure 3D) ▶ . Cellular pellets obtained from lung lavage of dust-exposed animals were gray in appearance, confirming delivery of the dust in the respiratory tract. There was no significant shift of the cell size distribution between different exposure groups (data not shown). Recovery of macrophages was generally higher for the 3-day-exposure animals, but the exact reason for this is unknown (factor DAYS, P < 0.001). Inhalation of EHC-93 or ozone reduced slightly the yield of macrophages across days of exposures but with equivocal effect of the pollutant combination (EHC × OZONE, P < 0.001; 0 mg/m3 versus 40 mg/m 3 EHC-93 within 0 ppm O3 and within 0.8 ppm O3; Tukey, α = 0.05).
Figure 3.

Assessment of lung injury by bronchoalveolar lavage after inhalation of urban dust and ozone for 4 hours, followed by 20-hour recovery in clean air. A: Soluble proteins were analyzed by Coomassie blue dye-binding assay. aOZONE main effect, P < 0.001. B: Fibronectin was measured by Western blotting and imaging densitometry and shown as a percentage of control. aOZONE main effect, P < 0.001. C: Number of neutrophils. aOZONE main effect, P < 0.001. D: Number of macrophages. aEHC × OZONE factor interaction, P < 0.001. Tukey, 0 mg/m3versus 40 mg/m 3 EHC-93 within 0 ppm O3 and within 0.8 ppm O3 (α = 0.05). The significant DAYS main effect, P < 0.001, indicated overall higher recoveries of macrophages for the 3-day versus 1-day exposure groups (not associated with treatments). All results are expressed as mean ± SE; n = 8 to 12 animals.
Macrophage Physiology
Viability of lavage cells, measured by reduction of alamarBlue, was not affected after the inhalation treatments (Figure 4A) ▶ . The phagocytic activity of macrophages was depressed after exposure to ozone (factor OZONE, P = 0.004); the effect of particles was not significant (Figure 4B) ▶ . Although exposure to the EHC-93 particles did not modify lung lavage cytology, a number of cellular functions were affected after the inhalation treatments. The LPS-induced production of nitric oxide was reduced in cells isolated from animals exposed either to particles or ozone although not significantly affected by the pollutant combination (Figure 5A ▶ ; factor interaction EHC × OZONE, P < 0.001; 0 ppm versus 0.8 ppm O3 within 0 mg/m 3 EHC-93 and 0 mg/m3 versus 40 mg/m 3 within 0 ppm O3; Tukey, α = 0.05). Secretion of TNF-α followed a complex pattern, with apparent increase for 1-day exposure and decrease after 3-day exposure (pollutant effects not significant); the time effect was significant across treatments (Figure 5B ▶ ; factor DAYS, P < 0.001). Secretion of MIP-2 was increased in macrophages from animals exposed to EHC-93, with no visible effect of ozone (Figure 5C ▶ ; factor EHC, P = 0.035). Secretion of ET-1 was elevated after inhalation of ozone; ET-1 was clearly elevated for the pollutant combination, but the low response to ozone alone for 1-day exposure did not translate into a significant factor interaction by multiway ANOVA (Figure 5D ▶ ; factor OZONE, P < 0.001).
Figure 4.

Integrity of macrophages after inhalation exposure of animals to particles and ozone. A: Viability was measured by reduction of alamarBlue and shown as a percentage of control. Results are expressed as mean ± SE; n = 12 animals. There were no effects of treatments. B: Phagocytosis of fluorescein-labeled E. coli by freshly isolated macrophages (2-hour incubation), shown as a percentage of control. Results are expressed as mean ± SE; n = 12 to 18 animals. aOZONE main effect, P < 0.004.
Figure 5.

Functional assays of macrophages after inhalation exposure of animals to particles and ozone. All data are presented as percentage of control. A: Nitric oxide production as determined by the assay of nitrite in 24-hour culture supernatants of macrophages stimulated with LPS. Results are expressed as mean ± SE; n = 12 to 18 animals. aEHC × OZONE factor interaction, P < 0.001. Tukey, 0 ppm versus 0.8 ppm O3 within 0 mg/m 3 EHC-93 and 0 mg/m3 versus 40 mg/m 3 EHC-93 within 0 ppm O3 (α = 0.05). B: TNF-α measured in 24-hour culture supernatants by ELISA. Results are expressed as mean ± SE; n = 5 to 12 animals. The DAYS main effect, P < 0.001, indicated overall higher amounts secreted for 1-day versus 3-day exposure groups. The data suggest a temporal pattern in the TNF-α response. C: MIP-2 was measured in 24-hour culture supernatants by ELISA. Results are expressed as mean ± SE; n = 9 to 10 animals. aEHC main effect, P = 0.035. D: ET-1 was measured in 24-hour cell culture supernatants by ELISA. Results are expressed as mean ± SE; n = 6 to 10 animals. aOZONE main effect, P < 0.001.
Plasma ET-1
Plasma levels of ET-1 were systematically increased after 20 hours in those rats exposed for 4 hours to particles alone or to particles in combination with ozone (Figure 6 ▶ ; factor EHC, P = 0.018). Inhalation of ozone did not amplify or reduce the effect of particle exposure on plasma ET-1. A small but perceptible baseline shift of plasma ET-1 was observed through the period of experimentation (11 months), which we have attributed to possible procedure variability related to plasma sample processing and ELISA performance. As a technical note, the use of EDTA as anticoagulant is preferable to heparin. Also, immediate processing of the plasma samples (acid/acetone precipitation of protein and recovery of ET-1 by evaporation of the supernatant) as opposed to storage of frozen plasma improves detection of ET-1 in the ELISA. A statistical interaction was observed between ozone exposure and the date of analysis of the samples (factor interaction OZONE × DATE, P = 0.04). We have assessed whether the drift of baseline was biasing the interpretation of the biological effects of particles. Data were retested without two outlier values (low value in ozone alone and high value in dust alone), and significant simple effects remained for the factors EHC and DATE, but the two-way factor interaction was removed. We have concluded that the baseline drift did not confound the interpretation of the EHC-93 effect.
Figure 6.

Alteration of levels of immunoreactive ET-1 in plasma after inhalation of urban dust and ozone for 4 hours, followed by 20-hour recovery in clean air. Results are presented as percentage of air control and expressed as mean ± SE; n = 9 to 15 animals. aEHC main effect, P = 0.018 (multiway ANOVA performed on raw data, pg of ET-1/ml of plasma).
Discussion
The observations of histological changes after exposure of animals to the individual pollutants EHC-93 or ozone were generally substantiated by the results of analyses in lung lavage fluids. The fact that rats exposed to the urban dust EHC-93 alone did not exhibit noticeable histological, cytological, protein, or fibronectin changes indicated a relative innocuity of the dust in terms of acute structural lung injury. Surprisingly, the urban dust in the EHC-93 plus ozone animals did not amplify those biochemical and cytological changes in lung lavage fluids that were measured for an exposure to ozone alone. In fact, data suggested that the particles might have lowered the plasma protein transudation, although this effect was not statistically significant. It is not clear whether particles in the EHC-93-plus-ozone treatment group could have increased coagulation factors or inhibited fibrinolysis in the alveolar lumen or the septa, which would have masked an ozone-induced or even a particle-specific increase of alveolar wall permeability. Nevertheless, inhalation exposure to the urban dust in combination with ozone did clearly potentiate the increase in septal cellularity, as determined histologically. Morphometric data indicated that particles in the EHC-93-plus-ozone animals affected lung structure essentially in the central acinus, a target site of ozone, and that there was no distal displacement of the ozone-induced injury. The results are in line with our previous cytokinetic measurements and extend the concept of a direct impact of complex particles on the progression of a primary lesion. 5
The analyses of cytokines produced by lung lavage cells suggest that inhalation of the particles could have amplified septal changes through modulation of paracrine mechanisms. Inhalation of the urban dust alone was associated with elevated secretion by isolated macrophages in culture of MIP-2, a chemokine to neutrophils, 6,7 but this effect was not accompanied in vivo by substantial influx of neutrophils in the alveoli as assessed by lung lavage technique. Elevation of MIP-2 may not have been sufficient by itself to attract significant amounts of neutrophils in the alveolar lumen if the integrity of the alveolar epithelium in animals exposed to the dust alone limited penetration of the chemokine. Macrophages isolated from animals exposed to EHC-93 plus ozone produced larger amounts of both MIP-2 and ET-1, by comparison with cells from air control rats. ET-1 is chemotactic to monocytes and activates neutrophils 13,24 and is mitogenic to fibroblasts. 25 The intensity of the inflammatory changes observed in lavage fluids need not correlate, in a dose-dependent fashion, with the intensity of the individual cytokine responses determined in the cell cultures. The interaction between the mix of cytokines could be more relevant than the absolute levels of a given cytokine. Also, because the epithelial lining was modified in animals exposed to ozone plus urban dust, it is conceivable that a number of mediators produced by macrophages in the alveolar lumen could have penetrated the interstitium, enhancing the septal changes either by recruitment of neutrophils or via mitogenic stimulation of monocytes, fibroblasts, and type II cells. Particles reaching the interstitium through the damaged epithelium may have also induced local cytokine production. Secretion of immunoreactive TNF-α by cultures of lung lavage cells was not significantly affected by the pollutants, although it should be kept in mind that the assay was insensitive to intracellular or membrane-bound TNF-α.
The impairment of macrophage functions and lung defense by inhaled pollutants is a classical paradigm of pulmonary toxicology. 10,11 Exposure of the animals to ozone reduced the phagocytic activity of lung lavage cells, in line with previous reports. 26,27 It has been shown, more than a decade ago, that intratracheal injection of urban particulate matter can promote bacterial infectivity in the lungs of mice. 12 In the present study, both ozone and EHC-93, when inhaled alone, resulted in a decrease of the LPS-stimulated production of nitric oxide, a bactericidal agent produced by macrophages, 28,29 but inhibition of nitric oxide was lifted in the co-exposure group. We recognize that overnight culture of lung lavage cells could have complicated the measured patterns of cell responses. For one, cells isolated from all four treatment groups presented with different combinations of cytokine responses, but it is not clear how cell physiology evolved during an overnight incubation. Also, the cell populations in culture were mixed with reference to the anatomical site from which they were actually recovered, eg, airways versus parenchyma and proximal versus distal, and eventual site-specific effects and responses were most probably diluted by subsequent mixing of the whole macrophage population. Consequently, in the EHC-93-plus-ozone treatment group, suppression of nitric oxide in macrophages of the distal lung might have still been biologically relevant, but complex cell changes initiated in the central acinus, and carried over in the cell cultures, could have masked the effect. Notwithstanding these difficulties in the interpretation of cytokine and nitric oxide data, the observations do reveal that exposure to particles will modulate a number of macrophage functions, notably lung defense-related functions and the production of inflammatory and mitogenic cytokines. Lung lavage techniques and cytology may not be fully sensitive to cellular responses associated with a limited focal injury. This should justify the application of immunocytochemical and in situ hybridization procedures to clarify the site-specific and cell-specific responses.
In our study, increase of the plasma levels of ET-1 was strictly associated with inhalation of the urban dust. The magnitude of this effect was not modified significantly in animals exposed simultaneously to ozone, indicating that structural lung injury was not a prerequisite for the particle effect on plasma ET-1. Furthermore, we did not observe a difference between repeated exposure versus single exposure to the dust. This pattern suggests a role of bioavailable agents leached from particles, and with relatively rapid distribution and clearance, in the induction of the plasma ET-1 shifts. Inhalation of cigarette smoke has been reported to increase plasma endothelin levels. 30 Cigarette smoke contains chemical agents that are also present in EHC-93, such as organic species and metals, 9 but the specific agents responsible for the effect of urban dust remain to be identified. The lungs are a primary site for the filtration of circulating ET-1, 31,32 and it is conceivable that a diffuse effect of soluble components of particles on the capillary bed of the lung parenchyma could have affected the capture and/or turnover of circulating ET-1. Alternatively, the pulmonary endothelial cells could have been induced to release ET-1 in the circulation, but evidence at this time is essentially phenomenological.
The mechanisms and the specific agent(s) responsible for the effect of inhaled urban particulate matter on circulating ET-1, the possible responses of other vasoconstrictors and vasodilators, as well as the dynamics and the physiological correlates of such effects, all remain to be defined. In particular, the levels of circulating ET-1 measured after a 20-hour recovery of the animals in clean air possibly underestimate the levels reaching the heart at the onset of the effect. Exposure of healthy rats to the EHC-93 dust in our study produced a shift of plasma ET-1 on the order of 15% after 20 hours, but the magnitude of plasma ET-1 changes need not be large to be physiologically relevant. Ideally, the physiological significance of such changes should be determined in a cardiovascular-compromised animal model. Despite overlapping ranges of plasma ET-1 between healthy and sick human individuals, a 25% increase in plasma ET-1 has high predictive value for chronic heart failure, 33 and a decrease of ET-1 on the order of 20% in chronic heart failure patients was associated with improvement of symptoms, leading investigators to postulate a direct role of ET-1 in the pathophysiology. 34 Plasma ET-1 is also a predictor of 1-year mortality after acute myocardial infarct, suggesting that an acute increase of ET-1 may be cardiotoxic by promoting infarct extension and arrhythmia. 35 The nuanced response of plasma ET-1 in our animal model, that is, an increase after acute inhalation exposure to the urban dust EHC-93 but not after ozone, is interesting in view of the fact that particulate matter, but not ozone, has been associated epidemiologically with acute cardiac hospitalization. 4 In short, observation of changes in circulating ET-1 after acute inhalation exposure to urban particulate matter, without noticeable lung lesions, offers a new lead for experimental investigation of the biological bases for cardiac health impacts of air pollutants detected epidemiologically.
The biological effects of EHC-93 should be relevant to the general issue of the toxicity of ambient air particulate matter. The cytotoxicity and the potency of EHC-93 toward a number of stress genes in cell culture models were previously found to be within the ranges of those of a number of field particulate samples. 9 The present data confirm and extend our previous communication indicating that a high potency of inhaled environmental particles for direct lung structural lesions is not an absolute prerequisite for toxicity. 5 Furthermore, the data identify a new potential mechanism of systemic effects of particles. In conclusion, the potential pathophysiological implications of the acute biological effects described in our report are 1) interference of inhaled urban particulate matter and ozone with lung defense mechanisms, 2) adverse interaction of the particles in primary lung lesions, and 3) impact of particle inhalation on systemic levels of vasoactive agent(s) by pathways that do not require structural lung injury. In a broad sense, acute biological effects of respirable particulate matter from ambient air appear related in part to modulation of paracrine/endocrine mechanisms. We submit as a plausible working hypothesis that such effects, in the context of individuals with precarious homeostasis, eg, afflicted with respiratory or cardiovascular disorders, could precipitate clinical manifestations of the diseases, which in turn could translate into the health metrics of population studies, such as increased daily rates of hospital admissions 1,3,4 and, hence, premature death. 2
Acknowledgments
We are grateful to Scott Alic and Kendra Stetler for technical assistance with the ELISAs and to our colleague Dr. Richard Burnett for generous and stimulating discussions.
Footnotes
Address reprint requests to Dr. Renaud Vincent, Room 332, Environmental Health Centre, 0803C Tunney’s Pasture, Ottawa, Ontario, Canada K1A 0L2. E-mail: Renaud_Vincent@hc-sc.gc.ca.
Supported by Health Canada. Dr. Léo Bouthillier was a Fellow in Canadian Government Laboratories of the Natural Sciences and Engineering Research Council of Canada.
References
- 1.Bates DV, Sizto R: Relationships between air pollution levels and hospital admissions in Southern Ontario. Can J Public Health 1983, 74:117-122 [PubMed] [Google Scholar]
- 2.Dockery DW, Pope CA, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Speizer FE: An association between air pollution and mortality in six U.S. cities. N Engl J Med 1993, 329:1753-1759 [DOI] [PubMed] [Google Scholar]
- 3.Burnett RT, Dales RE, Raizenne ME, Krewski D, Summers PW, Roberts GR, Raad-Young M, Dann T, Brook J: Effects of low ambient levels of ozone and sulfates on the frequency of respiratory admissions to Ontario hospitals. Environ Res 1994, 65:172-194 [DOI] [PubMed] [Google Scholar]
- 4.Burnett RT, Dales RE, Krewski D, Vincent R, Dann T, Brook JR: Associations between ambient particulate sulfate and admissions to Ontario hospitals for cardiac and respiratory diseases. Am J Epidemiol 1995, 142:15-22 [DOI] [PubMed] [Google Scholar]
- 5.Vincent R, Bjarnason SG, Adamson IYR, Hedgecock C, Guénette J, Potvin M, Kumarathasan P, Goegan P, Bouthillier L: Acute pulmonary toxicity of urban particulate matter and ozone. Am J Pathol 1997, 151:1563-1570 [PMC free article] [PubMed] [Google Scholar]
- 6.Driscoll KE, Hassenbein DG, Carter J, Poynter J, Asquith TN, Grant RA, Whitten J, Purdon MP, Takigiku R: Macrophage inflammatory proteins 1 and 2: expression by rat alveolar macrophages, fibroblasts, and epithelial cells and in rat lung after mineral dust exposure. Am J Respir Cell Mol Biol 1993, 8:311-318 [DOI] [PubMed] [Google Scholar]
- 7.Driscoll KE, Maurer JK, Higgins J, Poynter J: Alveolar macrophage cytokine and growth factor production in a rat model of crocidolite-induced pulmonary inflammation and fibrosis. J Toxicol Environ Health 1995, 46:155-169 [DOI] [PubMed] [Google Scholar]
- 8.Nadeau D, Vincent R, Kumarathasan P, Brook J, Dufresne A: Cytotoxicity of ambient air particles to rat lung macrophages: comparison of cellular and functional assays. Toxicol In Vitro 1996, 10:161-172 [DOI] [PubMed] [Google Scholar]
- 9.Vincent R, Goegan P, Johnson G, Brook JR, Kumarathasan P, Bouthillier L, Burnett RT: Regulation of promoter-CAT stress genes in HepG2 cells by suspensions of particles from ambient air. Fund Appl Toxicol 1997, 39:18-32 [DOI] [PubMed] [Google Scholar]
- 10.Gardner DE: Alterations in macrophage functions by environmental chemicals. Environ Health Perspect 1984, 55:343-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlesinger RB: Comparative toxicity of ambient air pollutants: some aspects related to lung defense. Environ Health Perspect 1989, 81:123-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatch GE, Boykin E, Graham JA, Lewtas J, Pott F, Mumford JL, Loud K: Inhalable particles and pulmonary host defense: in vivo and in vitro effects of ambient air and combustion particles. Environ Res 1985, 36:67-80 [DOI] [PubMed] [Google Scholar]
- 13.Michael JR, Markewitz BA: Endothelins and the lung. Am J Respir Crit Care Med 1996, 154:555-581 [DOI] [PubMed] [Google Scholar]
- 14.Battistini BP, D’OrlJans-Juste P, Sirois P: Endothelins: circulation plasma levels and presence in other biologic fluids. Lab Invest 1993, 68:600-628 [PubMed] [Google Scholar]
- 15.Tsutamoto T, Wada A, Maeda Y, Adachi T, Kinoshita M: Relation between endothelin-1 spillover in the lungs and pulmonary vascular resistance in patients with chronic heart failure. J Am Coll Cardiol 1994, 23:1427-1433 [DOI] [PubMed] [Google Scholar]
- 16.Yoshibayashi M, Nishioka K, Nakao K, Saito Y, Matsumura M, Ueda T, Temma S, Shirakami G, Imura H, Mikawa H: Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Circulation 1991, 84:2280-2285 [DOI] [PubMed] [Google Scholar]
- 17.Teerlink JR, Löffler B-M, Hess P, Maire J-P, Clozel M, Clozel J-P: Role of endothelin in the maintenance of blood pressure in conscious rats with chronic heart failure. Circulation 1994, 90:2510-2518 [DOI] [PubMed] [Google Scholar]
- 18.Ruschitzka F, Schrader J, Luders S, Schulz E, Gronau C, Talartschik J, Eisenhauer T, Verwiebe R, Warneke G, Scheler F: Effects of endothelin on coagulation, prostaglandins and hemodynamics. Contrib Nephrol 1993, 101:30-36 [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto C, Kaji T, Sakamoto M, Koizumi F: Effect of endothelin on the release of tissue plasminogen activator and plasminogen activator inhibitor-1 from cultured human endothelial cells and interaction with thrombin. Thromb Res 1995, 67:619-624 [DOI] [PubMed] [Google Scholar]
- 20.Last JA, Pinkerton KE: Chronic exposure of rats to ozone and sulfuric acid aerosol: biochemical and structural responses. Toxicology 1997, 116:133-146 [DOI] [PubMed] [Google Scholar]
- 21.Vincent R, Vu D, Hatch G, Poon R, Dreher K, Guénette J, Bjarnason S, Potvin M, Norwood J, McMullen E: Sensitivity of lungs of aging Fischer 344 rats to ozone: assessment by bronchoalveolar lavage. Am J Physiol (Lung Cell Mol Physiol 15) 1996, 271:L555-L565 [DOI] [PubMed] [Google Scholar]
- 22.Goegan P, Johnson G, Vincent R: Effects of serum protein and colloid on the alamarBlue assay in cell cultures. Toxicol In Vitro 1995, 9:257-266 [DOI] [PubMed] [Google Scholar]
- 23.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR: Analysis of nitrate, nitrite and [15N]nitrate in biological fluids. Anal Biochem 1982, 126:131-138 [DOI] [PubMed] [Google Scholar]
- 24.Hafström I, Ringertz B, Lundeberg T, Palmblad J: The effect of endothelin, neuropeptide Y, calcitonin gene-related peptide and substance P on neutrophil functions. Acta Physiol Scand 1993, 148:341-346 [DOI] [PubMed] [Google Scholar]
- 25.Cambrey AD, Harrison NK, Dawes KE, Southcott AM, Black CM, du Bois RM, Laurent GJ, McAnulty RJ: Increased levels of endothelin-1 in bronchoalveolar lavage fluid from patients with systemic sclerosis contribute to fibroblast mitogenic activity in vitro. Am J Respir Cell Mol Biol 1994, 11:439-445 [DOI] [PubMed] [Google Scholar]
- 26.Oosting RS, VanGolde LMG, Verhoef J, Van Bree L: Species differences in impairment and recovery of alveolar macrophage function following single or repeated ozone exposures. Toxicol Appl Pharmacol 1991, 110:170-178 [DOI] [PubMed] [Google Scholar]
- 27.Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers D, Koren HS: Exposure of humans to ambient levels of ozone for 6.6 hours causes cellular and biochemical changes in the lung. Am J Respir Cell Mol Biol 1991, 4:72-81 [DOI] [PubMed] [Google Scholar]
- 28.Granger DL, Hibbs JB, Perfect JR, Durack DT: Specific amino acid (l-arginine) requirement for the microbiostatic activity of murine macrophages. J Clin Invest 1988, 81:1129-1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovchik JA, Lyons CR, Lipscomb MF: A role for γ interferon-induced nitric oxide in pulmonary clearance of Cryptococcus neoformans. Am J Respir Cell Mol Biol 1995, 13:116-124 [DOI] [PubMed] [Google Scholar]
- 30.Haak T, Jungmann E, Raab C, Usadel KH: Elevated endothelin-1 levels after cigarette smoking. Metabolism 1994, 43:267-269 [DOI] [PubMed] [Google Scholar]
- 31.Sirviö ML, Metsärinne, Saijonmaa O, Fyhrquist F: Tissue distribution and half-life of 125I-endothelin in the rat: importance of pulmonary clearance. Biochem Biophys Res Commun 1990, 167:1191-1195 [DOI] [PubMed] [Google Scholar]
- 32.Stewart DJ, Levy RD, Cernacek P, Langleben D: Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med 1991, 114:464-469 [DOI] [PubMed] [Google Scholar]
- 33.Galatius-Jensen S, Wroblewski H, Emmeluth C, Bie P, Haunsø S, Kastrup J: Plasma endothelin-1 in chronic heart failure: a predictor of cardiac death? Circulation 1994, 90:I-379 [Google Scholar]
- 34.Tsutamoto T, Hisanaga T, Fukai D, Wada A, Maeda Y, Maeda K, Kinoshita M: Prognostic value of plasma soluble intercellular adhesion molecule-1 and endothelin-1 concentration in patients with chronic congestive heart failure. Am J Cardiol 1995, 76:803-808 [DOI] [PubMed] [Google Scholar]
- 35.Omland T, Lie RT, Aakaad A, Aarsland T, Dickstein K: Plasma endothelin determination as a prognostic indicator of 1 year mortality after acute myocardial infarction. Circulation 1994, 89:1573-1579 [DOI] [PubMed] [Google Scholar]