

Abstract
Although numerous epidemiological studies have shown that inorganic arsenicals cause skin cancers and hyperkeratoses in humans, there are currently no established mechanisms for their action or animal models. Previous studies in our laboratory using primary human keratinocyte cultures demonstrated that micromolar concentrations of inorganic arsenite increased cell proliferation via the production of keratinocyte-derived growth factors. As recent reports demonstrate that overexpression of keratinocyte-derived growth factors, such as transforming growth factor (TGF)-α, promote the formation of skin tumors, we hypothesized that similar events may be responsible for those associated with arsenic skin diseases. Thus, the influence of arsenic in humans with arsenic skin disease and on mouse skin tumor development in transgenic mice was studied. After low-dose application of tetradecanoyl phorbol acetate (TPA), a marked increase in the number of skin papillomas occurred in Tg.AC mice, which carry the v-Ha-ras oncogene, that received arsenic in the drinking water as compared with control drinking water, whereas no papillomas developed in arsenic-treated transgenic mice that did not receive TPA or arsenic/TPA-treated wild-type FVB/N mice. Consistent with earlier in vitro findings, increases in granulocyte/macrophage colony-stimulating factor (GM-CSF) and TGF-α mRNA transcripts were found in the epidermis at clinically normal sites within 10 weeks after arsenic treatment. Immunohistochemical staining localized TGF-α overexpression to the hair follicles. Injection of neutralizing antibodies to GM-CSF after TPA application reduced the number of papillomas in Tg.AC mice. Analysis of gene expression in samples of skin lesions obtained from humans chronically exposed to arsenic via their drinking water also showed similar alterations in growth factor expression. Although confirmation will be required in nontransgenic mice, these results suggest that arsenic enhances development of skin neoplasias via the chronic stimulation of keratinocyte-derived growth factors and may be a rare example of a chemical carcinogen that acts as a co-promoter.
Arsenic, a ubiquitous element, represents a human health concern when concentrated in the environment from natural or anthropogenic processes. Arsenic contamination of water supplies has resulted in a very high incidence of skin lesions and cancers in exposed populations from Taiwan, China, Eastern Europe, India, Southwestern United States, and Cental and South America. The U.S. Environmental Protection Agency (EPA) estimates that over 350,000 people in the U.S. consume drinking water containing over 50 μg/L arsenic, the current EPA standard, 1 and there is significant regulatory pressure to lower the acceptable levels. Chronic exposure to inorganic arsenic in drinking water is most often associated with increased mortality from skin cancer, but recent studies have also linked arsenic exposure to neoplasias in internal organs, including the lung, liver, bladder, kidney, and prostate. 1-4 Arsenic-induced skin cancers occur in sun-exposed as well as unexposed areas and include intraepidermal carcinomas (commonly referred to as Bowen’s disease), basal cell carcinomas, squamous cell carcinomas, or combined lesions. 5-7 Additional manifestations of chronic arsenic dermatotoxicity include hyperpigmentation and hyperkeratosis. 8
Although several hypotheses have been proposed for the mechanism of arsenic-induced carcinogenesis, it remains unknown, with limited evidence available for either genetic or epigenetic mechanisms. Arsenic neither promotes neoplastic disease in classical single- or two-stage murine models 9-11 nor is mutagenic. 12 Support exists that increased cell proliferation is a central event, as treatment of human keratinocytes with arsenic induces ornithine decarboxylase activity 13 and growth factor expression 14 as well as alters the DNA-binding activities of the AP-1 and AP-2 transcription factors, which are involved in proliferative events. 15,16 Furthermore, treatment of in vitro skin models with arsenic induces keratins associated with proliferation and acanthosis. 17 At the genetic level, arsenic is co-mutagenic with ultraviolet radiation, x-rays, or alkylating agents, induces sister chromatid exchange in lymphocytes, 18 and causes gene amplification in mouse 3T3 cells. 19 Genetic studies have focused recently on alterations in methylation patterns by arsenic, suggesting that DNA hyper- or hypomethylation may lead to altered gene expression, particularly in the p53 tumor suppressor gene. 20,21 In this respect, authors from the present study have shown that high levels of immunoreactive p53 are expressed in Bowen’s disease skin biopsies. 6
Previously, observations that addition of low concentrations of arsenic to human keratinocyte cultures resulted in the overexpression of several cytokines and growth factors, 14 including transforming growth factor (TGF)-α and granulocyte/macrophage colony-stimulating factor (GM-CSF), suggested their involvement in arsenic-mediated skin diseases. Although secretion of these products from keratinocytes are critical to maintaining homeostasis and barrier integrity in the skin, 22 their overexpression can lead to various pathological processes, such as allergic contact dermatitis, irritant contact dermatitis, psoriasis, and neoplasia. In particular, overexpression of TGF-α, and to a lesser extent GM-CSF, has been associated with neoplastic transformation in the skin, 23,24 and keratinocytes transfected with a constitutive TGF-α transgene develop benign skin papillomas when grafted to nude mice. 25 Injection of TGF-α into initiated mouse skin induces DNA synthesis in epidermal cells, an activity analogous to early tumor formation, 26 and targeted overexpression of TGF-α to the epidermis elicits hyperplasia, hyperkeratosis, and spontaneous squamous cell carcinomas. 27,28 Recent studies have demonstrated that TGF-α can synergize with c-myc to accelerate spontaneous and chemical-induced neoplastic development. 29-31 Furthermore, TGF-α transgenic mice exhibit keratinocyte hyperproliferation and tumors in the pancreas, liver, and mammary epithelia, 32-34 suggesting that TGF-α overexpression has the unique ability to complement both initiation and promotion by serving as a tumor enhancer or co-promoter. However, TGF-α may not be a strict requirement for papilloma formation as TGF-α null mice may develop papillomas at the same incidence and multiplicity as that induced in the wild type. 35
Based on the aforementioned observations and our previous in vitro studies using primary human keratinocytes, we hypothesized that arsenic induces tumorigenesis by modulation of keratinocyte-derived growth-promoting cytokines leading to enhanced cell proliferation. To address this question, a model was developed using the transgenic mouse line Tg.AC, which has been used previously to evaluate the carcinogenic potential of a number of topically administered chemicals. 36 Additional studies were conducted to examine gene expression in skin samples from individuals chronically exposed to high levels of arsenic in their drinking water.
Materials and Methods
Animal Treatment
Female, homozygous Tg.AC mice containing the fetal beta-globin promoter fused to the v-Ha-ras structural gene (with mutations at codons 12 and 59) and linked to a simian virus 40 polyadenylylation/splice sequence 37 and nontransgenic FVB/N mice were obtained from Taconic Farms (Germantown, NY). Mice were maintained in our animal facility in compliance with approved guidelines for the humane treatment of laboratory animals and were fed Purina Pico Chow 5058 and water ad libitum. Groups of 8-week-old, age- and sex-matched wild-type FVB/N or Tg.AC transgenic mice were provided 0.02% arsenic as sodium arsenite (Sigma Chemical Co., St. Louis, MO) in their drinking water. Four weeks later, the dorsal skin was shaved with electric clippers, and 72 hours later the first of four doses (twice a week for 2 weeks) of 12-O-tetradecanoyl phorbol-13-acetate (TPA; Sigma Chemical Co.) was applied topically in 200 μl of acetone. Animals were shaved thereafter during TPA dosing as needed. Groups of control and arsenic-treated transgenic mice were administered intravenous injections of 50 μg of either monoclonal antibodies to mouse GM-CSF (Genzyme, Cambridge, MA) 2 weeks before TPA treatment, 2 hours before application of TPA, and 2 weeks after TPA treatment or polyclonal antibodies to mouse TGF-α (Peninsula Laboratories, Belmont, CA) 2 hours before application of TPA. Papilloma incidents were recorded three times per week for 20 weeks. For cell proliferation studies, animals were injected with 50 mg/kg bromodeoxyuridine (BrdU) 30 minutes before sacrifice. Animals were euthanized using CO2 narcosis, and skin samples were collected.
Human Skin Samples
Human skin samples were obtained from volunteers residing in the Pa Chang Valley on the Southwest Coast of Taiwan where arsenic concentrations in the well water reached levels as high as 1.82 ppm. 38 Biopsies (3 mm2) were aseptically obtained using a punch. For each individual, samples were obtained from nonlesioned areas and areas displaying evidence of arsenic-induced hyperpigmentation and hyperkeratosis. The nonlesioned skin samples served as controls. The samples were shipped in dry ice and stored at −70°C before RNA isolation.
RNA Extraction and Reverse Transcription Polymerase Chain Reaction
Human and mouse skin samples (shaved dorsal area) were homogenized in a small volume of RNAzol B solution (Biotecx Laboratories, Houston, TX), and total cellular RNA was extracted according to the manufacturer’s instructions. For analysis of cytokine/growth factor gene expression, human and mouse RNA from human skin was further processed with poly A to isolate mRNA. 14 Synthesis of cDNA was performed as previously described 14 using 1 to 3 μg of total or mRNA from each sample. Commercially available polymerase chain reaction (PCR) primers for human GM-CSF, tumor necrosis factor (TNF)-α, TGF-α, and glyceraldehyde-3-phosphate dehydrogenase (G3PDH) and mouse GM-CSF, c-myc, and G3PDH were purchased from Clontech Laboratories (Palo Alto, CA). Commercially available PCR primers for mouse TNF-α and epidermal growth factor receptor (EGFR) were purchased from Stratagene (La Jolla, CA). The sequence for the mouse TGF-α primer, obtained from GenBank and synthesized by Bioserve Biotechnologies (Gaithersburg, MD), contained the following sequence: 5′ GGACAGCTCGCTCTGCTAGCG 3′ and 5′ CTTCTCGTGTCTGCAGACGAG 3′ (amplified PCR fragment, 410 bp).
Five-microliter aliquots of the synthesized cDNA (corresponding to 1 to 3 μg of RNA) were added to 45 μl of PCR mix containing 5 μl of 10X PCR buffer, 1 μl of deoxynucleotides (1 mmol/L each), 0.5 μl of sense and antisense primers (0.15 μmol/L), and 0.25 μl of DNA polymerase (GeneAmp PCR, Perkin Elmer Cetus, Norwalk, CT). The reaction mixture was covered with an Ampli(Gem) wax tablet (Perkin Elmer Cetus). Amplification was initiated by 1 minute of denaturation at 94°C for 1 cycle followed by 25, 30, or 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 55°C for 1 minute using a GeneAmp PCR System 9600 DNA Thermal Cycler (Perkin Elmer Cetus). After the last cycle of amplification, the samples were incubated for 7 minutes at 72°C. RNA concentrations and PCR cycles were titrated to establish standard curves, to document linearity, and to permit semiquantitative analysis of signal strength as previously described. 14 For each set of primers, dilutions of cDNA were amplified for 20, 23, 25, 28, 30, 33, and 35 cycles, and PCR conditions were optimized with a commercially available kit (Stratagene) to define optimal conditions. When appropriate, the specificity of the PCR bands was confirmed by restriction enzyme analysis of the amplified cDNA, which generated restriction fragments of the expected size (data not shown). RNA isolation and PCR amplification to evaluate expression of the Tg.AC transgene was performed as previously described. 35
The PCR products were visualized by ultraviolet illumination after electrophoresis through 2.0% agarose (UltraPure, Sigma) at 60 V for 80 minutes and staining in Tris borate/EDTA buffer (89 mmol/L Tris, 89 mmol/L boric acid, 2.5 mmol/L EDTA, pH 8.2) containing 0.5 μg/ml ethidium bromide. Gels were photographed with type 55 positive/negative film (Polaroid, Cambridge, MA). The films were analyzed using the Eagle Eye II Image Analysis System (Stratagene) and NIH Image 1.54 software, and the area under the curve was normalized for G3PDH content.
Immunohistochemistry and Histology
Frozen sections of skin, 6 μm thick, were cut and placed onto positively charged slides prepared for immunohistochemistry. Slides were pretreated with normal goat serum for 30 minutes and incubated for 4 hours at 4°C with a 1:50 dilution of polyclonal anti-mouse TGF-α antibody (Peninsula Labs) followed by an additional 1 hour with a 1:100 dilution of goat anti-rabbit IgG biotinylated antibody (Vector Laboratories, Burlingame, CA). For BrdU and hematoxylin and eosin (H&E) staining, samples of dorsal skin from control and arsenic-treated mice were preserved in 10% buffered formaldehyde, and paraffin-embedded tissue sections (5 to 6 μm thick) were prepared. For BrdU staining, slides containing three sections of skin from each animal were deparaffinized and hydrated with Automation buffer (Biomeda, Burlingame, CA). The slides were incubated in 2 N HCL for 30 minutes at 37°C and neutralized in borate buffer. The BrdU antigenic sites were unmasked by digestion of the sections in 0.01% trypsin in 1% CaCl2 at 37°C for 3 minutes. Endogenous peroxidase activity was blocked by incubation with 3% H2O2 for 15 minutes. Sections were incubated with a 1:50 dilution of monoclonal anti-BrdU antibody (Becton Dickinson, Mountain View, CA) for 30 minutes followed by incubation for 20 minutes with a 1:100 dilution of biotinylated horse anti-mouse IgG (Vector Laboratories). BrdU was localized with the peroxidase substrate 6,6′-diaminobenzidine enhanced with NiCl2. All slides were counterstained with modified Harris’ hematoxylin and dehydrated for mounting.
Determination of Arsenic Levels in Tissues and Hair
Samples of lung, liver, kidney, and skin from control and arsenic-treated mice were quick-frozen in acid-free vials and stored at −70°C. Determination of arsenic tissue levels was performed by Radian Corp. (Morrisville, NC). Matrix spikes in tissue and method spikes in reagents were prepared for each sample at a concentration of 40 μg/L. After weighing and spiking, 2.0 ml of concentrated nitric acid was added to each sample, the vessels were capped, and samples were processed by microwave digestion. Each sample was transferred to a graduated cylinder, and deionized water was added to a final volume of 25 ml. The samples were analyzed by atomic absorption spectroscopy using a SpectrAA 600 with Zeeman background correction (Varian, San Fernando, CA) set to 193.7 nm with a flow rate of 3 ml/minute. The instrument was calibrated using a six-point standard curve prepared from an arsenic standard reference solution, and quality control standards were run during the analysis to confirm the calibration.
Statistical Analysis
Data shown are representative of at least three separate experiments. Statistical significance was determined by the Bonferroni adjustment of the Student t-test. Statistically significant differences were reported when the P value was <0.05.
Results
The high affinity of arsenic for sulfhydryl groups leads to its accumulation and retention in keratin-rich tissues such as hair and skin, which can be used as a relative indicator of exposure in humans. 39 After 14 weeks of exposure, the arsenic content in the hair was 170 μg/g, as compared with 2.6 μg/g for controls (Table 1) ▶ . Significant arsenic accumulation also occurred in the skin and kidneys of treated animals, whereas no detectable accumulation occurred in the liver or lung.
Table 1.
Arsenic Tissue Levels in TG.AC Mice
| Tissue examined | Arsenic content (μg/g) | |
|---|---|---|
| Control water | 0.02% sodium arsenite | |
| Hair | 2.6 ± 1.6 | 170.2 ± 17.6* |
| Skin | <MDL | 8.3 ± 3.2* |
| Kidney | <MDL | 1.9 ± 0.4* |
| Liver | <MDL | <MDL |
| Lung | <MDL | <MDL |
Mice were treated for 14 weeks with 0.02% sodium arsenite via drinking water. <MDL, less than the minimal detection level for arsenic of 0.738 μg/L.
*Significantly different from control at P < 0.05.
To determine whether proliferation of keratinocytes occurred after in vivo arsenic exposure, as was demonstrated previously to occur in vitro, 14,16,17 animals were administered BrdU and skin samples were processed for histological examination and quantitation of proliferating nuclei. After 10 weeks of treatment with sodium arsenite, the skin of mice revealed hyperkeratosis, which is consistent with that observed in humans after arsenic exposure, 7 as well as increased numbers of proliferating cells when compared with controls (Figure 1) ▶ . Kinetic studies demonstrated that the number of BrdU-positive nuclei in the skin of arsenic-treated animals was increased after 4 weeks of exposure and remained elevated through 10 weeks (Figure 2) ▶ .
Figure 1.

BrdU staining of Tg.AC mouse skin after exposure to 0.02% sodium arsenite in the drinking water. Magnification, ×340. Tg.AC mice were injected with BrdU 30 minutes before sacrifice, and tissues and slides were prepared and stained as described in Materials and Methods. A: In normal skin, the epithelium is thin with a moderate amount of keratin on the surface of the skin. B: Mouse skin after 10 weeks of sodium arsenite is moderately thickened (hyperkeratosis) with the stratum corneum containing a moderate increase in the cornified cell layer.
Figure 2.

Kinetics of BrdU expression in Tg.AC mice receiving control (□) and 0.02% sodium-arsenite-treated ([circo) drinking water. *Significantly different from controls at P < 0.05.
The kinetics of growth factor gene expression in Tg.AC skin are shown in Figure 3 ▶ . Expression of mRNAs for GM-CSF and TGF-α were slightly elevated after 4 weeks in non-TPA-treated, arsenic-treated mice and significantly increased at 10 weeks, corresponding to the increased cell proliferation. TNF-α and EGFR mRNA expression were also significantly elevated after 10 weeks of arsenic, whereas c-myc levels remained relatively constant over the studied time course.
Figure 3.

Kinetics of mRNA expression in Tg.AC mouse skin over time. Total RNA was isolated from dorsal skin of Tg.AC transgenic mice receiving sodium arsenite in the drinking water. A: RT-PCR was performed as described in Materials and Methods and used to determine relative differences between skin from control (c) or arsenic-exposed animals at 1, 4, and 10 weeks. B: Image analysis of ethidium-bromide-stained PCR products. Values represent means ± SEM peak intensities for the samples shown in A after normalization for G3PDH. *Significantly different from controls at P < 0.05. +, positive control; MW, molecular weight standard.
Histological examination of the skin (Figures 4 and 5 ▶ ▶ >)with TPA. 40,41
Figure 4.

Histopathology of mouse skin from animals either vehicle treated, sodium arsenite exposed (0.02% drinking water), TPA treated (four doses of 2.5 μg of TPA twice a week for 2 weeks) or combined treated. Full-thickness skin sections were cut parallel to the dorsal midline, and 6-μm sections were prepared and stained with H&E. The magnification is ×200 for all panels, and the hair cycle is in the resting stage (telogen) for all samples shown. A: A section of normal skin from an untreated animal. Note the presence of the fatty layer of tissue below the dermal mesenchyme. B: A section of skin taken from an animal exposed to 0.02% sodium arsenite for 6 weeks. Although the epidermal layer of cells does not appear to be more hyperplastic than that observed in the control section, hyperkeratosis is present, as indicated by the increased layers of stratum corneum (arrows). Note the absence of fat cells below the dermal mesenchyme. C: A section of skin from a mouse that had received control drinking water taken 24 hours after the last of four treatments of 2.5 μg of TPA. There is a continuum of the hyperplasia in the outer root sheath epithelium and of the interfollicular epidermis (arrows). The epithelial hyperplasia is accompanied by an increase in hyperkeratosis. D: A section of skin from a mouse exposed to 0.02% sodium arsenite for 6 weeks and taken 24 hours after the last of four treatments with 2.5 μg of TPA. In contrast to C, there is a pronounced hyperplasia of the outer root sheath and interfollicular epithelia (arrows), which is accompanied by a marked increase in hyperkeratosis.
Figure 5.
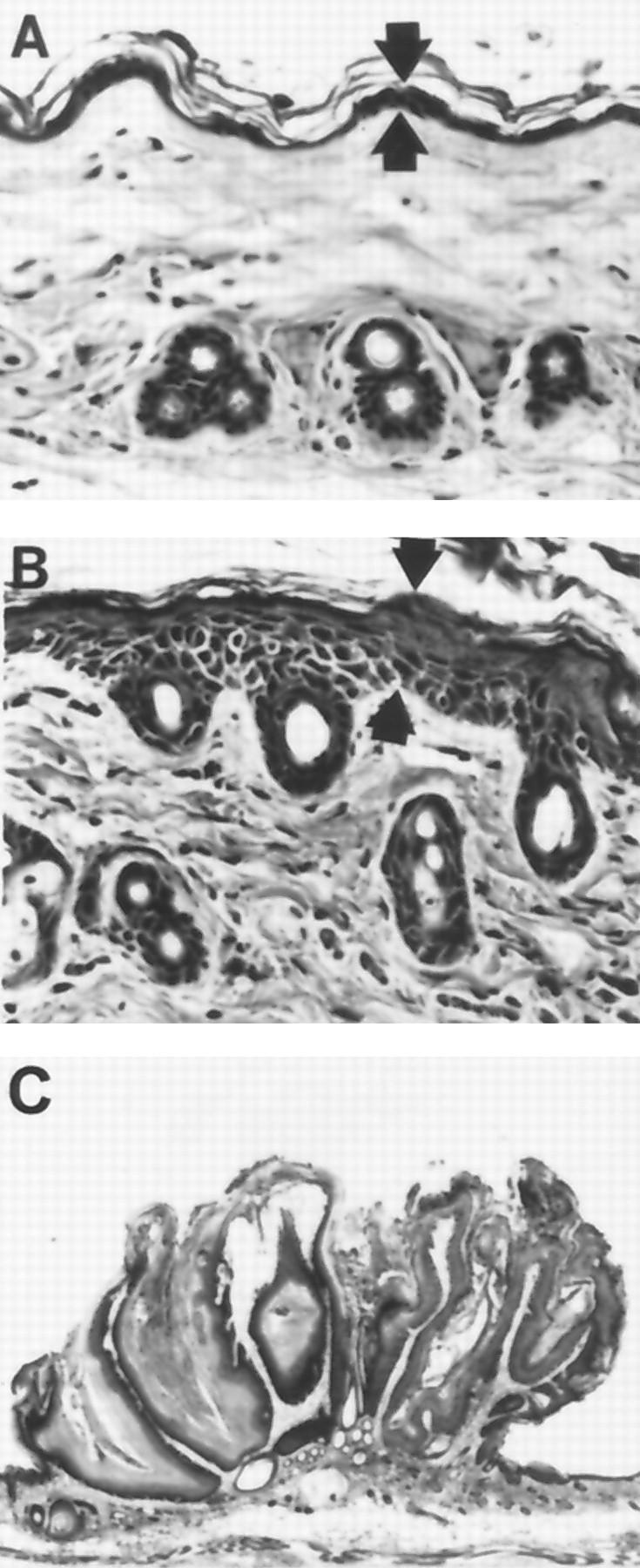
Histology (H&E) of murine skin from control and treated animals taken from full-thickness skin sections cut parallel to the dorsal midline. Tg.AC transgenic mice were provided 0.02% sodium arsenite in the drinking water starting at 12 weeks of age. A: In mice receiving control drinking water, the epithelium (arrows) is thin with a moderate amount of keratin on the surface of the skin. Magnification, ×340. B: After 4 weeks of sodium arsenite exposure, the epithelium (arrows) is minimally thickened with a moderate amount of keratin on the surface of the skin. Magnification, ×340. C: Squamous cell papilloma from the back of a mouse administered TPA and receiving arsenic for 12 weeks. Magnification, ×27.
Papilloma incidents were monitored up to 18 weeks after TPA treatment in Tg.AC transgenic mice after various treatments. Papillomas were not observed in non-TPA-treated Tg.AC mice receiving either control drinking water or water containing 0.02% sodium arsenite (Figure 6, A and B) ▶ . However, within 4 to 6 weeks after application of TPA (four times 2.5 μg), the incidence of papillomas in arsenic-exposed Tg.AC mice were severalfold higher than in TPA-treated mice that had not received arsenic (Figure 6, A and B) ▶ . The decrease in the number of papillomas that occurred 12 weeks after TPA treatment in arsenic-exposed mice was due to both early deaths from heavy papilloma burdens and regression of some papillomas. There were no papillomas observed in TPA-treated wild-type FVB/N mice receiving control or arsenic-treated drinking water for up to 3 months (data not shown). Groups of mice were also treated with a non-inducing dose of TPA (four times 1.25 μg), and papilloma incidence was monitored (Figure 6A) ▶ . Although there were no papillomas observed in transgenic mice receiving a non-inducing dose of TPA and maintained on control drinking water, arsenic-treated animals receiving the same dose of TPA developed an average of five papillomas per mouse within 6 weeks after TPA application. As TGF-α and GM-CSF were overexpressed in the skin after arsenic treatments, and these growth factors have been implicated in skin neoplasia, 23,27,28,42 we examined whether the administration of neutralizing antibodies against GM-CSF or TGF-α affected papilloma formation. Injection of antibodies to GM-CSF before and during application of TPA significantly reduced the average number of papillomas over the entire time period examined (Figure 6B) ▶ in both TPA-control-water- and TPA-arsenic-treated mice. Antibodies to TGF-α had no apparent effect on papilloma development (data not shown). To establish that arsenic-induced alterations in growth factor expression and papilloma formation were not due to arsenic activation of the v-Ha-ras transgene, we examined transgene expression in Tg.AC mouse skin after exposure to control or arsenite-treated drinking water for up to 8 weeks. As shown in Figure 7 ▶ , there was no evidence of transgene expression in arsenic-exposed mice. In contrast, cells derived from a TPA-induced Tg.AC carcinoma contained a high level of transgene mRNA (Figure 7 ▶ , lane 2).
Figure 6.

Papilloma incidence in Tg.AC transgenic mice provided 0.02% sodium arsenite in the drinking water starting at 12 weeks of age. A: Mice were pretreated with arsenite or water for 4 weeks and sub-grouped, and 1.25 (non-inducing; NITPA) or 2.5 μg of TPA were applied twice per week for 2 consecutive weeks. ×, control water; □, TPA plus water; ▪, TPA plus arsenic; ○, NITPA plus water; •, NITPA plus arsenic; n = 20 mice per treatment group. B: Same as A, using 2.5 μg of TPA, plus additional groups were treated with monoclonal antibodies to mouse GM-CSF 2 weeks before TPA treatment, 2 hours after each application of TPA, and 2 weeks after TPA treatment. ×, control water; □, TPA plus water; ▪, TPA plus As; ▵, TPA plus water plus anti-GM-CSF antibodies; ▴, TPA plus As plus anti-GM-CSF antibodies; n = 13 to 14 mice per treatment group. Papillomas were not observed in non-TPA-promoted, arsenic-treated transgenic mice or TPA-promoted, arsenic-treated wild-type FVB/N mice (data not shown).
Figure 7.

v-Ha-ras transgene mRNA expression in Tg.AC mouse skin. Total RNA was isolated and RT-PCR performed on dorsal skin of Tg.AC transgenic mice as previously described. 35 Transgene expression was evaluated at 2, 4, 6, or 8 weeks in mice receiving control (C) or 0.02% sodium-arsenite-treated drinking water (A). MW, molecular weight standard; +, positive control.
Immunohistology conducted on randomly selected, clinically normal skin sites from non-TPA-treated Tg.AC mice revealed that TGF-α-immunoreactive protein was markedly increased in mice that received arsenic in their drinking water for 14 weeks compared with control drinking water (Figure 8) ▶ . Increased staining was particularly evident in the hair follicles. GM-CSF immunostaining was only moderately enhanced in skin from arsenic-treated mice and not detected in mice that received control drinking water (data not shown). Thus, both the overexpression of TGF-α and tissue accumulation of arsenic can be localized to the hair follicle region, the site where the v-Ha-ras oncogene has been shown to be expressed early during TPA-induced tumorigenesis. 43
Figure 8.

Immunohistochemical localization of TGF in the skin of Tg.AC transgenic mice. Magnification, ×360. Samples of skin from control and arsenic-treated mice were quick-frozen, and 6-μm sections were prepared for immunohistochemistry as described in Materials and Methods. Shown are representative samples from Tg.AC transgenic mice after 14 weeks of exposure to either control (a) or arsenite-treated (b) drinking water. Note intense staining in follicular region of skin from arsenite-treated mice (arrow).
To determine whether in vivo arsenic exposure modulates growth factor expression in human skin, biopsy samples were obtained from individuals from the Pa Chang Valley of Taiwan who were exposed to arsenic via their drinking water, and TGF-α, GM-CSF, and TNF-α mRNA transcripts were examined by reverse transcription (RT)-PCR (Figure 9) ▶ . GM-CSF and TGF-α mRNA transcripts were highly expressed in skin from areas exhibiting hyperpigmentation and hyperkeratosis in arsenic-exposed individuals, as compared with nonlesioned skin. These differences became more evident when the values were normalized relative to the density of each corresponding band for G3PDH (Figure 9B) ▶ . There were no statistical differences in TNF-α expression.
Figure 9.

TGF-α, GM-CSF, TNF-α, and G3PDH mRNA expression in lesioned skin from Bowen’s disease patients drinking arsenic-contaminated drinking water from the Pa Chang Valley in Taiwan. Control skin samples were from individuals in the same village who had visible skin lesions. Poly(A+) mRNA was isolated from skin biopsies as described in Materials and Methods. A: RT-PCR was performed as described in Materials and Methods to determine relative differences between samples of lesioned and nonlesioned skin. Lanes 1 to 4, nonlesioned skin; lanes 5 to 10, lesioned skin. B: Image analysis of ethidium-bromide-stained PCR products after normalization relative to the density of each corresponding band for G3PDH. Values represent means ± SEM for the samples shown in A. *Significantly different from controls at P < 0.05.
Discussion
Our previous observations demonstrating the overexpression of growth factors and increased cell proliferation in primary human keratinocytes treated in vitro with arsenic 14 are supported by the current studies using Tg.AC transgenic mice and skin samples from arsenic-exposed patients. These findings are consistent with the hypothesis that arsenic exposure causes low-level, chronic stimulation of keratinocyte-derived growth factors that act to facilitate skin cancer through serving as a co-promoter, although studies with nontransgenic mice will be necessary before arsenic can be categorized unequivocally as a co-promoter for mouse skin carcinogenesis. These data neither support nor refute the possibility that arsenic also causes genetic changes leading to cancer. Similar to that seen in arsenic-exposed humans, 6,7,38 thickening of the stratum corneum and hyperkeratosis were observed in the skin of Tg.AC mice after arsenic exposure. These samples also contained abundant levels of TGF-α mRNA transcripts as well as immunoreactive protein concentrated in the hair follicle region. Overexpression of growth factors occurs in many human cancers, including squamous cell carcinomas of the skin and Bowen’s disease. 44,45 Evidence also exists that overexpression of EGFR and its ligand TGF-α participate in an autocrine loop that results in abnormal proliferation and accumulation of EGFR in keratinocytes from Bowen’s disease lesions. 45,46 Increased levels of TGF-α have also been demonstrated in a number of more benign hyperproliferative disorders of the skin, such as psoriasis and solar keratosis. 47
Several lines of evidence indicate that overexpression of TGF-α can stimulate tumor growth. TGF-α transgenic mice develop higher frequencies and larger, more dysplastic papillomas and squamous cell carcinomas, compared with the parental strain, after initiation with 7,12-dimethylbenz[a]anthracene and promotion with TPA. 48 Furthermore, transgenic mice with keratinocyte-targeted TGF-α overexpression, as well as nude mice engrafted with keratinocytes transfected with the human TGF-α transgene, demonstrate hyperkeratosis, hyperplasia, and the development of spontaneous squamous papillomas. 25,26 Like arsenic-induced papillomas, many of which regress 14 weeks after TPA promotion, these papillomas also frequently regress over time. Introduction of the v-Ha-ras oncogene into murine keratinocytes increases TGF-α expression and proliferation in vitro, 49 and TGF-α is overexpressed in skin papillomas formed when nude mice are engrafted with these keratinocytes. 50 Furthermore, p53 has been shown to induce transcriptional activation of the TGF-α promoter in a human tumor cell line. 51 However, TGF-α null mice develop papillomas, 35 indicating other growth factors can substitute, and TGF-α transgenic mice treated with TPA develop papillomas that exhibit classic Ha-ras gene mutations and not p53 gene mutations. 48
Synergy may exist in skin tumor formation between TGF-α overexpression and Ha-ras activation, 47 as well as chronic arsenic exposure. Application of TPA (2.5 μg twice a week for 2 weeks) to the dorsal shaved skin of Tg.AC transgenic mice induced a modest incidence of papillomas within 4 weeks (mean of three per mouse; J. Spalding, unpublished observation). When Tg.AC transgenic mice were provided sodium arsenite in the drinking water and 4 weeks later subjected to this TPA regimen, the incidence of skin papillomas was significantly higher than in TPA-treated mice that did not receive arsenic. Differences in papilloma frequencies were particularly evident when a non-inducing dose of TPA (four times 1.25 μg) was applied. Although no papillomas were observed in animals receiving control water and the low dose of TPA, arsenic-exposed animals receiving the same non-inducing TPA dose developed an average of five papillomas per mouse 6 weeks after TPA application. As papillomas were not observed in arsenic-exposed, TPA-treated, wild-type FVB/N mice or Tg.AC mice that did not receive TPA (data not shown) and as arsenic treatment alone did not induce transgene expression (Figure 7) ▶ , it would appear that arsenic can serve as a co-promoter to enhance papilloma formation but not as a complete tumor promoter or initiator. TPA-like tumor promoters, in addition to stimulating processes such as protein kinases, also increase growth factor expression, but unlike arsenic the response is assumed to be acute and more potent.
In addition to its importance in dendritic cell development in the skin, GM-CSF is a potent keratinocyte mitogen. 52 Whereas GM-CSF is only minimally, if at all, expressed in normal skin and is not found in unstimulated keratinocyte cultures, 23 increased expression has been shown in many skin diseases, and it is readily induced in a paracrine fashion by cytokines, including interleukin (IL)-1, IL-2, TGF-α, and TNF-α. 53 Direct intradermal injection of GM-CSF into human skin causes keratinocyte enlargement and thickening of the epidermis. 54 Recent studies have shown increased GM-CSF mRNA expression in lesional 55 and lesion-free 56 psoriatic skin as well as skin from patients with atopic dermatitis. 57 A role for GM-CSF in dermal carcinogenesis has also been postulated based on evidence that mice show dose- and time-dependent increases in GM-CSF in the skin after topical treatment with tumor-promoting agents. 41,58 We had shown previously that arsenic amplifies GM-CSF expression via paracrine stimulation by TGF-α in keratinocyte cultures. 39 Like TGF-α, increased immunoreactive GM-CSF and GM-CSF transcripts were found in the skin after arsenic treatment. Although treatment with TGF-α antibodies before TPA application did not have a protective effect against papilloma formation, antibodies to GM-CSF reduced the average number of papillomas to near background levels in animals receiving arsenic as well as control water. This suggests that TGF-α may play a critical role in keratinocyte proliferation but that GM-CSF is more directly involved in the growth and development of papillomas. Alternatively, neutralizing antibodies to TGF-α may be relatively ineffective as TGF-α is produced constitutively, is normally membrane bound, and has a substantial half-life.
Distribution studies after chronic arsenic exposure showed that arsenic accumulates in keratin-rich tissues such as the skin and hair 59 (Table 1) ▶ , whereas immunohistological studies demonstrated growth factor concentration to the hair follicle region. Interestingly, animals exposed to arsenic experienced a delay in their hair growth cycle compared with the control mice. Several mice did not exhibit hair growth for up to 4 months after arsenite treatment had been terminated, suggesting that irrevocable follicular toxicity had occurred. Arsenic forms stable bonds with thiol groups of proteins and has affinity for the SH groups of cysteine allowing for accumulation in the hair and skin, which are rich in cysteine residues. Consistent with these observations, it is believed that most skin papillomas also originate in hair follicles where the v-Ha-ras transgene is expressed in TPA-treated Tg.AC mice. 44 Pluripotent stem cell populations arising from the root sheath of the hair follicle may be particularly receptive to increased levels of growth-promoting cytokines after transgene activation. In addition, changes in DNA methylation may result in altered growth factor receptor expression or activation of transcription factors that promote keratinocyte proliferation. Taken together, the studies described herein provide evidence for a mechanistic-based hypothesis for arsenic carcinogenecity, suggesting that, in addition to genetic factors, neoplastic growth results from the ability of arsenic to concentrate in hair follicles and alter the expression of autocrine and paracrine growth signals. These data also suggest that elevated levels of skin-associated TGF-α and GM-CSF may serve as an early biomarker for skin diseases associated with arsenic exposure.
Acknowledgments
We thank Drs. J. Carl Barrett and Ainsley Weston for their thoughtful review of this manuscript, Jason Doherty and Rhonda Hines for their technical assistance, and Teresa Cummins for manuscript preparation.
Footnotes
Address reprint requests to Dr. Dori R. Germolec, Environmental Immunology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Heath, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail: germolec@niehs.nih.gov.
These studies were presented in part as an invited review in a special issue (Arsenic: a paradoxical human carcinogen. Rev Mutat Res 1997, 386:209–217).
References
- 1.US Environmental Protection Agency: Special Report on Ingested Inorganic Arsenic: Skin Cancer and Nutritional Essentiality. Risk Assessment Forum. Washington, DC, US Environmental Protection Agency, 1987
- 2.Nriagu JO: Arsenic in the Environment. II. Human Health and Ecosystem Effects. 1994. Wiley and Sons, New York
- 3.Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto P, Duggan HM, Wood R, Kornett MJ, Smith MT: Cancer risks from arsenic in drinking water. Environ Health Perspect 1992, 97:259-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiou HY, Hsueh YM, Liaw KF, Horng SF, Chiang MH, Pu YS, Lin JS, Huang CH, Chen CJ: Incidence of internal cancers and ingested inorganic arsenic: a 7-year follow-up study in Taiwan. Cancer Res 1995, 55:1296-1300 [PubMed] [Google Scholar]
- 5.Maloney ME: Arsenic in dermatology. Dermatol Surg 1996, 22:301-304 [DOI] [PubMed] [Google Scholar]
- 6.Chai C-Y, Yu H-S, Yen H-T, Tsai K-B, Chen S-S, Yu C-L: The inhibitory effect of UVB irradiation on the expression of p53 and Ki-67 proteins in arsenic-induced Bowen’s disease. J Cutan Pathol 1997, 24:8-13 [DOI] [PubMed] [Google Scholar]
- 7.International Agency for Research on Cancer (IARC): Arsenic and arsenic compounds. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans: Overall Evaluations of Carcinogenicity. An Update of IARC 1987, Lyon, France. Monographs 1 to 42, suppl 7, pp 100–106
- 8.Chen C-J, Chuang Y-C, Lin T-M, Wu H-Y: Malignant neoplasms among residents of a Blackfoot disease-endemic area in Taiwan: high-arsenic artesian well water and cancers. Cancer Res 1985, 45:5895-5899 [PubMed] [Google Scholar]
- 9.Kanisawa M, Schroder HA: Life term studies on the effect of trace elements on spontaneous tumors in mice and rats. Cancer Res 1969, 29:892-895 [PubMed] [Google Scholar]
- 10.Boutwell R: Feed additives: a carcinogenicity evaluation of potassium arsenite (KAsO2) and arsanillic acid. J Ag Food Chem 1963, 11:381-386 [Google Scholar]
- 11.Baroni C, Van Esch G, Saffiotti U: Carcinogenesis tests of two inorganic arsenicals. Arch Environ Health 1963, 7:668-674 [DOI] [PubMed] [Google Scholar]
- 12.Rossman TG, Stone D, Moling M, Troll W: Absence of arsenite mutagenicity in E. coli and Chinese hamster cells. Environ Mutagen 1980, 2:371-379 [DOI] [PubMed] [Google Scholar]
- 13.Brown JL, Kitchin KT: Arsenite, but not cadmium, induces ornithine decarboxylase and heme oxygenase activity in rat liver: relevance to arsenic carcinogenesis. Cancer Lett 1996, 98:227-231 [PubMed] [Google Scholar]
- 14.Germolec DR, Yoshida T, Gaido K, Wilmer JL, Simeonova PP, Kayama F, Burleson F, Dong W, Lange RW, Luster MI: Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol Appl Pharmacol 1996, 141:308-318 [DOI] [PubMed] [Google Scholar]
- 15.Kachinskas D, Phillips MA, Qin Q, Stokes JD, Rice RH: Arsenate perturbation of human keratinocyte differentiation. Cell Growth Differ 1994, 5:1235-1241 [PubMed] [Google Scholar]
- 16.Burleson FG, Simeonova PP, Germolec DR, Luster MI: Dermatotoxic chemical stimulation of c-jun and c-fos transcription and AP-1 DNA binding in human keratinocytes. Research Commun Mol Pathol Pharmacol 1996, 93:131-148 [PubMed] [Google Scholar]
- 17.Klimecki WT, Borchers AH, Egbert RE, Nagle RB, Carter DE, Bowden GT: Effects of acute and chronic arsenic exposure of human-derived keratinocytes in an in vitro human skin equivalent system: a novel model of human arsenicism. Toxicol In Vitro 1997, 11:89-98 [DOI] [PubMed] [Google Scholar]
- 18.Jha AN, Noditi M, Nilsson R, Natarajan AT: Genotoxic effects of sodium arsenic on human cells. Mutation Res 1992, 284:215-221 [DOI] [PubMed] [Google Scholar]
- 19.Lee T-C, Tanaka N, Lamb PW, Gilmer TM, Barrett JC: Induction of gene amplification by arsenic. Science 1988, 241:79-81 [DOI] [PubMed] [Google Scholar]
- 20.Mass M, Wang L: Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutation Res 1997, 386:263-278 [DOI] [PubMed] [Google Scholar]
- 21.Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP: Association of arsenic-induced malignant transformation with DNA hypo-methylation and aberrant gene expression. Proc Natl Acad Sci USA 1997, 94:10907-10912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luger TA, Schwarz T: Epidermal cell-derived cytokines. Bos JD eds. Skin Immune System. 1990, :pp 257-291 FL, CRC Press, Boca Raton [Google Scholar]
- 23.Vasunia KB, Miller ML, Puga A, Baxter CS: Granulocyte-macrophage colony-stimulating factor (GM-CSF) is expressed in mouse skin in response to tumor-promoting agents and modulates dermal inflammation and epidermal dark cell numbers. Carcinogenesis 1994, 15:653-660 [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb AB, Chang CK, Posnett DN, Fanelli B, Tam JP: Detection of transforming growth factor α in normal malignant and hyperproliferative human keratinocytes. J Exp Med 1988, 167:670-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finzi E, Kilkenny A, Strickland JE, Balaschak M, Bringman T, Derynck R, Aaronson S, Yuspa SH: TGF-α stimulates growth of skin papillomas by autocrine and paracrine mechanisms but does not cause neoplastic progression. Mol Carcinog 1988, 1:7-12 [DOI] [PubMed] [Google Scholar]
- 26.Furstenberger G, Rogers M, Schnapke R, Bauer G, Hofler P, Marks F: Stimulatory role of transforming growth factors in multistage skin carcinogenesis: possible explanation for the tumor-inducing effects of wounding in initiated NMRI mouse skin. Int J Cancer 1989, 43:915-921 [DOI] [PubMed] [Google Scholar]
- 27.Vassar R, Fuchs E: Transgenic mice provide new insights into the role of TGF-α during epidermal development and differentiation. Genes Dev 1991, 5:714-727 [DOI] [PubMed] [Google Scholar]
- 28.Dominey AM, Want XJ, King LE, Nanney LB, Gagne TA, Sellheyer K, Bundman DS, Longley MA, Rothnagel JA, Greenhalgh DA, Roop DR: Targeted overexpression of transforming growth factor α in the epidermis of transgenic mice elicits hyperplasia, hyperkeratosis, and spontaneous, squamous papillomas. Cell Growth Differ 1993, 4:1071-1082 [PubMed] [Google Scholar]
- 29.Thorgeirsson SS, Santoni-Rugui E: Transgenic mouse models in carcinogenesis: interaction of c-myc with transforming growth factor α and hepatocyte growth factor in hepatocarcinogenesis. Br J Clin Pharmacol 1996, 42:43-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amundadottir LT, Nass SJ, Berchem GJ, Johnson MD, Dickson RB: Cooperation of TGF-α and c-myc in mouse mammary tumorigenesis: coordinated stimulation of growth and suppression of apoptosis. Oncogene 1996, 13:757-765 [PubMed] [Google Scholar]
- 31.Takagi H, Sharp R, Takayama H, Anver MR, Ward JM, Merlino G: Collaboration between growth factors and diverse chemical carcinogens in hepatocarcinogenesis of transforming growth factor-α transgenic mice. Cancer Res 1993, 53:4329-4336 [PubMed] [Google Scholar]
- 32.Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC: Overexpression of TGF-α in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 1990, 61:1121-1135 [DOI] [PubMed] [Google Scholar]
- 33.Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT: TGF-α overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 1990, 61:1137-1146 [DOI] [PubMed] [Google Scholar]
- 34.Matsui Y, Halter SA, Holt JT, Hogan BLM, Coffey RJ: Development of mammary hyperplasia and neoplasia in MMTV-TGF-α transgenic mice. Cell 1990, 61:1147-1155 [DOI] [PubMed] [Google Scholar]
- 35.Humble MC, Szczesnial CJ, Luetteke NC, Spalding JW, Cannon RE, Hansen LA, Lee DC, Tennant RW: TGF-α is dispensable for skin tumorigenesis in Tg.AC mice. Toxicol Pathol 1998 (in press) [DOI] [PubMed]
- 36.Tennant RW, Spalding J: Predictions for the outcome of rodent carcinogenicity bioassays: identification of trans-species carcinogens and noncarcinogens. Environ Health Perspect 1996, 104:1095-1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leder A, Kuo A, Cardiff RD, Sinn E, Leder P: v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci USA 1990, 87:9178-9182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tseng WP, Chu HM, How SW, Fong JM, Lin CS, Yeh S: Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. J Natl Cancer Inst 1968, 40:453-463 [PubMed] [Google Scholar]
- 39.Molin L, Wester PO: The estimated daily losses of trace elements from normal skin by desquamation. Scand J Clin Lab Invest 1976, 36:679-688 [DOI] [PubMed] [Google Scholar]
- 40.Cardiff RD, Leder A, Kuo A, Pattengale PK, Leder P: Multiple tumor types appears in transgenic mouse with a RAS oncogene. Am J Pathol 1993, 142:1199-1207 [PMC free article] [PubMed] [Google Scholar]
- 41.Spalding JW, Momma J, Elwell MR, Tennant RW: Chemically induced skin carcinogenesis in a transgenic mouse line (TG.AC) carrying a v-Ha-ras gene. Carcinogenesis 1993, 14:1335-1341 [DOI] [PubMed] [Google Scholar]
- 42.Robertson FM, Bijur GN, Oberyszyn AS, Pellegrini AE, Boros LG, Sabourin LK, Oberyszyn TM: Granulocyte-macrophage colony stimulating factor gene expression and function during tumor formation. Carcinogenesis 1994, 15:1012-1029 [DOI] [PubMed] [Google Scholar]
- 43.Hansen LA, Tennant R: Follicular origin of epidermal papillomas in v-Ha-ras transgenic Tg.AC mouse skin. Proc Natl Acad Sci USA 1994, 91:7822-7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Partridge M, Green MR, Langdon JD, Feldmann M: Production of TGF-α and TGF-β by cultured keratinocytes, skin, and oral squamous cell carcinomas: potential autocrine regulation of normal and malignant epithelial cell proliferation. Br J Cancer 1989, 60:542-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant JJH, Howes G, McKee PH: Transforming growth factor-α expression in situ epidermal neoplasia. Clin Exp Dermatol 1995, 20:208-212 [DOI] [PubMed] [Google Scholar]
- 46.Imamoto A, Beltran LM, DiGiovanni J: Evidence for autocrine/paracrine growth stimulation by transforming growth factor-α during the process of skin tumor promotion. J Mol Carcinog 1991, 4:52-60 [DOI] [PubMed] [Google Scholar]
- 47.Elder JT, Fisher GJ, Lindquist PB, Bennett GL, Pittelkow MR, Coffey RJ, Ellingsworth L, Derynck R, Voorhees JJ: Overexpression of transforming growth factor-α in psoriatic epidermis. Science 1989, 243:811-814 [DOI] [PubMed] [Google Scholar]
- 48.Shibata MA, Ward JM, Merlino G: Enhanced sensitivity to tumor growth and development in multistage skin carcinogenesis by transforming growth factor-α- induced epidermal growth factor receptor activation but not p53 inactivation. Mol Carcinog 1997, 18:160-170 [DOI] [PubMed] [Google Scholar]
- 49.Lee E, Punnonen K, Cheng C, Glick A, Dlugosz A, Yuspa SH: Analysis of phospholipid metabolism in murine keratinocytes transformed by the v-ras oncogene: relationship of phosphatidylinositol turnover and cytokine stimulation to the transformed phenotype. Carcinogenesis 1992, 13:2367-2373 [DOI] [PubMed] [Google Scholar]
- 50.Glick AB, Sporn MB, Yuspa SH: Altered regulation of TGF-β1 and TGF-α in primary keratinocytes and papillomas expressing v-Ha-ras. Mol Carcinog 1991, 4:210-219 [DOI] [PubMed] [Google Scholar]
- 51.Shin TH, Patterson AJ, Kudlow JE: p53 stimulates transcription from the human transforming growth factor α promoter: a potential growth-stimulatory role for p53. Mol Cell Biol 1995, 15:4694-4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braunstein S, Kaplan G, Gottlieb AB, Schwartz M, Walsh G, Abalos RM, Fajardo TT, Guido LS, Krueger JG: GM-CSF activates regenerative epidermal growth and stimulates keratinocyte proliferation in human skin in vivo. J Invest Dermatol 1994, 103:601-604 [DOI] [PubMed] [Google Scholar]
- 53.Ansel J, Perry P, Brown J, Damm D, Phan T, Hart C, Luger T, Hefeneider S: Cytokine modulation of keratinocyte cytokines. J Invest Dermatol 1990, 94:101S-107S [DOI] [PubMed] [Google Scholar]
- 54.Kaplan G, Walsh G, Guido LS, Meyn P, Burkhardt RA, Abalos RM, Barker J, Frindt PA, Fajardo TT, Celona R, Cohn ZA: Novel responses of human skin to intradermal recombinant granulocyte/macrophage colony-stimulating factor: Langerhans cell recruitment, keratinocyte growth, and enhanced wound healing. J Exp Med 1992, 175:1717-1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lontz W, Sirsho A, Liu W, Lindberg M, Rollman O, Torma H: Increased mRNA expression of manganese superoxide dismutase in psoriasis skin lesions and in cultured human keratinocytes exposed to IL-1β and TNF-α. Free Radical Biol Med 1995, 18:349-355 [DOI] [PubMed] [Google Scholar]
- 56.Uyemora K, Yamamura M, Fivenson DF, Modlin RL, Nickoloff BJ: The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol 1993, 101:701-705 [DOI] [PubMed] [Google Scholar]
- 57.Pastore S, Fanales-Belasio E, Albanesi C, Chinni LM, Giannetti A, Girolomoni G: Granulocyte macrophage colony-stimulating factor is overproduced by keratinocytes in atopic dermatitis. J Clin Invest 1997, 99:3009-3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koury MJ, Balmain A, Pragnell IB: Induction of granulocyte-macro-phage colony-stimulating activity in mouse skin by inflammatory agents and tumor promoters. EMBO J 1983, 2:1877-1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lingren A, Vater M, Dencker L: Autoradiographic studies on the distribution of arsenic in mice and hamsters administered 74AS-arsenite or arsenate. Acta Pharmacol Toxicol 1982, 51:253-259 [DOI] [PubMed] [Google Scholar]