

Abstract
Small adenocarcinomas of the colorectum showing no evidence of origin from an adenoma have been called de novo carcinomas, a name that implies an origin via a different molecular genetic mechanism than the usual colorectal carcinoma which develops from an adenoma. Using microsatellite analysis, 35 early (pT1) de novo and 36 pT1 ex-adenoma carcinomas were compared using 8 microsatellite loci at 6 different chromosomal loci (1p, 2p, 8p, 5q, 17p, and 18q) known or hypothesized to be important for colorectal carcinogenesis. The rate of loss of heterozygosity (LOH) at the 17p locus (near the p53 gene) was significantly higher in the de novo than in the ex-adenoma group (73 vs. 37%, P = 0.004). The rates of LOH at the other loci (including the APC and DCC genes) and the rate of MSI were not significantly different in the two groups. These results indicate that de novo carcinomas of the colorectum develop via a similar carcinogenetic pathway as conventional ex-adenoma carcinomas; however, their higher rate of LOH at 17p is evidence for a biologically more advanced lesion with more frequent p53 mutations, consistent with clinicopathological data indicating that de novo carcinomas are more aggressive than ex-adenoma carcinomas.
Among all neoplastic processes, the stepwise development of colorectal carcinoma is perhaps the most exhaustively described with respect both to its morphology and to its molecular pathological events. 1 It is now accepted that most, if not all, colorectal carcinomas arise through a series of morphologically definable steps beginning with the formation of an adenoma. As its size increases, the development of high grade dysplasia and then invasive carcinoma is increasingly likely, so that the endoscopic removal of large (>1 cm in diameter) polyps is recommended for prevention of colorectal cancer. 2
In Japan over the last 10 to 15 years, the improvement and widespread use of endoscopy has been a crucial factor in the frequent detection of early gastric carcinomas. Endoscopically, early gastric carcinomas are seen predominantly as flat, plaque-like lesions and are seldom observed to arise from well-defined adenomas as in the colorectum. 3 The Japanese began to detect similar flat, plaque-like lesions in the colorectum (analogous to those in the stomach) which, because they had no endoscopic or histological evidence of polypoid adenoma formation, were called de novo carcinomas. 4 Their clinical significance rests on the fact that these de novo carcinomas are small, inconspicuous lesions and are therefore difficult to detect in comparison to polypoid lesions. In addition, some studies have indicated that they may be more aggressive (ie, show higher rates of lymphatic vessel invasion and lymph node metastases) than carcinomas developing from polypoid adenomas. 5,6
Unfortunately, although it may be an important clinicopathological entity, the concept of de novo carcinoma has not gained much acceptance, particularly in the Western world, because the name de novo implies that these lesions develop via an alternate pathway of carcinogenesis without a precursor lesion. Some molecular studies have been performed, primarily in Japan, to attempt to test the hypothesis that these de novo carcinomas arise along a separate pathway. While some differences have been seen, particularly with respect to a lower percentage of K-ras mutations in de novo carcinomas, 7 the small numbers studied and the varying definitions used for these lesions in Japan and the West do not clearly establish the existence of an alternate carcinogenetic pathway for these lesions.
In a previous study, we found that p53 overexpression detected by immunohistochemistry was significantly more frequent in a group of de novo carcinomas than in a group of carcinomas that had morphological evidence of origin from an adenoma (ex-adenoma carcinomas). 8 In the present study we compared the rates of microsatellite instability (MSI) and loss of heterozygosity (LOH) at gene loci that have been shown to be particularly important for colorectal carcinogenesis: APC (adenomatosis polyposis coli), DCC (deleted in colon carcinoma), and p53 genes, and three additional sites (1p, 2p, and 8p) that have been hypothesized to be sites of tumor suppressor genes important for colorectal carcinogenesis. Our goal was to see if evidence of a separate pathway of colorectal carcinogenesis for these de novo carcinomas exists and whether there might be any genotypic explanation for their apparently more aggressive phenotype.
Materials and Methods
Lesions
The study was carried out as a comparison of two groups of early (pT1) colorectal carcinomas which were defined as either de novo or ex-adenoma carcinomas as in our previous study. 8 In the first group (de novo carcinomas) were carcinomas with invasion into but not beyond the submucosa, 9 1 cm or less in diameter with no adenoma (low-grade dysplasia) elements in their vicinity (Figure 1) ▶ . All of these cases were extensively sectioned (at least 20 serial sections) to ensure, as completely as possible, that no adenoma elements were focally present. The requirement that the de novo carcinomas be 1 cm or less in diameter also minimized the chance that these were ex-adenoma carcinomas in which the adenomatous elements had been destroyed by the tumor. In 19 of the 35 de novo cases, no remaining mucosal component of the carcinoma was evident, whereas in the remaining cases high-grade dysplasia or carcinoma was still identifiable in the mucosa. The comparison group (ex-adenoma carcinomas), also pT1 carcinomas, consisted of 36 carcinomas with definite adenoma elements in their vicinity (Figure 2) ▶ .
Figure 1.
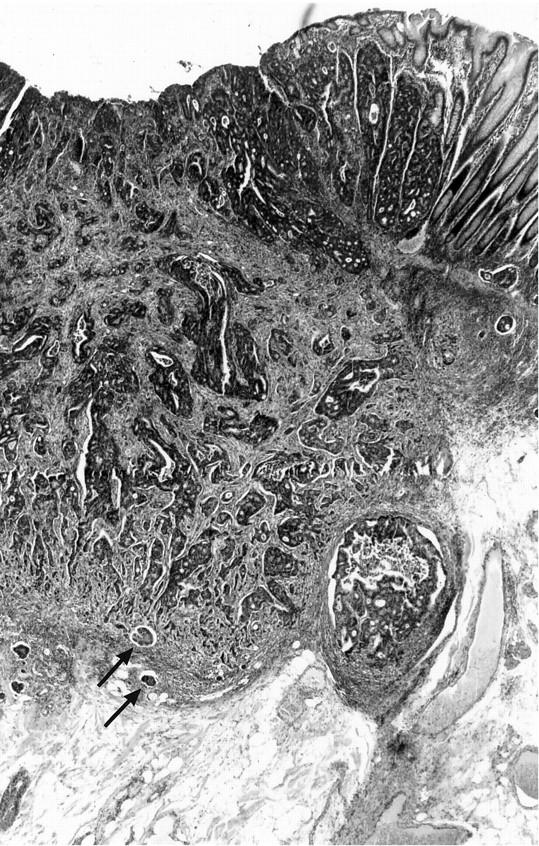
Example of a de novo colorectal carcinoma. This lesion was seen endoscopically as a reddish plaque, 0.8 cm in diameter with central depression. In this histological section, carcinoma is seen invading the submucosa with lymphatic vessel invasion (arrows) at the invasive tumor front. No adenoma elements are seen in the vicinity of the tumor and there is a sharp transition from the carcinoma to the normal-appearing mucosa at its edge (H&E stain, original magnification, ×25).
Figure 2.
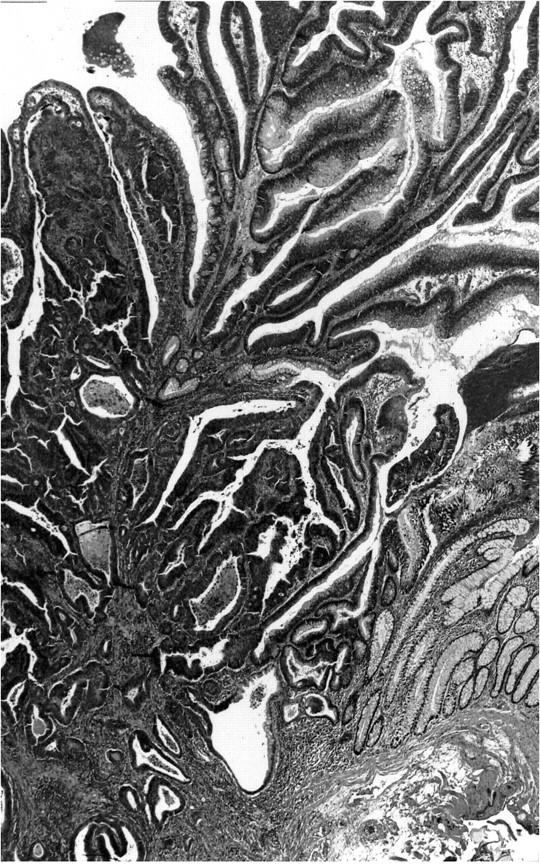
Example of an ex-adenoma carcinoma. This lesion was a polypoid adenoma 1.5 cm in diameter and contained an area of carcinoma invading the submucosa seen in the left hand side of the picture. Remaining adenoma elements are seen in the upper right and nonneoplastic-appearing mucosa in the lower right (H&E stain, original magnification, ×25).
Patient Groups
Clinicopathological data for the study groups are shown in Tables 1 and 2 ▶ ▶ . In the de novo carcinoma group (Table 1) ▶ there were 35 patients, 17 males and 18 females, with an average age of 64.4 (range, 30–83). All but one of the de novo carcinomas were located in the left colon or rectum. These lesions had an average size of 0.79 cm (range, 0.3–1.0 cm). With the exception of two cases, all had been removed by polypectomy. The majority were moderately differentiated. The comparison group of ex-adenoma carcinomas (n = 36) had clinicopathological characteristics comparable to the de novo group, as Table 2 ▶ shows. The average age of these patients was 68.2 (range, 39–91), there were 21 males and 15 females, all but one of the specimens had been removed by polypectomy, and all but three were located in the left colon or rectum. Histologically, the majority of these tumors were also moderately differentiated; a slightly higher number than in the de novo group (8 vs. 2) were poorly differentiated. The average size of these ex-adenoma lesions (carcinoma and adjacent adenoma) was 1.95 cm (range, 0.7–3.2 cm). None of the patients in either group had a known familial colon carcinoma syndrome such as familial adenomatous polyposis (FAP) or hereditary non-polyposis colon carcinoma (HNPCC).
Table 1.
De Novo Carcinomas: Clinicopathological Data
| Case | Sex | Age | Specimen | pT | G | Size (cm) | Site |
|---|---|---|---|---|---|---|---|
| 1 | F | 77 | PE | 1 | 2 | 0.8 | Sigmoid |
| 2 | F | 71 | PE | 1 | 2 | 1.0 | Rectum |
| 3 | F | 71 | PE | 1 | 2 | 1.0 | Sigmoid |
| 4 | F | 66 | PE | 1 | 2 | 0.7 | Sigmoid |
| 5 | M | 59 | PE | 1 | 1 | 0.7 | Sigmoid |
| 6 | M | 63 | PE | 1 | 2 | 0.5 | Rectum |
| 7 | F | 66 | PE | 1 | 2 | 0.5 | Left flexure |
| 8 | M | 48 | PE | 1 | 2 | 0.7 | Rectum |
| 9 | M | 68 | RS | 1 | 2 | 1.0 | Sigmoid |
| 10 | F | 73 | PE | 1 | 3 | 1.0 | Sigmoid |
| 11 | M | 71 | PE | 1 | 2 | 0.6 | Sigmoid |
| 12 | F | 57 | PE | 1 | 2 | 1.0 | Sigmoid |
| 13 | F | 75 | PE | 1 | 2 | 0.9 | Rectum |
| 14 | F | 80 | PE | 1 | 2 | 1.0 | Rectum |
| 15 | M | 83 | PE | 1 | 2 | 0.7 | Rectum |
| 16 | F | 69 | PE | 1 | 2 | 0.7 | Sigmoid |
| 17 | F | 68 | PE | 1 | 2 | 1.0 | Sigmoid |
| 18 | M | 54 | PE | 1 | 2 | 1.0 | Sigmoid |
| 19 | M | 51 | PE | 1 | 2 | 0.6 | Sigmoid |
| 20 | M | 61 | PE | 1 | 2 | 0.6 | Sigmoid |
| 21 | M | 48 | PE | 1 | 2 | 1.0 | Sigmoid |
| 22 | F | 72 | PE | 1 | 2 | 1.0 | Sigmoid |
| 23 | F | 74 | PE | 1 | 2 | 0.6 | Sigmoid |
| 24 | M | 57 | PE | 1 | 1 | 0.8 | Sigmoid |
| 25 | F | 73 | PE | 1 | 2 | 0.6 | Rectum |
| 26 | F | 55 | PE | 1 | 2 | 0.9 | Right flexure |
| 27 | M | 58 | PE | 1 | 1 | 0.9 | Rectum |
| 28 | M | 82 | RS | 1 | 1 | 0.9 | Sigmoid |
| 29 | F | 70 | PE | 1 | 2 | 0.7 | Descending |
| 30 | F | 30 | PE | 1 | 1 | 0.6 | Sigmoid |
| 31 | M | 65 | PE | 1 | 1 | 0.9 | Sigmoid |
| 32 | M | 66 | PE | 1 | 2 | 0.3 | Sigmoid |
| 33 | M | 47 | PE | 1 | 2 | 0.9 | Rectum |
| 34 | M | 75 | PE | 1 | 3 | 0.7 | Descending |
| 35 | F | 51 | PE | 1 | 2 | 0.9 | Rectum |
PE, polypectomy; RS, resection; G, differentiation.
Table 2.
Ex-Adenoma Carcinomas: Clinicopathological Data
| Case | Sex | Age | Specimen | pT | G | Size (cm) | Site |
|---|---|---|---|---|---|---|---|
| 1 | 67 | F | PE | 1 | 2 | 1.6 | Rectum |
| 2 | 70 | M | PE | 1 | 2 | 2.0 | Sigmoid |
| 3 | 82 | F | PE | 1 | 2 | 1.5 | Rectum |
| 4 | 55 | F | PE | 1 | 2 | 2.0 | Rectum |
| 5 | 67 | F | PE | 1 | 2 | 1.9 | Right flexure |
| 6 | 65 | F | PE | 1 | 2 | 2.0 | Rectum |
| 7 | 54 | F | PE | 1 | 1 | 2.0 | Rectum |
| 8 | 74 | F | PE | 1 | 2 | 1.9 | Sigmoid |
| 9 | 66 | F | PE | 1 | 2 | 2.0 | Sigmoid |
| 10 | 64 | F | PE | 1 | 2 | 2.0 | Sigmoid |
| 11 | 70 | M | PE | 1 | 2 | 3.0 | Sigmoid |
| 12 | 42 | M | PE | 1 | 1 | 2.0 | Sigmoid |
| 13 | 75 | M | PE | 1 | 2 | 1.5 | Sigmoid |
| 14 | 73 | M | RS | 1 | 2 | 2.8 | Ascending |
| 15 | 81 | M | PE | 1 | 2 | 3.2 | Sigmoid |
| 16 | 67 | M | PE | 1 | 1 | 1.9 | Sigmoid |
| 17 | 60 | M | PE | 1 | 2 | 3.0 | Rectum |
| 18 | 64 | M | PE | 1 | 2 | 2.0 | Sigmoid |
| 19 | 79 | M | PE | 1 | 2 | 1.7 | Sigmoid |
| 20 | 75 | F | PE | 1 | 1 | 0.7 | Sigmoid |
| 21 | 57 | F | PE | 1 | 2 | 1.9 | Sigmoid |
| 22 | 64 | F | PE | 1 | 2 | 1.5 | Sigmoid |
| 23 | 83 | M | PE | 1 | 2 | 2.6 | Sigmoid |
| 24 | 60 | F | PE | 1 | 1 | 1.3 | Sigmoid |
| 25 | 65 | M | PE | 1 | 2 | 2.5 | Sigmoid |
| 26 | 78 | M | PE | 1 | 2 | 1.5 | Sigmoid |
| 27 | 91 | M | PE | 1 | 2 | 2.0 | Sigmoid |
| 28 | 74 | M | PE | 1 | 2 | 2.5 | Rectum |
| 29 | 82 | M | PE | 1 | 2 | 1.2 | Rectum |
| 30 | 72 | M | PE | 1 | 1 | 2.5 | Sigmoid |
| 31 | 71 | F | PE | 1 | 2 | 1.4 | Rectum |
| 32 | 39 | M | PE | 1 | 3 | 2.4 | Rectum |
| 33 | 65 | M | PE | 1 | 2 | 1.1 | Sigmoid |
| 34 | 75 | F | PE | 1 | 2 | 2.0 | Transverse |
| 35 | 60 | M | PE | 1 | 1 | 1.1 | Sigmoid |
| 36 | 69 | M | PE | 1 | 1 | 2.1 | Sigmoid |
PE, Polypectomy; RS, Resection; G, differentiation.
DNA Extraction
Paraffin blocks from the lesions of the two groups were obtained from the Department of Pathology, Klinikum Bayreuth, Bayreuth, Germany and the Department of Pathology, Klinikum rechts der Isar, Technical University of Munich. Sections measuring 5 μm in thickness were cut from these tissue blocks onto glass slides and briefly stained (10 seconds) with hematoxylin. Under microscopic inspection, invasive carcinoma and normal- appearing mucosal areas from each case were scraped off the slides into Eppendorf tubes containing buffer. The DNA was then extracted by a previously described method. 10 The carcinomatous areas selected for microdissection were taken from areas invading the submucosa and were composed of at least 70% tumor cells to minimize contamination by normal cells.
Chromosomal Loci
Eight microsatellite loci (all (CA)n repeats) on 6 different chromosomal arms (shown in Table 3 ▶ ) were selected for analysis. These included three regions known to be involved in colon carcinogenesis: D5S346–5q21 and D5S107–5q11.2-q13.3, 11 directly adjacent to and in the vicinity of the APC gene, respectively, D18S34 (18q12.2) and D18S487 (18q21) 12 on 18q at the site of the DCC tumor suppressor gene, and D17S520 13 at 17p12 near the p53 gene. Three additional chromosomal areas which had been described as potentially playing a role in colon carcinoma development and progression were also investigated and included the D1S447 locus at 1p32, 14 the D2S123 locus at 2p16–21, 15 and the D8S137 locus 16 at 8p21-p12.
Table 3.
Microsatellite Loci used for MS Analysis
| Chromosome | Locus | Repeat | Sequence 5′ to 3′ |
|---|---|---|---|
| 17p12 | D17S520 | (CA)n | GGA GAA AGT GAT ACA AGG GA |
| GG AGA AAG TGA TAC AAG GGA | |||
| 5q21 | D5S346 | (CA)n | AGC AGA TAA GAC AGT ATT ACT AGT T |
| AC TCA CTC TAG TGA TAA ATC GGG | |||
| 5q11.2-q13.3 | D5S107 | (CA)n | GGC ATC AAC TTG AAC AGC AT |
| GAT CCA CTT TAA CCC AAA TAC | |||
| 18q12.2 | D18S34 | (CA)n | CTC ATG TTC CTG GCA AGA AT |
| CAG AAA ATT CTC TCT GGC TA | |||
| 18q21 | D18S487 | (CA)n | ACA ATC AGA AAC CCG CCA |
| AGC TGA CTT AGG TAG ATT TTC CTC G | |||
| 1p32 | D1S447 | (CA)n | TTA GTC TGA GTT TGT GGG GG |
| GTT TTA ACT TCA TGG CTG CC | |||
| 2p16-21 | D2S123 | (CA)n | GGA CTT TCC ACC TAT GGG AC |
| AAA CAG GAT GCC TGC CTT TA | |||
| 8p21-p12 | D8S137 | (CA)n | AAA TAC CGA GAC TCA CAC TAT A |
| GCT AAT CAG GGA ATC ACC CAA |
Microsatellite Analysis
Two milliliters of a 1:10 dilution from each of the extracted DNA samples were amplified by polymerase chain reaction (PCR) using primer pairs for the eight loci. One oligonucleotide of each the primer pairs was labeled with 6-FAM, TET, or HEX. The internal size standard, GS 500 (Applied Biosystems, Norfolk, CT), was labeled with TAMRA. PCR was carried out in a 25-ml mixture containing 10 mmol/L Tris-HCl (pH 8.3), 1.0 or 1.5 mmol/L MgCl2, 0.01% (w/v) gelatin, 200 μM dNTP, 0.04 μM of each primer, 1.25 U Taq polymerase and 2 μl of extracted DNA. After an initial denaturation step for 4 minutes at 94°C, 35 cycles consisting of 30 sec at 94°C, 30 sec. at 55–60°C, 30 sec. at 72°C followed by a final extension step of 4 minutes. at 72°C. The resulting products were diluted 1:10 to 1:2, after which 1 ml was mixed with 2 ml formamide, 0.5 ml gel loading buffer, and 0.5 ml size standard, followed by gel electrophoresis on a Long Ranger Hydrolink gel (Biozym, Oldendorf, Germany) in 1× TBE buffer in an automated fluorescent DNA sequencer (ABI 377, Applied Biosystems). The data were automatically collected and analyzed by Genescan software (Perkin Elmer Corp., Branchburg, NJ) to quantify peak size, height, and area under the curves. The ratio of the allele peak areas was calculated for the paired normal and tumor samples. The resulting allele ratio for the tumor sample was then divided by the allele ratio of the normal sample and, if above 1.00, converted to give a result ranging from 0.00–1.00. When this result was 0.60 or lower (40% reduction of a tumor allele), the case was scored as allelic imbalance and interpreted as a loss of heterozygosity (LOH) (Figure 3) ▶ . MSI was scored when, in at least one locus, the carcinoma showed at least one definite new peak compared to its paired normal DNA sample. LOH was not scored in cases positive for MSI. Abnormalities (MSI or LOH) were confirmed by a second electrophoresis and automated analysis. All of the cases were amplified by PCR at each of the eight loci. Cases in which the normal DNA sample was homozygous were classified as uninformative.
Figure 3.

a: Example of microsatellite analysis at locus D17S520 in a case of de novo carcinoma. The upper panel shows the normal DNA sample with two clearly identifiable peaks representing the two normal alleles. The lower panel shows tumor DNA, also with two peaks, but one is markedly smaller than the corresponding peak in the normal DNA sample. The ratio (T/N) of allele ratios was 0.39, a reduction of 61%, an allelic imbalance interpreted as LOH. b: Example of microsatellite analysis at locus D5S346 in a case of ex-adenoma carcinoma. Again, the upper panel shows the normal DNA sample with two clearly identifiable normal alleles. The lower panel shows tumor DNA with only one peak, ie, an LOH.
Immunohistochemistry for p53
Immunohistochemistry for p53 protein was performed as previously described 8 with the D01 monoclonal antibody (Oncogene Science, Cambridge, MA) used at a 1:100 dilution after microwave treatment (2 × 5 minutes, 750 Watts) with antigen retrieval solution (HK0500–5K, BioGenex GmbH, Mainz, Germany). Development was with the APAAP method (2×) using Neufuchsin (Dianova GmbH, Hamburg, Germany) as the reaction indicator. A case was scored as positive for overexpression when more than 10% of the cells in the entire carcinomatous component showed a positive nuclear reaction. Of the de novo carcinomas, 27 were part of the earlier p53 immunohistochemical study 8 and 8 were new cases not previously reported. Of the ex-adenoma cases, the results of p53 immunohistochemistry were previously reported for 24 cases, and 12 were previously unreported cases. (Cases selected for this study had sufficient areas of invasive carcinoma and adjacent normal-appearing mucosa that could be reliably separated from one another by microdissection.)
Statistical Analysis
Statistical analysis for comparison of the two groups was performed using the χ 2 test or the Fisher two-tailed test. A P value <0.05 was considered statistically significant.
Results
Microsatellite Instability
Results of the microsatellite analysis are summarized for each locus in Table 4 ▶ . Three of the ex-adenoma carcinomas (two cases at D1S47 and one case at D17S520) and five of the de novo carcinomas showed MSI (one case each at D8S137 and D2S123, two cases at D18S34, and one case at both D5S107 and D17S520), an overall rate of 11%. Seven of the eight cases had MSI at only one locus. The remaining case, a de novo carcinoma, had MSI at two loci.
Table 4.
Results of Microsatellite Analysis
| MS Locus | Type | Cases | Inf | MSI | LOH | P |
|---|---|---|---|---|---|---|
| D17S520 | De novo | 35 | 31 | 1 | 22 | 0.00431 |
| ex-adenoma | 36 | 31 | 1 | 11 | ||
| D5S346 | De novo | 35 | 30 | 0 | 12 | N.S. |
| ex-adenoma | 36 | 30 | 0 | 11 | ||
| D5S107 | De novo | 35 | 28 | 1 | 9 | N.S. |
| ex-adenoma | 36 | 30 | 0 | 8 | ||
| D18S34 | De novo | 35 | 30 | 2 | 14 | N.S. |
| ex-adenoma | 36 | 27 | 0 | 14 | ||
| D18S487 | De novo | 35 | 25 | 0 | 16 | N.S. |
| ex-adenoma | 36 | 32 | 0 | 15 | ||
| D1S447 | De novo | 35 | 28 | 0 | 4 | N.S. |
| ex-adenoma | 36 | 29 | 2 | 1 | ||
| D2S123 | De novo | 35 | 23 | 1 | 1 | N.S. (0.055) |
| ex-adenoma | 36 | 26 | 0 | 7 | ||
| D8S137 | De novo | 35 | 28 | 1 | 7 | N.S. |
| ex-adenoma | 36 | 27 | 0 | 9 |
N.S., not significant.
Loss of Heterozygosity
The percentage of cases with LOH, calculated as (cases of LOH)/(informative cases − cases with MSI), varied among the loci with the D17S520 (17p), D18S487 (18q), D18S34 (18q), D5S346 (5q), D5S107 (5q), and the D8S137 (8p) loci showing the highest rates, in that order. The D1S447 (1p) locus for both the de novo and ex-adenoma groups and the D2S123 (2p) locus for de novo group had rates of LOH below 20%, suggesting a lack of association with carcinogenesis for these loci.
A comparison of the rates of LOH between the de novo and ex-adenoma carcinoma groups showed a significant difference at the D17S520 locus of 73% vs. 37%, respectively (P = 0.0043). The D2S123 locus had a slightly but not significantly higher rate of LOH in the ex-adenoma group (5% vs. 27% respectively, P = 0.055). There were no significant differences between the groups in the rates of LOH at the remaining loci.
Within the de novo group, we also compared those cases with a remaining intramucosal carcinomatous component (16 cases) with the 19 cases with no remaining identifiable intramucosal elements. There were no significant differences between these subgroups in the rates of LOH or MSI at any of the 8 loci examined.
p53 Protein Overexpression
Although some of the cases in both the de novo and ex-adenoma groups were new in comparison to our previous study, the difference in rates of p53 overexpression seen in these cases was similar to that observed in the previous study. With 23 of 35 (66%) of de novo and 15 of 36 (42%) of ex-adenoma cases showing overexpression, there is a significantly higher rate (P = 0.042) in the de novo group. A correlation of p53 immunohistochemistry results with microsatellite analysis at the D17S520 locus for the two groups is shown in Table 5 ▶ . This correlation was better for the ex-adenoma than for the de novo group. Eight of the de novo cases had no LOH at D17S520, but 7 of these were positive for p53 overexpression by immunohistochemistry, compared with only 4 of the 19 ex-adenoma cases without LOH at this locus. Of the 22 de novo cases with LOH at D17S520, 8 were negative by p53 immunohistochemistry, compared with the ex-adenoma cases, only 1 of 11 of which showed LOH.
Table 5.
Correlation between p53 IHC and MS Analysis at D17S520
| MS Analysis | Total | IHC Result | |
|---|---|---|---|
| Negative | Positive | ||
| De novo cases | |||
| Uninformative | 4 | 2 | 2 |
| MSI | 1 | 1 | 0 |
| Normal pattern | 8 | 1 | 7 |
| LOH | 22 | 8 | 14 |
| Total | 35 | 12 | 23 |
| Ex-adenoma cases | |||
| Uninformative | 5 | 5 | 0 |
| MSI | 1 | 0 | 1 |
| Normal pattern | 19 | 15 | 4 |
| LOH | 11 | 1 | 10 |
| Total | 36 | 21 | 15 |
Discussion
The question of whether de novo carcinoma of the colorectum represents a distinct clinicopathological entity is important from both theoretical and clinicopathological points of view. The name given to these lesions suggests that they arise from normal colorectal mucosa without any intervening precancerous steps, a pathway which has been suggested by some authors. 17,18 In light of the morphological and molecular biological evidence that has accumulated over the past 10–20 years for the stepwise development of carcinoma in the colorectum, 1,19,20 such a concept is extremely difficult, if not impossible, to accept. For this reason, despite the clear morphological evidence collected in Japan, 17,21 the existence of very small but already invasive colorectal carcinomas not associated with an adenoma has not been widely accepted in the Western world. 22 This is not a purely academic question, because these lesions are both difficult for the endoscopist to detect and apparently more aggressive than conventional ex-adenoma carcinomas, as evidenced by higher rates of regional lymph nodes metastases reported for them in the few available studies. 4,23
To clarify the origin of these lesions, a few molecular pathological studies of de novo colorectal carcinomas have been performed, mostly in Japan. Hasegawa, 24 using single strand confirmational polymorphism (SSCP) analysis, found 12 of 30 de novo versus 11 of 30 “polyp-forming cancer” with mutations of p53. Using sequence analysis, Aoki 25 reported p53 mutations in 4 of 6 and APC mutations in 2 of 6 cases of de novo carcinomas. Perhaps the clearest finding to date has been that of a relative lack of mutations in the K-ras gene in de novo carcinomas and other flat lesions of the colorectum, as reported in several studies. 26 However, these findings are difficult to interpret, particularly for a Western reader, because there is no uniform definition of “de novo carcinoma” and because intramucosal lesions that have not yet invaded the submucosa are considered to be carcinomas in Japan and have been included in all of these studies. With this in mind, we defined our lesions so that they fulfilled the definition of “de novo” as it is used in Japan but also were carcinomas (ie, with invasion of the submucosa) according to the Western definition. 9 In addition, we selected only invasive areas of tumor from each group to provide the best possible comparison between them.
The most striking result we observed was the significant difference in rates of LOH between de novo and ex-adenoma carcinomas at the D17S520 (17p) locus located near the p53 gene. This result fits with findings of our previous study 8 showing a significantly higher rate of p53 protein overexpression in de novo carcinomas compared to ex-adenoma carcinomas. The incidence of p53 mutations has been shown to increase with tumor progression in the colorectum, 20 so the higher rate of LOH at 17p together with a higher rate of p53 protein overexpression indicates a more biologically advanced lesion and fits with the observed aggressiveness of these lesions. As mentioned above, Hasagawa 24 also studied p53 mutations in de novo carcinoma by PCR-SSCP analysis, and, even taking into account only the lesions which had invaded the submucosa, found a slightly but not significantly higher mutation rate in his de novo carcinomas (9/18 de novo versus 7/17 in “polyp forming” cancers). The discrepancy between his study and ours may be due to the smaller number of cases in his study, the different method of analysis, or perhaps a difference in the microdissection technique (for example, selecting intramucosal rather than clearly invasive components).
An additional finding with regard to p53 in our cases resulted from a comparison of microsatellite analysis at the 17p locus and immunohistochemistry for p53 (Table 5) ▶ . The ex-adenoma group showed a better correlation between the two techniques than the de novo group, due primarily to the 8 de novo cases that were negative for p53 over-expression by immunohistochemistry but still had an LOH at 17p12. There are two possible explanations for this discrepancy. First, the LOH at 17p12 may affect a gene other than p53 and so have no effect on the p53 gene itself. Second, the LOH may be a sign for a mutation of p53 which fails to produce a protein recognized by the D01 antibody. It is also interesting that 7 de novo cases (compared to 4 ex-adenoma cases) had p53 protein overexpression but no LOH or MSI at 17p12. These cases might have been the result of a point mutation in p53 too small to be detected by the microsatellite analysis method.
A second important finding was that rates of LOH in the two groups were remarkably similar at the 5q, 18q, and 8p loci. This indicates that rates of LOH at the APC loci for both groups are in keeping with the rate described for adenomas and carcinomas in previous studies. 27 A similar rate of LOH at 5q for de novo and ex-adenoma carcinomas is somewhat surprising because mutations in APC are known to be particularly important for adenoma development 28 and might not be expected in de novo carcinoma, which supposedly does not have a precursor adenoma stage. At the 18q (DCC) loci, the rate of approximately 50%, which we detected in both groups, fits well for an early colorectal carcinoma, because this is slightly higher than the rate reported for large adenomas (40%) but lower than the rate of about 75% reported for colorectal carcinomas as a whole. 27 The 8p chromosomal arm is thought to contain one or perhaps two tumor supressor genes important for colorectal carcinogenesis. 16 The rate of 30% that we observed at this locus again fits well for early carcinoma, because it is slightly lower than has been reported for colorectal carcinomas as a whole (45%) 16,29 but significantly higher than for adenomas (10%).
For the 1p locus, the rate of LOH in both groups was below 20%, indicating that this locus is not important for development of the tumors in either group. This is despite the fact that this chromosomal arm has been previously described as being important in sporadic tumor development in the colorectum. 14 A possible explanation might be a difference in the techniques used, because this previous study used fluorescent in situ hybridization to detect chromosomal deletions.
The low rate of microsatellite instability which we saw (overall 11%) is just below the range of 12–20% which has been reported for sporadic colorectal carcinoma. 30 In addition, none of our cases showed MSI at a majority of the loci tested, a pattern which has been called “widespread MSI.” Our results were not surprising in light of the fact that nearly all of the lesions in both groups were located in the left colon, where rates of both widespread and overall MSI are very low in comparison to the proximal colon. 31 The predominance of left-side lesions in both groups may have been a result of the exclusion of cases from patients with a family history of colon cancer or, perhaps, through earlier detection of these small lesions due to symptoms such as blood in the stool. De novo and ex-adenoma carcinomas in the right colon might possibly show a different pattern of genetic alterations than the low rate of MSI and the relatively high rates of LOH at the p53, DCC, and APC loci which we saw in these left-side colonic lesions.
What do our results signify for the concept of de novo carcinogenesis? Similar rates of LOH at the APC and DCC loci indicate that mutations of these genes are equally important for de novo and ex-adenoma carcinoma development. The significantly higher rate of LOH at 17p and the higher rate of p53 protein overexpression in the de novo groups are clues that the p53 gene or a gene in its vicinity on 17p plays an important role in the development of the distinctive histomorphology of these lesions. Although p53 mutations are generally considered to be relatively late events in colorectal carcinogenesis, one could hypothesize that a p53 mutation might occasionally take place in a colorectal carcinoma precursor lesion before morphologically recognizable polypoid adenoma elements have formed or reached a significant size. Given the evidence from previous studies showing a relative lack of K-ras mutations in de novo carcinomas, 7 the important determinant may be that the p53 mutation occurs before, rather than after, the K-ras mutation. Thus, the de novo carcinoma that we see histologically is the result of a transformation to an invasive phenotype before the polypoid adenoma, which usually precedes this step, has had a chance to develop. A good candidate as a precursor lesion for the de novo carcinoma is the flat adenoma, which not only has a similar, inconspicuous endoscopic appearance, 23 but also has been shown to have infrequent K-ras mutations. 26 Although this argues against a “de novo” pathway in the sense of a lack of stepwise progression, it does support the concept that, in agreement with the Japanese clinicopathological literature, colorectal carcinomas can develop in an endoscopically inconspicuous manner due to the lack of an association with a recognizable polypoid adenoma. In the future this concept could have important implications for the endoscopic prevention of advanced colorectal carcinomas.
Acknowledgments
The authors thank Ms. Sabine Scheel for excellent technical assistance.
Footnotes
Address reprint requests to James D. Mueller, M.D., Department of Surgery, Klinikum rechts der Isar, Technical University of Munich, Ismaninger Str. 22, D-81675 Munich, Germany. E-mail: jd.mueller@lrz.tu-muenchen.de.
Supported by a grant from the Deutsche Krebshilfe 70–1950-Mu-I.
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL: Genetic alterations during colorectal-tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 2.Winawer SJ, Zauber A, Ho M, O’Brien M, Gottlieb L, Sternberg S, Waye J, Shapiro M, Bond J, Panish J, Ackroyd F, Shike M, Kurtz R, Hornsby-Lewis L, Gerdes H, Steward E, Workgroup NPS: Prevention of colorectal cancer by colonscopic polypectomy. New Engl J Med 1993, 392:1977-1981 [DOI] [PubMed] [Google Scholar]
- 3.Kodama Y, Inokuchi K, Soejima K, Matsusaka T, Okamura T: Growth patterns and prognosis in early gastric carcinoma: superficially spreading and penetrating growth types. Cancer 1983, 51:320-326 [DOI] [PubMed] [Google Scholar]
- 4.Shimoda T, Ikegami M, Fusisaki J, Matsui T, Aizawa S, Ishikawa E: Early colorectal carcinoma with special reference to its development de novo. Cancer 1989, 64:1138-1146 [DOI] [PubMed] [Google Scholar]
- 5.Kasumi A, Kratzer GL, Takeda M: Observations of aggressive, small, flat, and depressed colon cancer: report of three cases. Surg Endosc 1995, 9:690-694 [DOI] [PubMed] [Google Scholar]
- 6.Bethke B, Walter T, Stolte M: Endoskopische polypektomie: Vergleich der Häufigkeit kolorektaler Ex-Adenoma- und De Novo-Frühkarzinome. Coloproctology 1995, 17:176-183 [Google Scholar]
- 7.Minamoto T, Sawaguchi K, Mai M, Yamashita N, Sugimura T, Esumi H: Infrequent K-ras activation in superfical type (flat) colorectal adenomas, and carcinomas. Cancer Res 1994, 54:2841-2844 [PubMed] [Google Scholar]
- 8.Mueller J, Mueller E, Hoepner I, Jutting J, Bethke B, Stolte M, Hofler H: Expression of bcl-2 and p53 in de novo and ex-adenoma colon carcinoma: a comparative immunohistochemical study. J Pathol 1996, 180:259-265 [DOI] [PubMed] [Google Scholar]
- 9.Union Internationale Contre le Cancer. TNM Atlas: An Illustrated Guide to the TNM/pTNM Classification of Malignant Tumors. Heidelberg, Springer-Verlag, 1992
- 10.Keller G, Rotter M, Vogelsang H, Bischoff P, Becker KF, Mueller J, Brauch H, Siewert JR, Hofler H: Microsatellite instability in adenocarcinomas of the upper gastrointestinal tract: relation to clinicopathological data and family history. Am J Pathol 1995, 147:593-600 [PMC free article] [PubMed] [Google Scholar]
- 11.Samowitz W, Thliveris A, Spirio L, White R: Alternatively spliced adenomatous polyposis coli (APC) gene transcripts that delete exons mutated in attentuated APC. Cancer Res 1995, 55:3732-3734 [PubMed] [Google Scholar]
- 12.Jen J, Kim H, Piantaadosi S, Liu ZF, Levitt RC, Sistonen P, Kinzler KW, Vogelstein B, Hamilton SR: Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med 1994, 331:213-221 [DOI] [PubMed] [Google Scholar]
- 13.Iino H, Fukayama M, Maeda Y, Koike M, Mori T, Takahashi T, Kikuchi-Yanoshita R, Miyaki M, Mizuno S, Watanabe S: Molecular genetics for clinical management of colorectal carcinoma: 17p, 18q, and 22q loss of heterozygosity and decreased DCC expression are corrleated with metastatic potential. Cancer 1994, 73:1324-1331 [DOI] [PubMed] [Google Scholar]
- 14.Di Vinci A, Infusini E, Peveri C, Risio M, Rossini FP, Giaretti W: Deletions at chromosome 1p by fluorescence in situ hybridization are an early event in human colorectal tumorigenesis. Gastroenterology 1996, 111:102-107 [DOI] [PubMed] [Google Scholar]
- 15.Heinen CD, Richardson D, White R, Groden J: Microsatellite instabilty in colorectal adenocarcinoma cell lines that have full-length adenomatous polyposis coli protein. Cancer Res 1995, 55:4797-4799 [PubMed] [Google Scholar]
- 16.Fujiwara Y, Emi M, Ohata H, Kato Y, Nakajima T, Mori T, Nakamura Y: Evidence for the presence of two suppressor genes on chromosome 8p for colorectal cancer. Cancer Res 1993, 53:1172-1174 [PubMed] [Google Scholar]
- 17.Kuramoto S, Oohara T: Minute cancers arising de novo in the human large intestine. Cancer 1987, 61:829-834 [DOI] [PubMed] [Google Scholar]
- 18.Bedenne L, Faivre J, Boutron MC, Piard F, Cauvin JM, Hillon P: Adenoma-carcinoma sequence or “de novo” carcinogenesis? A study of adenomatous remnants in a population-based series of large bowel cancers. Cancer 1992, 69:883-888 [DOI] [PubMed] [Google Scholar]
- 19.Morson BC: Polyps and cancer of the large bowel. Yardley JH eds. In The Gastrointestinal Tract. 1977, :pp 101-108 Williams and Wilkins, Baltimore [PubMed] [Google Scholar]
- 20.Cho KR, Vogelstein B: Genetic alterations in the adenoma-carcinoma sequence. Cancer 1992, 70:1727-1731 [DOI] [PubMed] [Google Scholar]
- 21.Wada R, Matsukuma S, Abe H, Kuwabara N, Suda K, Arakawa A, Kitamura S: Histopathological studies of superficial-type early colorectal carcinoma. Cancer 1996, 77:44-50 [DOI] [PubMed] [Google Scholar]
- 22.Colton CG, Sivak MJ: Flat adenomas and cancers. Gastrointest Endosc 1995, 42:182-184 [DOI] [PubMed] [Google Scholar]
- 23.Minamoto T, Sawaguchi K, Ohta T, Itoh T, Mai M: Superficial-type adenomas and adenocarcinomas of the colon and rectum: a comparative morphological study. Gastroenterology 1994, 106:1436-1443 [DOI] [PubMed] [Google Scholar]
- 24.Hasegawa H, Ueda M, Furukawa K, Watananbe M, Teramoto T, Mukai M: p53 gene mutations in early colorectal carcinoma: de novo vs. adenoma-carcinoma sequence. Int J Cancer 1995, 64:47-51 [DOI] [PubMed] [Google Scholar]
- 25.Aoki T, Takeda S, Yanagisawa A, Kato Y, Ajioka Y, Watanabe H, Kudo S, Nakamura Y: APC and p53 mutations in de novo colorectal adenocarcinomas. Hum Mutat 1994, 3:342-346 [DOI] [PubMed] [Google Scholar]
- 26.Fujimori T, Satonaka K, Yamamura-Idei Y, Nagasako K, Maeda S: Non-involvement of ras mutations in flat colorectal adenomas and carcinomas. Int J Cancer 1994, 57:51-55 [DOI] [PubMed] [Google Scholar]
- 27.Cho K, Vogelstein B: Supressor gene alterations in the colorectal adenoma-carcinoma sequence. J Cellular Biochem 1992, 16G:137-141 [DOI] [PubMed] [Google Scholar]
- 28.Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K, Kanamaru R, Kanegae Y, Saito I, Nakamura Y, Shiba K, Noda T: Rapid colorectal adenoma formation initiated by conditional targeting of the APC gene. Science 1997, 278:120-123 [DOI] [PubMed] [Google Scholar]
- 29.Cunningham C, Dunlop M, Bird C, Wyllie A: Deletional analysis of chromosome 8p in sporadic colorectal adenomas. Br J Cancer 1994, 70:18-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cawkwell L, Li D, Lewis FA, Martin I, Dixon MF, Quirke P: Microsatellite instability in colorectal cancer: improved assessment using fluorescent polymerase chain reaction. Gastroenterology 1995, 109:465-471 [DOI] [PubMed] [Google Scholar]
- 31.Kim H, Jen J, Vogelstein B, Hamilton SR: Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994, 145:148-156 [PMC free article] [PubMed] [Google Scholar]