

Abstract
Intron-size distributions for most multicellular (and some unicellular) eukaryotes have a sharp peak at their “minimal intron” size. Across the human population, these minimal introns exhibit an abundance of insertion-deletion polymorphisms, the effect of which is to maintain their optimal size. We argue that minimal introns affect function by enhancing the rate at which mRNA is exported from the cell nucleus.
Decades of research on the mechanisms of pre-mRNA splicing have revealed a remarkably intricate process. These complexities include exon versus intron recognition (Berget 1995), co-transcriptional splicing (Goldstrohm et al. 2001), alternative splicing (Dredge et al. 2001; Grabowski and Black 2001), exonic splicing enhancers (Blencowe 2000; Nissim-Rafinia and Kerem 2002), intronic splicing enhancers (McCullough and Berget 2000), and tight coupling between splicing and efficient mRNA export from the nucleus (Luo and Reed 1999; Zhou, et al. 2000). Given such complexity, it is not hard to imagine that different introns are processed differently, not only between species but also within species. That being the case, can we segregate intron sequences according to differences in how they are processed? If so, might these differences be reflected in the nature of the sequence polymorphisms that are found in the population? We will argue that the answer to both questions is yes.
On the surface, this is an implausible idea, because intron sequences are poorly conserved. Known splicing motifs (e.g., GT-AG, branch points) are only a few bases in size, whereas intron lengths can be hundreds of kilobases. In the transition between cold-blooded to warm-blooded vertebrates, many introns experienced a twofold increase in GC content (Bernardi 2000). The fact that intron sequence contents are so pliable is the reason why introns are often considered “junk.” On the other hand, the enzymatic degradation of the excised introns must be a significant biochemical burden for the cell, especially if most of the human genome is transcribed (Wong et al 2000, 2001). Why would the cell go to so much trouble? Why not just get rid of the introns? People who do experiments on transgenic mice have an answer, for they have long known that some introns are essential for a high level of expression (Choi et al. 1991; Palmiter et al. 1991). If introns can influence expression levels, they are certainly not junk. Where might one go to find such introns?
One of the most conspicuous features of eukaryotic genomes is that a significant fraction of the introns are often clustered around a species-specific peak at the low end of the size distribution. We call them “minimal” introns because there are none smaller. Our objective is to show that the evolutionary persistence of such an optimal intron size is owing to functional constraints. However, there are many practical difficulties. Minimal intron sequence contents are degenerate (Lim and Burge 2001). Not every intron is size constrained, and any benefits to having an optimal intron size are likely to be marginal. We reasoned that our chances of success would be best in a genome in which large introns are prevalent, like human, because evolution would have already selected those introns that need to remain small from those that do not. Because of the recent expansion in the human population (Harpending and Rogers 2000), even a slight benefit, as might be expected from a small change in the intron size, would have a high probability of being fixed in the population. One might thus expect to see an abundance of minor alleles that embody the process of intron size optimization.
Specifically, we present resequencing data on a collection of 93 minimal introns sampled across a diverse human population. The data reveal an abundance of insertion-deletion (indel) polymorphisms that are clearly trying to maintain the optimal intron size. From an analysis of the yeast expression data, we will show that minimal introns can enhance mRNA synthesis rates. In essence, we present an example of selection based on conservation of intron size, as opposed to conservation of sequence content. In fact, studies of recombination rates in Drosophila melanogaster have indicated that there are selective pressures on intron size (Carvalho and Clark 1999). Perhaps the perception that introns are junk is an artifact of an overly narrow focus on conservation of sequence content as the only signature of selection.
RESULTS
Minimal Introns Are Found in Most Multicellular Eukaryotes
The distributions for intron size in Homo sapiens, Arabidopis thaliana, D. melanogaster, and Caenorhabditis elegans are displayed in Figure 1. All of these data are based on cDNA-to-genomic alignments, not gene-prediction programs. The mean number of introns per gene is 12.1, 6.2, 4.7, and 7.7, respectively. A significant fraction of the introns is always clustered about a species-specific minimum size, reflected by the sharp “spike” in the distribution centered around a mean (±SD) intron size of 92±14, 89±12, 61±10, and 48±9 bp, respectively. The idea that introns might have a minimum size, independent of sequence content, is not new (Wieringa et al. 1984). Presumably, it is a reflection of the physical constraints imposed by the cellular machinery, and the dimensions of this machinery are species specific. Minimal introns are also observed in Mus musculus, Gallus gallus, Xenopus laevis, Fugu rubripes, and Oryza sativa, albeit with annotation data parsed from GenBank. Yeast also has minimal introns, at 92±20 and 49±11 bp, for Saccharomyces cerevisiae (Ares et al. 1999; Spingola et al. 1999) and Schizosaccharomyces pombe (Wood et al. 2002), respectively.
Figure 1.

Intron size for Homo sapiens (a), Arabidopis thaliana (b), Drosophila melanogaster (c), and Caenorhabditis elegans (d). There is always a species-specific minimum intron size, at which a significant fraction of the introns tend to cluster. This “spike” in the distribution is centered around the mean (±SD) intron sizes of 92±14, 89±12, 61±10, and 48±9 bp, respectively. For larger introns, the size distribution is highly species-specific. In the extreme case, H. sapiens, there is a broad “hump” attributable to transposon insertions inside the introns. Color indicates GC content: Red is GC-rich; blue, AT-rich.
In contrast, there is an enormous variability from species to species in the distributions for larger introns. In the most extreme case, H. sapiens, there is a broad “hump” in the intron size distribution, extending out to hundreds of kilobases. Sequence content analysis with RepeatMasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html) reveals a gradual transition, from introns with no detectible transposons, at <1 Kb, to introns with one or more transposons, at >1Kb. It is thus reasonable to make a distinction between “minor” humps owing to introns <1Kb, and “major” humps owing to introns >1Kb. Given that the absence of a major hump can be owing to acquisition biases against large sequence contigs, the only statement that we are comfortable with is that there is a major hump in H. sapiens, M. musculus, G. gallus, and X. laevis.
Minimal Introns Are Not Randomly Distributed Among Genes
Considering that so many H. sapiens introns have been expanded by transposon insertions, one has to wonder why the minimal intron peak persists. Are there benefits to the organism for maintaining some introns at an optimal size? Given the predominance of the neutral theory of evolution (Kimura 1983), we must first eliminate any neutral or semineutral explanations. Perhaps some introns were never bombarded by transposons. Even if an intron was bombarded, it could have deleted back to the minimum because of the mutational bias for deletions over insertions, which is well known from comparisons of processed pseudogenes with their functional paralogs (Ophir and Graur 1997; Petrov 2001). Combined with the selection against introns too small for the splicing machinery, it would appear that persistence of minimal introns can be explained, without using any special functional constraints. However, something else must also be going on, as there is a glaring inconsistency in the data.
A distinguishing characteristic of the above-mentioned processes is that they do not favor any particular intron or gene. Therefore, if minimal introns are a fraction fm of the total, and if there are R introns per gene, the probability that a gene has a minimal intron would be 1 −(1 − fm)R. More precisely, we define a minimal intron as anything that lies within three standard deviations of the optimum, which in H. sapiens amounts to 13.6% of the introns. Because fm varies with GC content, we compute fm in four groups (similar results are obtained with eight groups) based on GC content in 10-Kb windows at each end of the gene. We then integrate over all observed Rs. The computation is performed on 882 genes from a previous analysis (Wong et al. 2000), containing every gene with a cDNA sequence that could be aligned in its entirety to finished genomic sequence. The neutral expectation is that minimal introns will be found in 56.2% of the genes, but the reality is 44.4%. The difference is statistically significant, P(binomial)=5 × 10−13, which means that minimal introns tend to cluster in certain genes.
In our four primary data sets—H. sapiens, A. thaliana, D. melanogaster, and C. elegans—the magnitude of the difference between the observed and expected number of genes with at least one minimal intron is −11.7%, −2.0%, −6.5%, and −4.7%, respectively. Evidently, the nonrandom distribution of minimal introns among genes is most readily observable in those species with a greater number of extremely large introns. Assuming that these introns are the result of transposon bombardment, this implies that transposon activity acts as a probe of how sensitive each gene is to the presence of minimal introns. Without significant transposon activity over the evolutionary history of a species, minimal introns remain randomly distributed. Thus, the genome we should resequence is H. sapiens, because evolution has already separated those introns that need to remain small from those that do not.
Minimal Introns Are Full of Indel Polymorphisms
According to Kimura (1993), “polymorphism is just a transient phase of molecular evolution.” We reasoned that, if evolution is really trying to maintain some species-specific minimal intron size, it might be possible to catch this process in action from an analysis of minimal intron polymorphisms across a diverse population. Without further justification, we will let the data speak for themselves.
Our resequencing efforts were focused on introns with sizes close to the human optimum of 92±14 bp. We resequenced 93 of these introns in a population of diverse ethnicity (Collins et al. 1998), over an average of 45.7 individuals (91.4 chromosomes). These introns were small, so there were no transposons in them. Their mean (±SD) size was 94±14 bp. To minimize the potential biases arising from differences in mutation and recombination rates, most of which are correlated with local GC content, we selected introns that span the full range of GC content, as depicted in Figure 2. We identified 42 polymorphic sites in all, 30 single-base substitutions and 12 indels, with the indels in nine different introns. To compare our results with the published data, we adjusted for variations in sample depths and sequenced lengths. If K polymorphic sites are found in a region of length L after sequencing n chromosomes, the commonly used population genetics parameter (Cargill et al. 1999; Halushka et al. 1999) is the normalized number of variant sites,
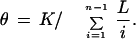 |
We decompose θ into separate components, θ(subst) = 6.75 × 10−4 for substitutions, and θ(indel) = 2.70 × 10−4 for indels. Strikingly, this substitution rate is not significantly different from the substitution rate of 7.51 × 10−4 reported for the human genome; Sachidanandam et al. 2001). It means that intron sequence content is not what was being conserved.
Figure 2.
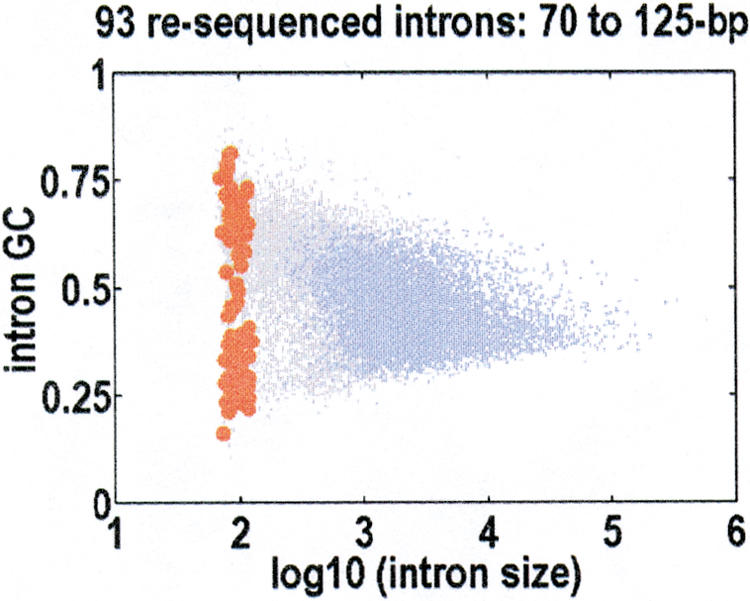
Resequenced introns, shown against the joint distribution for intron size and GC content. Introns with a detectible transposon are colored blue; introns with no detectible transposons, gray. The 93 introns that we resequenced are red. They are selected to sample the full range of GC contents, and their mean size is 94±14 bp.
However, 28.6% of our minimal introns polymorphisms were indels, which is significantly more than usual. To make this point, we need a background rate for human indels. Such a rate is not readily available, as most large-scale polymorphism discovery projects are focused on the easier-to-genotype substitution polymorphisms. Many of the putative indels are in poly-N tracts, where N is any nucleotide, and these are usually caused by sequencing errors. For example, 13% of chromosome 22 polymorphisms were indels, but this ratio was only 4% when poly-N tracts were ignored (Mullikin et al. 2000). We note that in our minimal intron indels, the longest poly-N tract was a run of only nine Gs, as shown in Table 1. For a background rate, we used the data from the Environmental Genome Project (http://www.genome.washington.edu/projects/egpsnps). These data focus on introns of every size but are restricted to the first few hundred bases flanking the exons, much like our minimal intron data, which also never stray far from the exons. Averaged over 90 genes, 7.9% of 392 intron polymorphisms were indels. Taking 7.9% as the null hypothesis, the observation of 28.6% indel polymorphisms in minimal introns is statistically significant, with P(binomial) = 6 × 10−5.
Table 1.
Insertion-deletion (indel) polymorphism summary
| L(intron) | dL | f(minor) | GC(gene) | GC(intron) | Primates | intron sequence |
|---|---|---|---|---|---|---|
| 73 | 1 | 0.033 | 0.387 | 0.164 | 95–99% | gtaatttaaaacttaaaattatttatttgattgtatttttattcatgtgctt aaagaattttTcttttttgtag |
| 80 | 1 | 0.054 | 0.595 | 0.787 | N/A | gtgagtcccagggtggggctggggaccgtgggacGggggggggtcccagccc tgccctcacgcccaccccaccgcccccag |
| 81 | 1 | 0.034 | 0.595 | 0.778 | N/A | gtggggcggggcccaggcggggaGgggggcccacgcagcggagcagccccaa catcccgcggccatctcccacccccaacag |
| 90 | 1 | 0.022 | 0.595 | 0.700 | N/A | gtgagtgggcaggacaggggcctggggtaggggacagcaagtgaCccccccc tccacagcccagtctgacccaccccttccgtggccgcag |
| 101 | −3 | 0.011 | 0.361 | 0.317 | 88–100% | gtaagaaagcaggtgtctgcaaaaagtcatgtatcgatttattgtttgtaat gatacAGTagtatagcagataactaagacatattttcttgaatttgcag |
| 120 | −3 | 0.021 | 0.516 | 0.292 | 100% | gtattttgtcactcttgaaagtttttattgggtaagaggttcatgccctttg |
| −4 | 0.010 | tcctcattttTTCttcttgttattttatcTTTAtttaCTTTTtccacttca | ||||
| −5 | 0.010 | tgTtttttttcctttag | ||||
| −1 | 0.010 | |||||
| 125 | −1 | 0.021 | 0.411 | 0.376 | 92–98% | gtaaatgTtctcctctttgttcaactcttaagtttcacatccagaagtttcat acactgacaagttgtggctttgatctggtttttgcgtaaccttaaatatga ctttttttttccccaccccag |
| 79 | −2 | 0.354 | 0.374 | 0.234 | 100% | gtagtaaattacttaaattcaatttttccttgaaatAAgtgtgattagtaac ccattattatttcctttttattttcag |
| 81 | −1 | 0.375 | 0.516 | 0.370 | 100% | gtaggaagagtgggagttttgcaaatggacaacTtaaagatggggaagaga atcaaactacacttttttccttttttctag |
From left to right, we list the intron size, the size change with respect to the major allele, the minor allele frequency, the gene level GC content, the intron level GC content, the degree of conservation relative to their primate orthologs, and lastly, the intron sequence itself with the indel in uppercase. A total of 42 polymorphic sites were identified. There were 30 single-base substitutions and 12 indels, at normalized rates of θ(subst) = 6.75 × 10−4 and θ(indel) = 2.70 × 10−4.
The observed indels lie in two distinct clusters. There are 10 rare indels of minor allele frequency f < 0.06, plus two common indels of minor allele frequency f > 0.35. The direction of the intron size change, relative to the major allele, is shown in Figure 3. All the rare indels drive the introns back toward their optimal size of 92 bp. The exceptions are the two common indels, which likely arose from different population dynamics. We further note that the probability of 10 indels in a row with the correct sign, under a null hypothesis that the sign is random, is 1 × 10−3. To confirm that the major allele is the ancestral allele, we resequenced these indel-containing introns in a panel of 10 primates, ranging from chimpanzees to lemurs. Our polymerase chain reaction primers failed on the three most GC-rich introns, but in the other introns, the major allele agreed with the primate orthologs. The sole exception was in the most AT-rich intron, in which both human alleles were observed in different primates.
Figure 3.
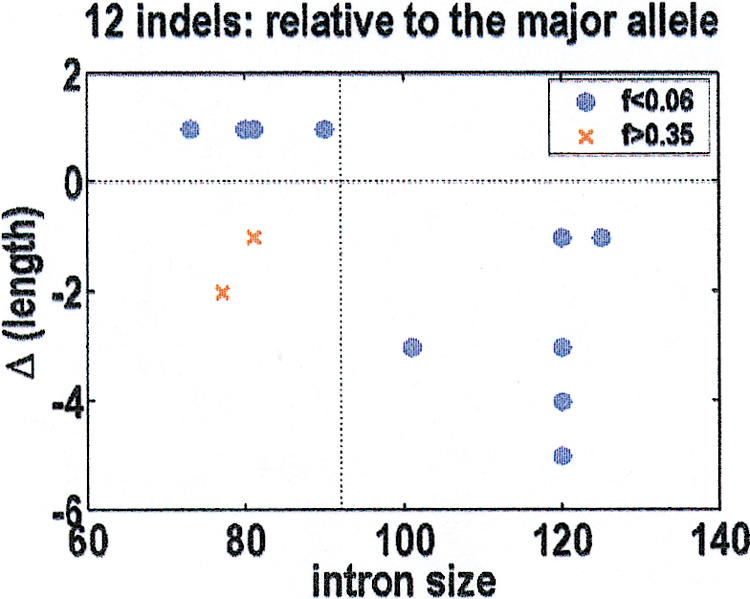
Insertion-deletion (indel) direction, relative to the major allele, as a function of intron size. For the 10 rare indels of minor allele frequency f < 0.06 (blue), the resultant size changes drive the introns back to their optimal size of 92 bp. The two common indels with f > 0.35 (red) are the only exceptions to this rule, presumably because they arose from a different population dynamics.
Minimal Introns Can Enhance the Export of Spliced mRNAs
These data indicate that at least for some genes, the presence of a minimal intron can be beneficial. Transgenic mice experiments (Choi et al. 1991; Palmiter et al. 1991) have long shown that some introns can affect expression levels. The most current explanation (Luo and Reed 1999; Zhou et al. 2000) is that “splicing generates a specific isolable complex that promotes rapid and efficient mRNA export.” Thus, our conjecture is that minimal introns can affect mRNA maturation by coupling more efficiently to the biochemically linked machineries of splicing and export, thereby increasing the rate at which mRNA is exported from the cell nucleus.
Support for this conjecture can be found in the yeast expression data (Holstege et al. 1998). One must be careful not to mix up S. cerevisiae and S. pombe, because their minimal intron peaks are at very different sizes, and their genomes contain different sets of splicing-related proteins (Kaufer and Potashkin 2000). For this purpose, S. cerevisiae is more appealing because, with the 229 of the 6188 genes that do have introns, there is generally only one intron per gene. Moreover, there is a striking dichotomy in the types of introns found in different types of genes. Ribosomal-protein genes have nonminimal introns, but nonribosomal genes have minimal introns. As we show in Table 2, mRNA synthesis rates were 3.4 times higher in nonribosomal genes with minimal introns than in nonribosomal genes without introns. Ribosomal-protein genes with and without nonminimal introns showed no such differences in mRNA synthesis rates. Although there may be other explanations for this observation besides mRNA export, these data are not inconsistent with our conjecture.
Table 2.
Saccharomyces cerevisiae expression data, adapted from Holstege et al. (1998)
| Nonribosomal | Ribosomal-protein | |
|---|---|---|
| No introns | 5.17 | 91.9 |
| With intron | 17.6 | 91.7 |
| Minimal intron | Nonminimal intron |
We classify the genes in two ways: based (1) on whether they are ribosomal-protein or nonribosomal genes and (2) on whether or not they have any introns. Ribosomal-protein genes tend to have nonminimal introns, and nonribosomal genes tend to have minimal introns. The 2 × 2 grid shows the averaged mRNA synthesis rate for those genes in each of the four categories.
Additional support for our conjecture is observed in Drosophila populations with a 66-bp intron presence-absence polymorphism in the jingwei (jgw) gene. Absence of this minimal intron reduces the expression level by almost a factor of two (Llopart et al. 2002).
DISCUSSION
The general understanding is that many mRNAs are actively transported out of the nucleus, not passively diffused. Incompletely processed mRNAs are poor substrates for this export machinery, indicating that mRNA export is a type of quality control to ensure that only functional mRNAs reach the cytoplasm (Cullen 2000). We envision a competition to get out of the cell nucleus, with at least three different export paths: one each for mRNAs with no introns, mRNAs with minimal introns, and mRNAs with nonminimal introns. Perhaps minimal introns function as “routing” tags that define a more secure export path. Comparisons between species might not be simple. Particular export paths may not be present in some species, and orthologous genes need not use the same export path. For example, in Encephalitozoon cuniculi (Katinka et al. 2001) and in the nucleomorph chromosomes of Guillardia theta (Douglas et al. 2001), all introns are minimal (23 to 52 bp in E. cuniculi and 42 to 52 bp in G. theta), but they are in ribosomal-protein genes, the opposite of the situation for S. cerevisiae.
Our conclusion is that those genes that are reliant on the improvement in the rate of mRNA export that having minimal introns provide would be more resistant to intron expansion. Furthermore, any intron that drifts away from this optimum will be returned to it at the first opportunity. Assuming this is the correct explanation, it is difficult to see how such a complicated system of interacting molecules could ever be reconstituted in a typical in vitro experiment. What we did, in effect, was let evolution perform the in vivo experiments for us and then query the results through a statistical analysis of the extant human polymorphisms. The interesting question is whether or not this methodology can be applied to other degenerate sequences with functional significance, such as promoter motifs associated with transcription regulation.
METHODS
We constructed high-quality databases of intron sequences, based exclusively on cDNA-to-genomic sequence alignments (Wong et al. 2000), for all of the multicellular eukaryotes for which a significant fraction of the genome had been finished. Resequencing was performed on the National Human Genome Research Institute (NHGRI)/Coriell Human Diversity Panel, which is a representation of all of the major ethnicities, including Northern European, Chinese, Indo-Pakistani, African American, Middle Eastern, Southwestern American Indian, Japanese, Mexican, and Puerto Rican (Collins et al. 1998). For ancestral alleles, we sequenced a primate panel from Coriell, which has chimpanzee, pigmy chimpanzee, lowland gorilla, orangutan, rhesus macaque, pig-tailed macaque, red-bellied tamarin, woolly monkey, black-handed spider monkey, and ring-tailed lemur. Polymerase chain reaction primers were designed from exon sequences flanking the selected introns. Sequencing was performed with dye-terminator chemistry on capillary sequencers. Every polymorphism, particularly the indels, was confirmed by visual inspection of the sequence traces. SNPs were submitted to GenBank/dbSNP with the handle UWGC (batches 2.12.2001.1 to 2.12.2001.4).
WEB SITE REFERENCES
http://ftp.genome.washington.edu/RM/RepeatMasker.html; The RepeatMasker program is available at this site.
http://www.genome.washington.edu/projects/egpsnps; Data from the Environmental Genome Project.
Acknowledgments
We thank Maynard Olson, Lars Bolund, and Changqing Zeng for comments and suggestions. This analysis was partially supported by a grant from the National Institute of Environmental Health Sciences (1 RO1 ES09909).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL junyu@u.washington.edu; FAX (206) 685-7344.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.224602.
REFERENCES
- Ares M, Jr, Grate L, Pauling MH. A handful of intron-containing genes produces the lion's share of yeast mRNA. RNA. 1999;5:1138–1139. doi: 10.1017/s1355838299991379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ. Exonic splicing enhancers: Mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG. Intron size and natural selection. Nature. 1999;401:344. doi: 10.1038/43827. [DOI] [PubMed] [Google Scholar]
- Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Nuclear RNA export pathways. Mol Cell Biol. 2000;20:4181–4187. doi: 10.1128/mcb.20.12.4181-4187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S, Zauner S, Fraunholz M, Beaton M, Penny S, Deng LT, Wu X, Reith M, Cavalier-Smith T, Maier UG. The highly reduced genome of an enslaved algal nucleus. Nature. 2001;410:1091–1096. doi: 10.1038/35074092. [DOI] [PubMed] [Google Scholar]
- Dredge BK, Polydorides AD, Darnell RB. The splice of life: Alternative splicing and neurological disease. Nat Rev Neurosci. 2001;2:43–50. doi: 10.1038/35049061. [DOI] [PubMed] [Google Scholar]
- Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Co-transcriptional splicing of pre-messenger RNAs: Considerations for the mechanism of alternative splicing. Gene. 2001;277:31–47. doi: 10.1016/s0378-1119(01)00695-3. [DOI] [PubMed] [Google Scholar]
- Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Prog Neurobiol. 2001;65:289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- Harpending H, Rogers A. Genetic perspectives on human origins and differentiation. Annu Rev Genomics Hum Genet. 2000;1:361–385. doi: 10.1146/annurev.genom.1.1.361. [DOI] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature. 2001;414:450–453. doi: 10.1038/35106579. [DOI] [PubMed] [Google Scholar]
- Kaufer NF, Potashkin J. Analysis of the splicing machinery in fission yeast: A comparison with budding yeast and mammals. Nucleic Acids Res. 2000;28:3003–3010. doi: 10.1093/nar/28.16.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge, UK: Cambridge University Press; 1983. [Google Scholar]
- Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart A, Comeron JM, Brunet FG, Lachaise D, Long M. Intron presence-absence polymorphism in Drosophila driven by positive Darwinian selection. Proc Natl Acad Sci. 2002;99:8121–8126. doi: 10.1073/pnas.122570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MJ, Reed R. Splicing is required for rapid and efficient mRNA export in metazoans. Proc Natl Acad Sci. 1999;96:14937–14942. doi: 10.1073/pnas.96.26.14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough AJ, Berget SM. An intronic splicing enhancer binds U1 snRNPs to enhance splicing and select 5′ splice sites. Mol Cell Biol. 2000;20:9225–9235. doi: 10.1128/mcb.20.24.9225-9235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullikin JC, Hunt SE, Cole CG, Mortimore BJ, Rice CM, Burton J, Matthews LH, Pavitt R, Plumb RW, Sims SK, et al. An SNP map of human chromosome 22. Nature. 2000;407:516–520. doi: 10.1038/35035089. [DOI] [PubMed] [Google Scholar]
- Nissim-Rafinia M, Kerem B. Splicing regulation as a potential genetic modifier. Trends Genet. 2002;18:123–127. doi: 10.1016/s0168-9525(01)02619-1. [DOI] [PubMed] [Google Scholar]
- Ophir R, Graur D. Patterns and rates of indel evolution in processed pseudogenes from humans and murids. Gene. 1997;205:191–202. doi: 10.1016/s0378-1119(97)00398-3. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Sandgren EP, Avarbock MR, Allen DD, Brinster RL. Heterologous introns can enhance expression of transgenes in mice. Proc Natl Acad Sci. 1991;88:478–482. doi: 10.1073/pnas.88.2.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA. Evolution of genome size: new approaches to an old problem. Trends Genet. 2001;17:23–28. doi: 10.1016/s0168-9525(00)02157-0. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Spingola M, Grate L, Haussler D, Ares M., Jr Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA. 1999;5:221–234. doi: 10.1017/s1355838299981682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieringa B, Hofer E, Weissmann C. A minimal intron length but no specific internal sequence is required for splicing the large rabbit β-globin intron. Cell. 1984;37:915–925. doi: 10.1016/0092-8674(84)90426-4. [DOI] [PubMed] [Google Scholar]
- Wong GKS, Passey DA, Huang YZ, Yang Z, Yu J. Is “junk” DNA mostly intron DNA? Genome Res. 2000;10:1672–1678. doi: 10.1101/gr.148900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GKS, Passey DA, Yu J. Most of the human genome is transcribed. Genome Res. 2001;11:1975–1977. doi: 10.1101/gr.202401. [DOI] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Luo MJ, Strasser K, Katahira J, Hurt E, Reed R. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]