

Abstract
Arsenic ranks as the number one toxic environmental contaminant. In humans, arsenic exposure is associated with various forms of cancer, cardiovascular and skin diseases, neuropathies of the central nervous system, and genotoxic and immunotoxic effects. Although a well recognized human carcinogen, arsenic itself is not a potent mutagen and has been thought to act through epigenetic mechanisms that modify DNA methylation patterns, perhaps in conjunction with DNA-damaging agents. To develop preliminary support for a more thorough examination of this hypothesis, we have measured the effect of submicromolar and low-micromolar concentrations of arsenite on the methylation status of DNA and the biochemical reactions that regulate it. We find that arsenic causes the depletion of S-adenosylmethionine, the main cellular methyl donor, and represses the expression of the DNA methyltransferase genes DNMT1 and DNMT3A. Possibly as a consequence of these two complementary mechanisms, long-term exposure to arsenic results in DNA hypomethylation.
Keywords: Arsenic, S-adenosylmethionine, DNA methyltransferases, methyl-Cytosine
INTRODUCTION
Arsenic, a metalloid released from both natural and anthropogenic sources, ranks as the number one toxic agent in the ATSDR Priority List of Hazardous Substances[1]. Chronic low-dose arsenic exposure in humans is epidemiologically associated with an increased risk of epithelial cancers of the skin, bladder, liver, kidney, prostate and lung and with cardiovascular and skin diseases, neuropathies of the central nervous system, and genotoxic and immunotoxic effects[2, 3]. Arsenic also induces tumors of the liver, lung, ovaries and adrenal glands in adult mice exposed in utero[3, 4]. To date, the mechanism of arsenic-mediated carcinogenesis remains a subject of debate, with several lines of evidence supporting causes such as perturbation of signaling cascades, oxidative stress and chromosomal aberrations[5]. Although a known carcinogen, arsenic is not a potent mutagen by itself, but it can act as a comutagen with DNA-damaging agents. For this reason, elucidation of the mechanism of arsenic-induced carcinogenesis has remained difficult. The lack of any prominent signal transduction mechanism or satisfactory animal model has led to the extensively held belief that arsenic is an epigenetic carcinogen; however, direct evidence for this contention is scarce. We and others have observed widespread disruption of transcriptional activity following arsenic exposure[6], with extensive changes in global gene expression, suggesting that diverse regulatory mechanisms of gene expression might be affected by arsenic exposure. Arsenic has been implicated in the disruption of cellular methylation reactions, particularly cytosine methylation at CpG islands[7], which might explain the extensive gene expression changes resulting from exposure, perhaps as a consequence of epigenetic remodeling of chromatin structure. Here, we present results that support the hypothesis that competition with cellular processes for methyl donors and repression of DNA methyltransferases are two effects of arsenic exposure that lead to a significant reduction in genomic DNA methylation.
MATERIALS AND METHODS
Cells and chemical treatments
Human HaCaT keratinocytes[8] were grown in medium 199, which contains very low levels of folic acid, supplemented with 10% fetal bovine serum and 1% antibiotics (Antibiotic Antimycotic Solution, Sigma Aldrich Co.). Cells were incubated at 37°C with 5% CO2 and grown to 80 – 90% confluence before treatment. Aqueous solutions of NaAsO2 (hereafter referred to as arsenite) (Sigma) were prepared as 1000X stocks. Cultures were treated with arsenite or control vehicle at the concentrations indicated in each individual experiment.
Quantification of total cellular SAM and SAH content
For the determination of S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) cells were treated with 25 μM arsenite for 1 h and 24 h and harvested in 0.4 M perchloric acid/PBS. SAM and SAH were separated from other cellular constituents by HPLC using a GeminiTM C18 250 X 3 mm (Phenomenex) column and a mobile phase gradient starting at 84% mobile phase A (8 mM Na Octanesulfonic acid/ 50 mM NaH2PO4/5% MeOH/pH 3.0). and 16% mobile phase B (5% Buffer A / 95% MeOH). At a flow rate of 1ml/minute the initial ratio of mobile phase A:B was maintained for 5 minutes followed by an increase in mobile phase B to 39% over 30 seconds that was maintained for 15 minutes. Concentrations of eluted SAM and SAH were determined using their absorbance at 254 nm in comparison to known standards. SAM concentrations were normalized to input cell numbers,
Quantification of methyl-dCMP content
To measure genomic cytosine methylation, HaCaT cells were cultured for 10 serial passages in medium 199 containing 0.2 μM arsenite while control cells were similarly grown for 10 passages in medium in the absence of arsenite. At each passage, 1X106 cells were replated in a 15-cm plate and the total number of cells collected per passage was recorded. For a positive demethylation control, cells were grown for one passage in medium containing 1 μM 5-deoxyazacytidine. The content of DNA methyl-dCMP was measure as described by others[9]. Briefly, Cells were harvested in TNE lysis buffer (20 mM Tris-HCl pH 7.5, 10 mM EDTA, 0.3 M NaCl) and lysed by the addition SDS to a final concentration of 0.5% (w/v). After proteinase K digestion, total genomic DNA was obtained by phenol extraction and isopropanol precipitation. Contaminating RNA was removed by incubation with RNase A (100 μg/ml) and RNAse T1 (2000 U/ml) for 2 hours at 37°C. After a second phenol extraction and DNA precipitation, recovered DNA was resuspend in 200 μl deoxyribonuclease I (DNase I) digestion buffer (10 mM Tris-HCl, pH 7.2, 0.1 mM EDTA, 4 mM MgCl2) and DNA was hydrolized with 10 U/100 μl DNase I for 14 hours at 37°C. DNA was further digested by incubating samples for 7 hours at 37°C in the presence of 50 μg/ml nuclease P1, 2 volumes of 30 mM sodium acetate (pH 5.2) and 1 mM zinc sulfate. Proteins were removed and DNA bases were recovered by a final chloroform:isoamyl alcohol (24:1) extraction and ethanol precipitation. Recovered DNA was resuspend in 100 μl ddH2O. Hydrolized DNA was analyzed for methyl-dCMP content by isocratic reverse-phase HPLC separation of individual deoxyribonucleotide monophosphates after dilution to 0.5 μg/μl for separation using a GeminiTM C18 250 X 3 mm (Phenomenex) column with a mobile phase consisting of 50 mM ammonium orthophosphate, pH 4.1.at a flow rate of 1 ml/minute. Individual dNMPs were detected at 278 nm and their peak height was normalized to the dAMP peak area (loading control). Amount of methyl-CMP present in each sample was expressed relative to the total dCMP present (methyl-dCMP + dCMP) and expressed relative to the untreated control levels.
RNA isolation and real-time RT-PCR
Total RNA was extracted using NucleoSpin RNA II columns (Macherey-Nagel) according to the manufacturer’s instructions. cDNA was synthesized by reverse transcription of 1 μg total RNA in a total volume of 20 μl containing 1X reverse transcriptase buffer (Invitrogen), 25 μg/ml oligo (dT)12–18 (Invitrogen), 0.5 mM dNTP mix (GeneChoice), 10 mM dithiothreitol (Invitrogen), 20 U of RNase inhibitor (RNasin®, Promega), and 100 U of SuperScript™ II reverse transcriptase (Invitrogen). Samples were denatured and annealed to the (dT)12–18 deoxyribonucleotide primer for 10 min at 70°C, and reverse-transcribed for 1 h at 42ºC. Before amplification, the reverse transcriptase was inactivated by heating to 70ºC for 15 min, and RNA was hydrolyzed by incubation with 2 U RNase H (Invitrogen) at 37°C for 20 min. The resulting cDNA products were diluted in a final volume of 200 μl and a 2-μl aliquot was used as template for subsequent quantification by real-time PCR amplification. Quantitative real-time PCR was performed in a 25 μl reaction mixture containing 1X BD QTaq polymerase reaction mix (BD Biosciences), 1X SybrGreen (Invitrogen) as a marker of DNA amplification, and 0.1 μM of each primer. Products were amplified with human DNMT1 primers (forward: 5’-GCACAAACTGACCTGCTTCA-3’ and reverse: 5’-GCCTTTTCACCTCCATCAAA -3’) giving a product of 213 bp, DNMT3A primers (forward: 5’-GACAAGAATGCCACCAAAGC-3’ and reverse: 5’-CGTCTCCGAACCACATGAC-3’) giving a product of 190 bp, and HMOX-1 primers (forward: 5’-CTCAAACCTCCAAAAGCC3-3’ and reverse: 5’-TCAAAAACCACCCCAACCC-3’) giving a product of 220 bp. Amplifications were performed using an Opticon II thermocycler (MJ research) where the reaction was heated to 95°C for 10 min and immediately cycled 35 times through a denaturing step at 95°C, an annealing step at 60°C and an elongation step at 72°C for 30 s each. Cycle threshold (Ct) of each sample was automatically determined to be the first cycle at which a significant increase in optical signal above an arbitrary baseline was detected. Amplification of β-actin cDNA in the same samples was used as an internal control for all PCR amplification reactions. Relative mRNA expression was quantified using the comparative Ct (Δ Ct) method and expressed as 2−ΔΔ. Each assay was done in triplicate and presented as the mean ± S.D.
RESULTS
Arsenic depletes cellular SAM concentrations
The cytotoxicity of arsenic is largely dependent on its oxidation state and methylation status. In many species, particularly humans, trivalent inorganic arsenic is metabolized intracellularly in a series of alternating oxidative-methylation and reduction steps, yielding a number of intermediate mono- and dimethylated products[6,7]. In both animal and cell culture model systems, arsenic is biotransformed by the transfer of a labile methyl moiety from SAM to arsenite and monomethylarsenite, yielding monomethyl- and dimethyl-arsenate, respectively. Environmental factors that interfere with the synthesis of SAM, such as folic acid deficiency, have a profound effect on cellular SAM concentrations. Conversely, environmental agents that utilize SAM, such as arsenic, can decrease SAM concentrations while simultaneously raising SAH concentrations, producing a net reduction in the SAM:SAH ratio. Thus, metabolism of arsenic has the capacity to deplete cells of SAM under conditions where arsenic availability is unlimited and SAM is utilized at a rate that exceeds its synthesis, such as during restricted folate availability.
To investigate the effect of arsenic treatment on the SAM:SAH ratio, HaCaT keratinocytes were cultured in Medium 199, which contains 1/400th the folic acid concentration of other cell culture media and can be considered as being folic acid-depleted, and were treated with 25 μM sodium arsenite for 24 hours. After exposure, SAM and SAH concentrations were measured using high pressure liquid chromatography. Arsenite treatment reduced cellular SAM levels relative to SAH levels, resulting in a 30% reduction in the SAM/SAH ratio (Fig. 1). This change was due to a drop in SAM concentration since there was no noticeable increase in SAH concentration. These results support our argument that short term exposure to arsenite can deplete SAM, which in turn may have further consequences on the methylation of DNA.
Fig. 1. Depletion of SAM by arsenic treatment.
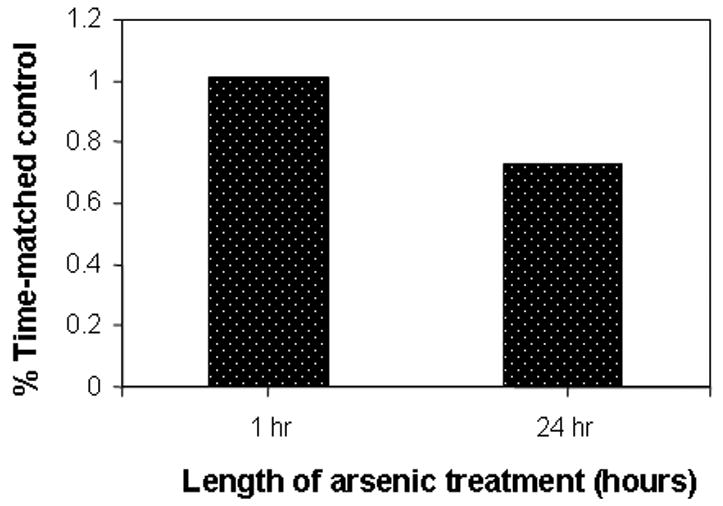
Asynchronous HaCaT cell cultures were treated with 25 μM arsenite and harvested at the indicated time as described in Materials and Methods. SAM and SAH concentrations were determined by HPLC and concentrations determined relative to known standards using their absorbance at 254 nM. Ratios are expressed relative to corresponding untreated controls.
Long-term exposure to arsenite causes genomic DNA hypomethylation
SAM is a cofactor universally required for cellular methylation reactions including DNA and histone methylation. As a consequence, extensive SAM depletion by arsenic metabolism could impose cofactor limitations on the activities of other cellular methyltransferase catalyzed reactions, including DNA and histone methyltransferases, and cause global cytosine hypomethylation, leading to epigenetic modification of gene expression. DNA is methylated predominatly at CpG dinucleotides and, although the precise patterns of DNA methylation are tissue-specific and can vary substantially throughout development[9], adult human somatic cells contain roughly 0.8% of all DNA bases as 5-methylcytosine, or about 4% of all genomic cytosines. Hence, since global DNA hypomethylation may be an immediate consequence of SAM reduction, we measured the effects of arsenic on genomic methylation. HaCaT cells were cultured for 10 serial passages in medium 199 containing 0.2 μM arsenite, a concentration corresponding to the limit of safe levels in drinking water[1] and the levels of 5-methyl-dCMP were determined after enzymatic hydrolysis of their DNA. Genomic methylation was significantly decreased by this prolonged arsenic exposure, which resulted in a 27% decrease in the level of 5-methyl-dCMP compared with cells cultured for the same number of passages in medium without arsenite (Fig. 2). This change corresponds to a loss of approximately 6X106 methylcytosine residues, sufficient to include all the CpG islands in the human genome---with an average of 200 CpG dinucleotides per gene. By comparison, the potent pharmacologic methylation inhibitor 5-azadeoxycytidine decreased the level of 5-methylcytosine by 38% in one cell doubling (Fig. 2). The concentration of arsenic producing this level of hypomethylation had little effect on the proliferation rate of the HaCaT cells, which grew for the same number of divisions and with a similar overall growth rate as the untreated controls. In addition, this arsenite concentration caused little induction of heme oxygenase 1, a sensitive marker of arsenic exposure (see Fig. 3).
Fig. 2. Genomic dCMP methylation.
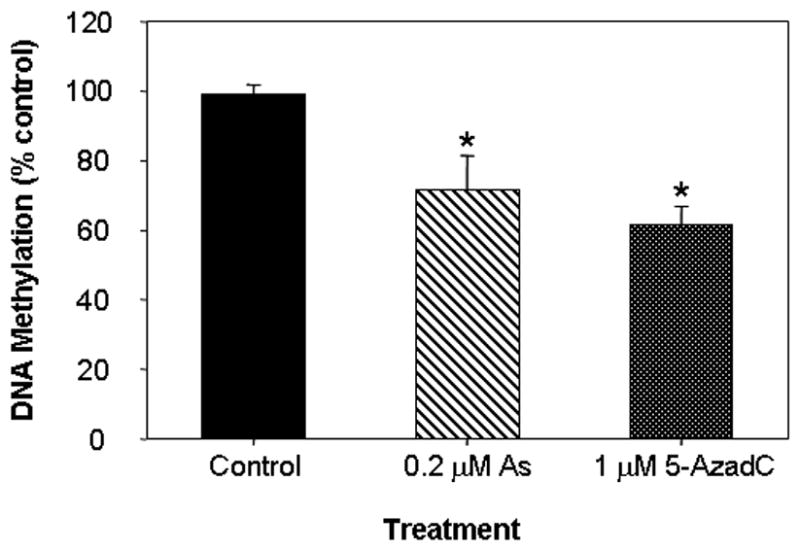
HaCaT cells were cultured with 0.2 μM arsenic for 10 passages or cultured in the presence of 5-azadeoxycytidine for 1 passage. Levels of genomic 5-methyl-dCMP were calculated as the fraction of total dCMP (5me-dCMP/(5me-dCMP+dCMP) and expressed relative to untreated control levels. The asterisk denotes significant differences from control (p<0.05) by one-way ANOVA.
Fig.3. Arsenic represses DNMT expression in asynchronous HaCaT cells.
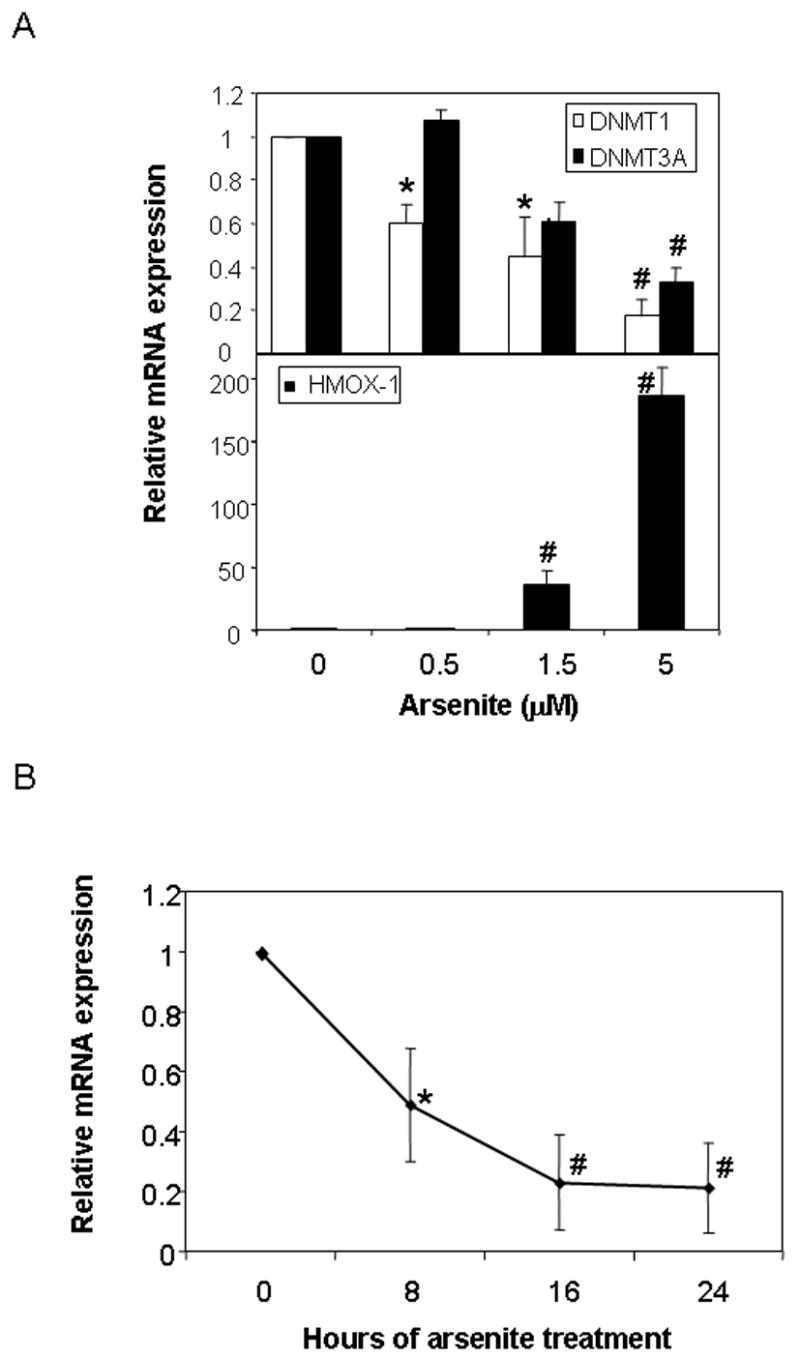
(A) Relative expression of DNMT1 and DNMT3A (upper panel) and HO-1 (lower panel) in exponentially growing HaCaT cells treated with the indicated concentration of arsenic for 72 hours. (B) Relative expression of DNMT1 following 5 μM arsenite for the indicated period of time. Gene expression was measured by real-time PCR and expression level calculated relative to β-actin expression. Values are presented relative to untreated control mRNA levels for each gene. Significant differences from control are denoted by *, for p<0.05 and #, for p<0.01, by one-way ANOVA.
Arsenite represses DNMT1 and DNMT3A mRNA expression
The DNA methyltransferase (DNMT) family of enzymes catalyzes the transfer of a methyl group to the 5’ position of cytosine using SAM as the methyl donor. In mammals, DNA methylation is catalyzed by four DNA methyltransferases, DNMT1, DNMT2, DNMT3A and DNMT3B; all of which have differing activities, substrate specificities and capacities for maintenance and de novo methylation[10,11]. Broadly, DNMT1 is localized to replication foci and is the methyltransferase predominantly responsible for methylating the newly synthesized DNA strand after replication, while the DNMT3 family appears to be predominantly associated with establishing de novo methylation patterns during embryogenesis. DNMT1 is the major methyltransferase activity in S-phase cells carrying the majority of de novo methylation activity in embryo lysates. To investigate whether the reduction in DNA methylation observed after long-term arsenite exposure could be due to repression of the DNMT genes, we measured the levels of DNMT mRNA after a 72-hour exposure of HaCaT cells to various concentrations of arsenite. Expression of either DNMT2 or DNMT3B mRNAs was undetectable in these cells, but both DNMT1 and DNMT3A mRNAs were detected at high levels in asynchronous exponentially growing cells and both were down regulated by the low-micromolar concentrations of arsenite that gave rise to a strong induction of HMOX-1 mRNA (Fig. 3A). A time course of DNMT1 repression demonstrated that the full extent of down-regulation was established by 16 hours following arsenic treatment (Fig 3B). These data suggest that decreased expression of the DNMT genes during repeated cycles of cell replication may be responsible for the hypomethylation caused by arsenite.
DISCUSSION
We show that treatment of human HaCaT keratinocytes with arsenite causes a decrease in the SAM:SAH ratio due to SAM depletion, most likely as a result of competing arsenite methylation reactions. Concomitantly, arsenite represses expression of the DNA methyltransferase genes DNMT1 and DNMT3A, coding for methyltransferase isoforms responsible for methylation of newly synthesized DNA during S-phase and embryonic development, respectively. One apparent consequence of these arsenite effects is the accumulation of globally hypomethylated DNA observed after several cell passages in the continued presence of low arsenite concentrations.
It is likely that arsenic regulates DNMT expression by interfering with the RB/E2F transcriptional axis. Expression of DNMT1 is regulated by E2F/DP1 and is S-phase specific, as needed to methylate the newly synthesized DNA[12]. Increased basal and cell cycle-specific DNMT1 expression has been reported in RB-null prostate epithelial cells in association with DNA hypermethylation and transcriptional silencing of gene promoter elements[13]. A likely explanation for this observation is that, in the absence of RB, active repression of E2F/DP1-dependent transcription does not take place, leading to the up-regulation of DNMT1. Conversely, along with repression of other cell cycle proteins, inactivation of E2F would down-regulate DNMT1 expression. Arsenic has been shown to induce RB hypophosphorylation[12], the key event in the inactivation of RB as a repressor. Hence, it is likely that, through Rb hypophosphorylation, arsenic inhibits E2F/DP1 activity and consequently DNMT1 transcription.
Several lines of evidence associate environmental factors with changes in DNA methylation and epigenetic inheritance. Chronic administration of dietary arsenic produced genome-wide hypomethylation and protooncogene-specific hypomethylation in mice[14,15]. Short-term transplacental arsenic exposure, in the absence of any other treatment, effectively produced a variety of internal tumors in adult offspring[3,4]. In these mice, arsenic exposure, at concentrations that does not greatly exceed those measured in the drinking water consumed by human populations, caused tumors of the liver, lungs ovaries and adrenal glands. The relationship between maternal exposure and cancer development in adult offspring clearly suggests the possibility of an epigenetic mode of transmission. Hypomethylation of DNA is thought to be an early epigenetic mechanism that coincides with malignant transformation and a mechanism that transmits inappropriate gene expression patterns transgenerationally. Given the central role of DNMT in maintaining chromatin structure, we conclude that exposure to physiologically relevant concentrations of arsenic mediates hypomethylation of chromatin by two complementary mechanisms: (1) competition for methyl donors and inhibition of DNA methyltransferase reactions that utilize SAM as a cofactor; and (2) repression of DNMT1 and DNMT3A gene expression.
Acknowledgments
We thank Dr. N.E Fusenig, Division of Differentiation and Carcinogenesis in Vitro, German Cancer Research Center, Heidelberg, Germany for a gift of HaCaT keratinocytes. This research was supported by NIEHS grants R01 ES10807, The NIEHS Center for Environmental Genetics grant P30 ES06096 and the NIEHS Superfund Basic Research Program grant P42 ES04908. J.F.R. is a Postdoctoral Trainee partly supported by NIEHS T32 ES07250, Environmental Carcinogenesis and Mutagenesis Training Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agency for Toxic Substances and Disease Registry. CERCLA Priority List of Hazardous Substances. Department of Health and Human Services. 2005 Available from: URL: http://www.atsdr.cdc.gov/
- 2.Waalkes MP, Ward JM, Diwan BA. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis. 2004;25:133–141. doi: 10.1093/carcin/bgg181. [DOI] [PubMed] [Google Scholar]
- 3.Waalkes MP, Liu J, Ward JM, Diwan BA. Animal models for arsenic carcinogenesis: inorganic arsenic is a transplacental carcinogen in mice. Toxicol Appl Pharmacol. 2004;198:377–384. doi: 10.1016/j.taap.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 4.Waalkes MP, Liu J, Ward JM, Diwan BA. Mechanisms underlying arsenic carcinogenesis: hypersensitivity of mice exposed to inorganic arsenic during gestation. Toxicology. 2004;198:31–38. doi: 10.1016/j.tox.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Huang C, Ke Q, Costa M, Shi X. Molecular mechanisms of arsenic carcinogenesis. Mol Cell Biochem. 2004;255:57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- 6.Kann S, Estes C, Reichard JF, Huang MY, Sartor MA, Schwemberger S, Chen Y, Dalton TP, Shertzer HG, Xia Y, Puga A. Butylhydroquinone protects cells genetically deficient in glutathione biosynthesis from arsenite-induced apoptosis without significantly changing their prooxidant status. Toxicol Sci. 2005;87:365–384. doi: 10.1093/toxsci/kfi253. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Liu J, Merrick BA, Waalkes MP. Genetic events associated with arsenic-induced malignant transformation: applications of cDNA microarray technology. Mol Carcinog. 2001;30:79–87. doi: 10.1002/1098-2744(200102)30:2<79::aid-mc1016>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsahoye BH. Measurement of genome-wide DNA cytosine-5 methylation by reversed-phase high-pressure liquid chromatography. Methods Mol Biol. 2002;200:17–27. doi: 10.1385/1-59259-182-5:017. [DOI] [PubMed] [Google Scholar]
- 10.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 11.Liu K, Wang YF, Cantemir C, Muller MT. Endogenous assays of DNA methyltransferases: Evidence for differential activities of DNMT1, DNMT2, and DNMT3 in mammalian cells in vivo. Mol Cell Biol. 2003;23:2709–2719. doi: 10.1128/MCB.23.8.2709-2719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyun PW, Hee CY, Won JC, Oh PJ, Kim K, Hyuckon IY, et al. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem Biophys Res Commun. 2003;300:230–235. doi: 10.1016/s0006-291x(02)02831-0. [DOI] [PubMed] [Google Scholar]
- 13.McCabe MT, Davis JN, Day ML. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 2005;65:3624–3632. doi: 10.1158/0008-5472.CAN-04-2158. [DOI] [PubMed] [Google Scholar]
- 14.Okoji RS, Yu RC, Maronpot RR, Froines JR. Sodium arsenite administration via drinking water increases genome-wide and Haras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis. 2002;23:777–785. doi: 10.1093/carcin/23.5.777. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Liu J, Zhao CQ, Diwan BA, Merrick BA, Waalkes MP. Association of c-myc overexpression and hyperproliferation with arsenite-induced malignant transformation. Toxicol Appl Pharmacol. 2001;175:260–268. doi: 10.1006/taap.2001.9253. [DOI] [PubMed] [Google Scholar]