

Abstract
We investigate the co-occurrence of domain families in eukaryotic proteins to predict protein cellular localization. Approximately half (300) of SMART domains form a “small-world network”, linked by no more than seven degrees of separation. Projection of the domains onto two-dimensional space reveals three clusters that correspond to cellular compartments containing secreted, cytoplasmic, and nuclear proteins. The projection method takes into account the existence of “bridging” domains, that is, instances where two domains might not occur with each other but frequently co-occur with a third domain; in such circumstances the domains are neighbors in the projection. While the majority of domains are specific to a compartment (“locale”), and hence may be used to localize any protein that contains such a domain, a small subset of domains either are present in multiple locales or occur in transmembrane proteins. Comparison with previously annotated proteins shows that SMART domain data used with this approach can predict, with 92% accuracy, the localizations of 23% of eukaryotic proteins. The coverage and accuracy will increase with improvements in domain database coverage. This method is complementary to approaches that use amino-acid composition or identify sorting sequences; these methods may be combined to further enhance prediction accuracy.
A corollary to the sequencing of a genome is the determination of the functions of its proteins. It is not yet feasible to characterize each protein directly by experiment, so instead we perform large-scale in silico analyses, using methods that assign attributes on the basis of sequence similarity and homology. These approaches implicitly assume that protein function evolves slowly relative to protein sequence, but nevertheless are a useful first prediction of function, which can be tested by experiment.
A key functional attribute of a protein is its subcellular localization. Methods such as green fluorescent protein (GFP) tagging (Sawin and Nurse 1996) and gene trap screens (Sutherland et al. 2001) are beginning to provide experimental details of localization for relatively large sets of proteins. However, there remains a need for fast, accurate, cheap, and complementary approaches that provide localization predictions for any organism. The following three classes of methods are prevalent currently:
(1) Sorting signals, which are short sequence segments that localize proteins to intra- or extracellular environments. These include (Nakai 2000) signal peptides, membrane-spanning segments, lipid anchors, nuclear import signals, and motifs that direct proteins to organelles such as mitochondria, peroxisomes, lysosomes, chloroplasts, the Golgi apparatus, and the endoplasmic reticulum.
Current methods (Nakai and Horton 1999; Drawid and Gerstein 2000) that predict subcellular localization from sorting signal data are not infallible. They rarely achieve true positive rates over 80% while simultaneously making <10% false positive or negative predictions (Menne et al. 2000; Moller et al. 2001). Furthermore, protein sequences predicted from draft genomes are often incomplete, lacking N-terminal regions that contain signal peptides (Lander et al. 2001; Venter et al. 2001), and in forthcoming years these sequences will represent a significant proportion of the eukaryotic protein databank. Consequently, we need complementary prediction methods that are independent of the presence of complete sequences and a bona fide N-terminal sequence.
(2) Amino-acid composition. Neural networks (Reinhardt and Hubbard 1998) and support-vector machines (Hua and Sun 2001) have been used to classify proteins into subcellular locales using amino-acid composition. On a test set, a prediction accuracy of just under 80% for eukaryotic proteins has been reported (Hua and Sun 2001). This approach is promising and has the advantage of very high coverage, but the test set excluded all multilocale and plant proteins. Consequently, the prediction accuracy may be lower when applied more generally. It is important to predict which proteins might shuttle between locales or are transmembrane proteins.
(3) Genomic context methods. A protein's localization to an organelle correlates with the distribution of phyla possessing its homologs (Marcotte et al. 2000); such correlations may be used for localization predictions. In this work, we develop another context method that is based on domain co-occurrences in proteins. We exploit a “rule-of-thumb” used by molecular biologists for many years, namely those proteins containing particular domains often share the same cellular localization (“locale”). For example, disulphide-rich structures, such as epidermal growth-factor–like or kringle domains, are found mostly in secreted proteins as disulphide bridges, and rarely are formed under reducing intracellular conditions, while ATPases and DNA-binding domains are found in intracellular compartments. In this study, we codify the “rule-of-thumb” into a probabilistic method that predicts proteins' locales.
Here, we define a domain family as a set of compact, structurally similar and homologous protein segments, and depending on the context, “domain” refers to either a domain family or an instance of a domain in a particular protein. The detection and classification of domains is straightforward (Ponting and Birney 2000), and it now is possible to annotate single proteins, complete proteomes (e.g., Lander et al. 2001; Venter et al. 2001), and entire sequence databases using collections of domain families such as Pfam (Bateman et al. 2000) and SMART (Schultz et al. 2000).
We consider three locales: secreted (representing extracellular and proteins in many organelles and the extracellular portions of most transmembrane proteins), cytoplasmic, and nuclear. We use data from 300 SMART domains that frequently co-occur in proteins (Ponting et al. 2000; Schultz et al. 2000) to estimate the probabilities that domains are secreted, cytoplasmic, nuclear, or multilocale. We then predict the probable locales of proteins that contain these domains.
RESULTS
SMART Domain Family Locale Probabilities
Of 523 SMART (Schultz et al. 2000) domains, we chose a subset of 329 genetically mobile domains that co-occur with at least two distinct domains in eukaryotic proteins. Evolutionarily related domain families, such as serine/threonine- and tyrosine-specific protein kinases, or the different types of epidermal growth-factor–like domains, were merged into single domains. Approximately half of these domains co-occur with over 10 other domains, and only 10% co-occur with only two or three other domains.
We analyzed the domains' patterns of co-occurrence in 57,909 eukaryotic proteins from SP-TrEMBL that contain at least one of the 329 domains. This represented ∼23% of SP-TrEMBL eukaryotic proteins at the date of the analysis. In total, 130,898 instances of domains from 329 SMART domain families were found, an average of 2.26 domains per protein. Removing repeats of the same domain in a protein left on average 1.30 domain families per protein. Twelve thousand, one hundred forty-five proteins contained domains from more than one family.
We set out to visualize the propensities of domains to be found together in proteins. A “domain projection” method was devised based on a pairwise distance measure for the co-occurrence of domain pairs (A, B). We took account of “bridging” domains, where, for instance, domains A and B are rarely or never found together yet each occur together with another domain, C. Three hundred of the domains formed a single connected component; the remaining 29 domains were not considered further. The data then were projected onto two dimensions (Fig. 1).
Figure 1.

Domain projection of 300 SMART domains, colored according to their SMART subcellular locales. The axes are the first two principal coordinates in the metric scaling projection. Open circles identify chromatin-related nuclear domains and domains that regulate Ras-like small GTPase functions. The “others” are domains classified in SMART as indiscriminate or multilocale. The labels refer to the misclassified domains in Table 2.
Manual classification based on literature surveys of the 300 SMART domain families in Figure 1 indicate 121 cytoplasmic, 76 nuclear, 70 secreted, and 33 other domains. The latter “indiscriminate” group contains repeats such as ankyrin, cystathionine β-synthase, leucine-rich, tetratricopeptide, and WD40, or domains such as fibronectin type III, immunoglobulin, IPT, transglutaminase-like, and von Willebrand factor A that are prevalent in multiple locales, as well as RNA-binding domains (e.g., double-stranded RNA-binding motif, K homology, RNA-recognition motif, S1, and S4) that are localized to both cytoplasmic and nuclear structures.
By coloring the domains in the Figure 1 according to their SMART locale, it is apparent that domains of known locale are distinguishable. There are three overlapping clusters corresponding closely to the nuclear, secreted, and cytoplasmic locales. There is almost no overlap between secreted and either cytoplasmic or nuclear locales, but there is some intersection between nuclear and cytoplasmic domains. Because the domain projection did not use information concerning the domains' locales, Figure 1 provides strong evidence that a protein's locale is predictable from its domain composition.
We identified two substructures within the Figure 1. Among the nuclear domains, those regulating chromatin structure (Jenuwein 2001) are clustered, perhaps because these domains have highly specific nuclear functions. There is also a more diffuse cluster of domains that regulate the functions of Ras-like small GTPases among the cytoplasmic domains.
Benchmarking
Three-state locale probabilities were assigned to the 300 domains. We divided the domains into two broad categories, depending on their specificity. We found 232 (77%) had a probability of over 0.9 of residing in a single locale, while 29 (10%) were strongly multilocale, having probabilities >0.33 in two locales (Table 1). Two domains, WSN and FN3, were predicted to be in both cytoplasmic and secreted proteins, whereas the remaining 27 were predicted in cytoplasmic and nuclear proteins. This suggests that evolutionary constraints on protein structure and function are more similar between cytoplasmic and nuclear environments than they are between extracellular and intracellular environments. It also reflects an extensive trafficking of molecules between the cytoplasm and the nucleus (Gorlich and Mattaj 1996).
Table 1.
Domains Predicted to Be Strongly Multilocale, with a Probability >0.33 in Two Compartments
| Cytoplasmic/Nuclear | |||
| Pc | Pn | Ps | |
|---|---|---|---|
| REC | 0.53 | 0.46 | 0 |
| HDc | 0.56 | 0.38 | 0.04 |
| GAF | 0.58 | 0.41 | 0 |
| G-alpha | 0.61 | 0.38 | 0 |
| UBCc | 0.62 | 0.37 | 0 |
| ZnF_UBP | 0.40 | 0.59 | 0 |
| AAA | 0.40 | 0.59 | 0 |
| AT_hook | 0.52 | 0.47 | 0 |
| TOP4c | 0.59 | 0.40 | 0 |
| TOP2c | 0.60 | 0.39 | 0 |
| SMR | 0.64 | 0.35 | 0 |
| KRAB | 0.33 | 0.66 | 0 |
| LER | 0.35 | 0.64 | 0 |
| ZnF_U1 | 0.37 | 0.62 | 0 |
| SANT | 0.39 | 0.60 | 0 |
| ZnF_NFX | 0.40 | 0.59 | 0 |
| RRM | 0.56 | 0.43 | 0 |
| CUE | 0.58 | 0.41 | 0 |
| PolyA | 0.33 | 0.66 | 0 |
| R3H | 0.36 | 0.64 | 0 |
| ZnF_TAZ | 0.36 | 0.63 | 0 |
| LON | 0.36 | 0.63 | 0 |
| CLH | 0.37 | 0.62 | 0 |
| GEL | 0.42 | 0.57 | 0 |
| TUDOR | 0.43 | 0.56 | 0 |
| ZnF_UBR1 | 0.46 | 0.63 | 0 |
| KH | 0.49 | 0.50 | 0 |
| Cytoplasmic/Secreted | |||
| WSN | 0.53 | 0.03 | 0.43 |
| FN3 | 0.63 | 0 | 0.36 |
Pc, Pn, Ps are the probabilities each domain is respectively cytoplasmic, nuclear, or secreted.
There were 35 (12%) domains whose most probable predicted locales conflicted with their SMART annotations (Table 2). Six of these domains were also strongly multilocale (Table 1), with a second-best locale that coincided with the SMART annotation. Twelve of these domains (4.1m, ARM, BPI2, Calx_beta, HTH_CRP, Ku78, MATH, MBT, MIR, SAND, TIR, and TSPc) were predicted as single locale with probability >0.9.
Table 2.
Domains Whose Predicted Locales Differ from Their SMART Annotations
| Domain | Prob | Reasonb |
|---|---|---|
| cytoplasmic → secreted | ||
| 4.1m | 1.00 | TM |
| Calx_beta | 0.99 | TM |
| TIR | 0.99 | TM |
| cytoplasmic → nuclear | ||
| AAAa | 0.59 | I |
| ARM | 0.99 | I |
| BIR | 0.83 | I |
| BTB | 0.74 | I |
| CASc | 0.76 | I |
| CARD | 0.75 | I |
| MIR | 0.95 | I |
| RING | 0.72 | I |
| SPRY | 0.88 | I |
| UBOX | 0.73 | I |
| VHP | 0.73 | I |
| ZnF_AN1 | 0.99 | I |
| ZnF_RBZ | 0.85 | I |
| ZnF_UBPa | 0.60 | I |
| nuclear → cytoplasmic | ||
| A1pp | 0.59 | I |
| AT_hooka | 0.53 | (i) |
| HLH | 0.76 | CI (PAS) |
| HOX | 0.77 | CI (LIM) |
| HSF | 0.73 | CI (REC) |
| HTH_CRP | 0.90 | CI (cNMP) |
| HTH_LUXR | 0.70 | CI (REC) |
| MBT | 0.95 | I |
| SFM | 0.63 | (ii) |
| SMRa | 0.64 | I |
| TOP2ca | 0.60 | CI (HATPase_c) |
| TOP4ca | 0.59 | CI (HATPase_c) |
| nuclear → secreted | ||
| Ku78 | 0.99 | CI (VWA) |
| SAND | 0.91 | (iii) |
| secreted → cytoplasmic | ||
| BPI2 | 0.98 | (iv) |
| LysM | 0.67 | TM |
| TSPc | 0.94 | CI (PDZ) |
| secreted → nuclear | ||
| MATH | 0.99 | I |
For example, cytoplasmic → nuclear means domains listed as cytoplasmic in SMART but predicted to be nuclear. Prob is the predicted locale probability of the domain. aDomain predicted as strongly multilocale. bTM: Domain occurs in transmembrane proteins. I: Indiscriminate domain for which there is literature evidence that the domain occurs in proteins found in more than one locale. CI: Domain is companion of an indiscriminate domain (listed). (i) AT_hook was wrongly predicted as cytoplasmic because of its close proximity in the domain projection plot to UBOX, an indiscriminate domain. (ii) SFM was wrongly designated as a nuclear domain in SMART. (iii) SAND was wrongly predicted as secreted because of an error in the domain architecture prediction by SMART of sequence Q9JLW9. (iv) BPI2 was predicted as nuclear rather than secreted as a result of a likely aberrant fusion with a PHD-containing sequence Q9LTR5.
Examination of the proteins containing these 35 domains showed that 18 cases are from “indiscriminate” domains that occur in two or more locales, rather than one, while a nineteenth domain (SFM) was correctly predicted by the projection method as cytoplasmic, but erroneously listed in SMART as nuclear. The method therefore predicts the locale of 284 of 300 domain families (95%) correctly. To evaluate how well the automated process works, these domains were not reassigned to their correct locales for the remainder of the analysis. Consequently, a number of avoidably incorrect predictions occurred.
Of the remaining 16 domains that were incorrectly predicted:
(1) Nine were because of their frequent co-occurrence with “indiscriminate” domains. This was, in effect, “guilt by association”. For example, the indiscriminate (cytoplasmic and nuclear) PAS, REC (CheY-like), and HATPase_c (histidine kinase-like) domains are found with HLH, HTH_LUXR, and TOP2c domains in, for instance, mammalian single-minded (O70284), algal transcriptional regulator YCF29 (P51343) and eukaryotic topoisomerase type II (O55078) sequences, respectively. Thus HLH, HTH_LUXR, and TOP2c domains were assigned as cytoplasmic when, from their well-established interactions with nuclear DNA, they are clearly situated in the nucleus.
(2) Four were from their presence in multidomain proteins that span the plasma membrane. For example, 4.1m is a cytoplasmic motif that occurs in, among others, neurexins. Here they are the only intracellular portions of transmembrane proteins containing other domains (for example, LamG and EGF) that are only found in extracellular environments.
(3) One, BPI2, is likely to have arisen from errors in sequence that give rise to aberrant fusions. BPI2 contrasts with domains such as KU, which is correctly predicted as secreted even though it is wrongly fused with a HOX domain in Caenorhabditis elegans C02F12.5 (Q11101) (Eisenhaber and Bork 1999), and FAF/UAS (cytoplasmic), another example of an aberrant fusion (SpTrEMBL code Q23467). The reason for these successes, when the assignment of BPI2 fails, is that for KU and FAF/UAS there is a significant contribution from other, accurate, domain co-occurrence information.
(4) The prediction of AT_hook was inaccurate because of its close proximity in the projection plot to an indiscriminate domain, whereas the prediction for the SAND domain was wrong because of a false positive prediction by SMART (see Table 2 legend).
Protein Locale Probabilities
Domain locale probabilities were used (see Methods, equation 4) to predict the cellular locales of 53,821 eukaryotic protein sequences from the SpTrEMBL database that contains at least one of the 300 domains. For 31,605 proteins (58%) the most likely locale had a probability of >0.9, and the protein was assigned a single locale. The remainder were assigned their two most probable locales (Fig. 2). The likely reason why protein locale predictions tend to be less definite than domain locale probabilities is that many multidomain proteins contain indiscriminate, and hence uninformative, domains that dilute locale specificity.
Figure 2.

Pie chart of protein (multi)locale assignments for the 57,909 SpTrEMBL proteins used in the domain projection showing the distribution of locale assignments. A protein was assigned to a single locale if it had a locale probability >0.9. The number of secreted/nuclear proteins is 50 (0.1%).
Only 50 proteins (0.1%) were assigned to the nuclear and secreted category (Fig. 2), consistent with the expectation that no protein possesses both nuclear and secreted functions. Furthermore, only nine of the 50 had a large (P>0.15) secreted locale probability. These represented a small number of false positive predictions where disulphide-rich (secreted) domains and cysteine-rich (nuclear) zinc fingers were predicted by SMART to overlap.
The accuracy of the protein locale predictions was assessed by comparison with the annotation-based locale assignments of Meta-A (Eisenhaber and Bork 1998, 1999). Meta-A predicts subcellular localization only for SWISSPROT sequences (a subset of the SpTrEMBL database) so we were only able to compare domain projection and Meta-A annotations for 2965 proteins annotated by both methods. In those cases where either method predicted more than one locale for a protein, an agreement was recorded if at least one locale was in common. We identified 262 proteins (8.9%) with conflicting predictions. In 1839 cases (62%), the most probable prediction was consistent. Detailed consideration of the 262 conflicting cases showed that, in 23 instances, either the SwissProt or the Meta-A annotation contradicted the literature evidence, and for a further six proteins there was evidence for multiple locales (Table 2). Consequently, protein locales are predicted with 92% apparent accuracy, which agrees well with the 95% prediction accuracy for domain locales. The domains implicated in the conflicts with Meta-A, together with example sequences, are listed in Table 3.
Table 3.
Domains Occurring in Proteins Whose Locale Predictions by Domain Projection and Meta-A Differed
| Domain | Q Example | L | Pr(L) | Meta-A |
|---|---|---|---|---|
| ArfGAPa | GLO3_YEAST | Cyt | 1.00 | Nuc |
| ARM | PLAK_XENLA | Nuc | 0.99 | Cyt |
| BPI1 | CETP_RABIT | Nuc | 0.99 | Sec |
| DnaJ | YRY1_CAEEL | Nuc | 0.87 | Sec |
| DYNc | MX1_MOUSE | Cyt | 1.00 | Nuc |
| Efhandb | FCA4_TRYBB | Cyt | 1.00 | Sec |
| EXOIIIc | YWO2_CAEEL | Nuc | 0.76 | Sec |
| FA58C | DISA_DICDI | Sec | 0.99 | Sec |
| HX | ALB2_PEA | Sec | 0.99 | Cyt |
| IPT | REL_MOUSE | Sec | 0.88 | Nuc |
| KISc | KIP1_YEAST | Cyt | 1.00 | Nuc |
| LH2 | LOXA_LYCES | Sec | 0.97 | Cyt |
| LIM | LI11_CAEEL | Cyt | 0.98 | Nuc |
| LysM | KTXA_KLULA | Cyt | 0.67 | Sec |
| MATH | TRA1_HUMAN | Nuc | 0.99 | Cyt |
| PAS | ARNT_MOUSE | Cyt | 1.00 | Nuc |
| PDZ | SPA1_MOUSE | Cyt | 0.99 | Nuc |
| Phosphatase | MCE1_MOUSE | Cyt | 0.89 | Nuc |
| PI3Kc | PIK1_YEAST | Cyt | 0.98 | Nuc |
| PLAc | PLB1_YEAST | Cyt | 1.00 | Sec |
| PP2Ac | PP11_SCHPO | Cyt | 1.00 | Nuc |
| Protein Kinase | CC2_HUMAN | Cyt | 0.94 | Nuc |
| SH2 | STA1_MOUSE | Cyt | 1.00 | Nuc |
| SH3 | MIA_BOVIN | Cyt | 1.00 | Sec |
| TGc | TGLD_RAT | Cyt | 0.91 | Sec |
| TIR | MY88_MOUSE | Sec | 0.90 | Cyt |
| TSPc | IRBP_BOVIN | Cyt | 0.94 | Sec |
| VWA | KU88_MOUSE | Sec | 1.00 | Nuc |
| ZnF_C2HC | CNBP_HUMAN | Nuc | 0.91 | Cyt |
| ZnF_C2HCd | GRP2_NICSY | Nuc | 1.00 | Sec |
| ZnF_ZZ | RF2P_DROME | Cyt | 1.00 | Nuc |
Meta-A wrongly predicts a nuclear localization for ArfGap zinc finger domains on the basis that all zinc fingers bind DNA.
Meta-A predicts a nuclear localization for centrosomal proteins. However, the centrosome is a nucleus-associated structure, rather than being a region of the nucleus.
The signal peptide predicted in SwissProt for YWO2_CAEEL is unlikely, as it is not predicted using other methods.
Predicted as secreted by Meta-A on the basis that it was originally described as a cell-wall structural protein. The six proteins mentioned in the text that have multiple locales and were predicted to have different locales by the domain projection and Meta-A methods are: SCD1_SCHPO, YB54_XENLA, YB56_XENLA, NUMB_DROME, RANG_YEAST, and SNF4_YEAST.
A comparative breakdown of the two methods' locale assignments for the 2965 proteins is given in Table 4. There is generally good agreement for secreted proteins. We investigated those instances where the domain projection method predicted proteins as either cytoplasmic/nuclear or nuclear/cytoplasmic, when Meta-A classifies them as nuclear. Of the 827 proteins classified as nuclear by Meta-A but cytoplasmic/nuclear by domain projection, 760 contain one or more of the domains HOX (536 cases), RRM (155), WD40 (143), and HLH (118). Only six of the 760 contain other domains. These four domains are either promiscuous or companions of promiscuous domains. Similarly, of the 237 proteins classified as nuclear by Meta-A but nuclear/cytoplasmic by domain projection, 168 contain one or more of the domains ZnF_C2H2 (64), AAA (50), RING (31), HATPase_c (23), or SANT (22).
Table 4.
Comparison of Domain Projection and Meta-A Classifications of 2965 Proteins that Could be Predicted by Both Methods
| Meta-A | Domain Projection | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| c | c,n | n,c | c,s | n | n,s | s | s,c | Total | |
| c | 109 | 46 | 34 | 11 | 15 | 0 | 29 | 2 | 246 |
| c+n | 10 | 30 | 48 | 6 | 5 | 0 | 0 | 0 | 99 |
| n | 156 | 827 | 257 | 14 | 573 | 4 | 8 | 1 | 1840 |
| s | 25 | 1 | 1 | 0 | 12 | 0 | 537 | 193 | 769 |
| c+s | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 10 | 11 |
| Total | 300 | 904 | 341 | 31 | 605 | 4 | 574 | 206 | 2965 |
n,c,s are abbreviations for nuclear, cytoplasmic, secreted. E.g., “n,c” means proteins predicted to be either nuclear or cytoplasmic by domain projection, but with a higher probability for the nuclear local. “c+n” means predicted as either nuclear or cytoplasmic by Meta-A, with no preference.
As a further check on the accuracy of the method, we performed a cross-validation exercise in which each of the 2965 proteins was excluded in turn from the data set, and the complete analysis repeated (i.e., domain projection and locale assignment of the excluded protein). The results were almost identical to the original analysis; in particular the number of errors was unchanged.
We also compared our predictions with those obtained from signal peptide and transmembrane helix searches and gene ontology (GO) annotations (Ashburner et al. 2000). There was moderate agreement (∼80%) with the signal peptide/TM predictions but poor agreement (∼50%) with GO annotations. However, signal peptide/TM predictions are relatively inaccurate (Menne et al. 2000; Moller et al. 2001) and there are substantial locale ambiguities in the GO annotations of domains. For example, the term “membrane” is the sole GO assignment for many different nuclear, cytoplasmic, and secreted domains (see http://golgi.ebi.ac.uk/ego/QuickGO?mode=display&entry=GO:0016020).
The “maximum” method (see Methods, equation 5) for assigning proteins to locales also worked quite well, although the assignment probabilities were more diffuse: The percentage of proteins assigned to a unique locale dropped from 58.7% to 51.4% but the number of incorrect predictions also fell from 8.9% to 6.7%. However, because this improvement was made at a considerable loss in specificity, and because the number of proteins assigned as nuclear, secreted doubled, we prefer to use the product method (see Methods, equation 4).
The complete analysis of 57,909 sequences and 329 domains (i.e., creation of dissimilarity matrix from a file of the domain composition of each protein, followed by principle coordinates projection and assignment of domains and proteins to their locales) took 35 CPU seconds on a Pentium III workstation running Debian Linux.
DISCUSSION
We have demonstrated that most mobile eukaryotic protein domains can be clustered on the basis of their domain co-occurrences, which may be projected into two-dimensional space in such a way that the subcellular localization of the domains is preserved. In general, domains only co-occur if they have the same locale, and approximately three-quarters of protein domains have a unique locale. The remainder are either indiscriminate domains, or occur in transmembrane proteins, or are artefacts caused by sequence or prediction errors. One of the method's strengths is that it is probabilistic and models multilocale domains and proteins with ease.
It is worth remarking that, rather than lying in many small, disconnected groups, over half of the SMART domains form one highly connected cluster. These domains form a small-world network, in which no pair of domains suffers more than seven degrees of separation, and the majority not more than two. This observation supports the view that a large fraction of domains are reused in many different contexts, but in a manner that tends to preserve subcellular localization.
In terms of domain reuse, our results show that the secreted and nuclear locales are almost entirely distinct (no domain has probabilities >0.25 in both locales), consistent with the hypothesis that few proteins perform functions in both locales. Secreted and cytoplasmic domains are well separated, even though many of these domains are found together in transmembrane proteins that span both locales. The distinction between cytoplasmic and nuclear domains is not so clear-cut, most likely because of the greater degree of molecular trafficking between the cytoplasm and nucleus, than between the cytoplasm and extracellular compartments. The method is not able to predict protein localization down to subcellular structures, including organelles, except for chromatin-related domains. Notwithstanding this, some domains such as ZnF_C4 and HOLI domains of nuclear receptors, which translocate between the cytoplasm and nucleus, are predicted to be only nuclear with high probability (>0.9).
Domain projection predicted the localization of 53,821 eukaryotic proteins to an accuracy of at least 92%, in a comparison with the Meta-A textual analysis algorithm. Coverage is limited mainly by the proportion of eukaryotic protein sequences (23%) containing at least one domain from the set of 300 SMART domain families. The method is likely to be of particular use in predicting the localization of proteins whose gene sequences are only known partially through expressed sequence tags or incomplete gene prediction. The majority of locales wrongly predicted by us arise from indiscriminate domains (i.e., those found in multiple locales) or else from domains that often co-occur with indiscriminate domains.
In a few cases, cellular localization was predicted incorrectly because of gene fusion sequence errors (Table 1). However, it is worth noting that not all aberrant fusions cause inaccuracies in locale assignment. For example, SpTREMBL entry Q9UNH1 represents an aberrant fusion in a mucosa-associated lymphoid tissue lymphoma (Dierlamm et al. 1999). This aberrant sequence is predicted to contain both BIR (nuclear) domains and IG-like C2-type (secreted) domains. This example demonstrates how the influence of other proteins with manifold domain combinations can compensate for a small number of erroneous sequences.
Coverage could be extended significantly, for those proteins containing domains not linked to the set of 300 domains used here in those cases where these isolated domains have a well-defined locale. Prediction accuracy is expected to improve as the number of domains in SMART and other domain databases increases. This is particularly the case for those proteins that at present are only known to contain indiscriminate domains such as HOX, where we cannot distinguish reliably between the nuclear and cytoplasmic locales. Accuracy also would improve if the secreted and intracellular portions of transmembrane proteins were treated as independent sequences. However, this approach was not pursued because, as mentioned previously, it would have been hampered by the relatively low accuracy of transmembrane segment prediction. The domain projection approach is more accurate than, but complementary to, methods based on predicting signal peptides and transmembrane helices, and to predictors based on amino-acid composition. Thus, in principle, the locale probabilities of all of these methods could be combined to produce further improvements in prediction accuracies.
Data relating to this work may be found at http://www.well.ox.ac.uk/∼rmott/DOMAINS; the localization prediction method will be implemented shortly in SMART (http://smart.embl-heidelberg.de).
METHODS
Domain Co-Occurrence Measures
We first cluster domain families represented in the SMART database by their co-occurrences in eukaryotic proteins and then investigate how the clusters correlate with locale. Here, domain co-occurrence is measured by the probability Pr(A|B) that a protein contains domains of type A given that it contains others of type B. This probability is estimated as the number of known proteins containing A and B divided by the number containing B. A symmetric pairwise dissimilarity for domains A, B then is defined as
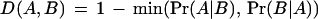 |
Thus D(A,B) is 1 if the domains never co-occur, 0 if they always occur together, and lies between otherwise. We investigated using several alternatives to this definition, namely 1 − max(Pr(A|B), Pr(B|A)), 1 − (Pr(A|B)+Pr(B|A)/2, −log((Pr(A|B) + Pr(B|A))/2). Although they produce broadly similar domain projections, we found that these measures give markedly inferior predictions of protein locale.
D(A,B) has the drawback that it is a short-range measure: Any pair of domains that never co-occur will have a dissimilarity of 1, regardless of whether or not the domains are present in proteins that have the same locales. Furthermore, D(A,B) is not a metric—it does not obey the triangle inequality, and therefore is hard to visualize by projection into a Euclidean space. However, we can create a metric d(A,B) from this dissimilarity to infer relationships between domains for which D(A,B) = 1, but which still preserves the short-range structure.
We treat the domain families as nodes in a weighted, undirected graph in which there is an edge between A, B if, and only if, D(A,B) < t, where t is a threshold value, set at 0.98. Next, we identify all connected components in the graph. In our data set, 300 domains form a single connected component, the remainder in isolated small components, which are ignored for the remainder of the analysis. Within each component we find the shortest path between every pair of nodes, that is, a sequence s of adjacent nodes [A, x1 … xn, B] connecting A, B such that
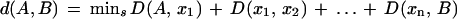 |
By construction, d(A,B) is a metric, as it is the length of a shortest path between A and B. By using either Floyd's (1962) or Dijkstra's (1959) algorithms, it is possible to compute d for all pairs of nodes in a graph of n nodes efficiently.
Domain Projection
We project the domains onto a two-dimensional Euclidean space (Fig. 1) using metric scaling (Torgeson 1958) applied to d, creating a “domain projection”. To determine whether domains that are found in the same locale are clustered, we colored the domain projection according to known SMART locales. The idea of shortest-path reconstruction followed by projection has been used previously in different contexts (Newell et al. 1995; Tenenbaum et al. 2000).
Assignment of Domain Family Locale Probabilities
We use kernel density estimation to attach locale probabilities to the domains. Throughout this section, we use the two-dimensional projected Euclidean distance between domains, which we denote by d2(A,B). Let NL1,NL2, … NLn be the list of domains with a particular SMART locale L. We then define the probability that the (possibly unlocalized) domain A is from locale L as
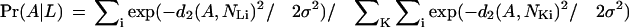 |
1 |
That is, Pr(A|L) is the sum of n normal distributions, each with variance σ2, centered on a domain in locale L. A is excluded from the sums to avoid self-effects. The standard deviation σ controls the degree of smoothing. After some experimentation, we found that σ2 = 0.025 was a good choice.
Assignment of Multidomain Protein Locale Probabilities
The probability that a protein Q, containing distinct domains AQ1, AQ2, … AQn, is in one of the three locales L is defined as
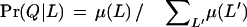 |
2 |
Here μ(L) is Πi Pr(AQi|L), the product of the locale domain-based probabilities Pr(AQi|L), taken over the number i = 1, 2, … , n of different domains. The index L‘ varies over the three locales. By considering domains repeated in a protein only once rather than by their multiplicity, we avoid an overweighting by single domain types.
We also investigated an alternative assignment method,
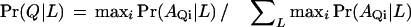 |
2* |
That is, the domain with maximum probability of locale L is taken as the evidence for the protein residing in L.
Benchmarking Domain- and Protein-Based Locale Probabilities
Domains were classified as secreted, cytoplasmic or nuclear, based on their SMART annotations derived from detailed literature searches. Locale assignments were based on experimental data for the majority of domains; exceptional cases, such as the PDZ domain in the secreted molecule interleukin-16 and the SH3 domain in the extracellular melanoma derived growth regulatory protein, were ignored. Domains that occur in multiple locales were labeled “other”.
Domain projection was benchmarked against the Meta annotation prediction of subcellular localization derived from the annotation of SwissProt (Eisenhaber and Bork 1998; Eisenhaber and Bork 1999; Bairoch and Apweiler 2000). Meta-A is a lexical analyzer that uses keywords to infer locale. Results also were compared with locale assignments of signal peptide, (Krogh et al. 2001) and transmembrane (Nielsen et al. 1997) prediction algorithms, and with the GO consortium (Ashburner et al. 2000), as applied to SMART via their mapping to InterPro (Apweiler et al. 2000).
WEB SITE REFERENCES
http://golgi.ebi.ac.uk/ego; GO web site.
http://smart.embl-heidelberg.de; SMART web site.
http://smart.ox.ac.uk; SMART web site.
http://www.well.ox.ac.uk/∼rmott/DOMAINS; author web page.
Acknowledgments
This work was funded by the Wellcome Trust (RM), Medical Research Council (CPP), and Bundesministerium for Bildung und Forschung (PB and JS).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rmott@well.ox.ac.uk; FAX +44 1865 287664.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.96802.
REFERENCES
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, et al. InterPro—an integrated documentation resource for protein families, domains and functional sites. Bioinformatics. 2000;16:1145–1150. doi: 10.1093/bioinformatics/16.12.1145. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, De Wolf-Peeters C, Hagemeijer A, Van den Berghe H, Marynen P. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. 1999;93:3601–3609. [PubMed] [Google Scholar]
- Dijkstra EW. A note on two problems in connection with graphs. Numerische Mathematik. 1959;1:269–271. [Google Scholar]
- Drawid A, Gerstein M. A Bayesian system integrating expression data with sequence patterns for localizing proteins: Comprehensive application to the yeast genome. J Mol Biol. 2000;301:1059–1075. doi: 10.1006/jmbi.2000.3968. [DOI] [PubMed] [Google Scholar]
- Eisenhaber F, Bork P. Wanted: Subcellular localization of proteins based on sequence. Trends Cell Biol. 1998;8:169–170. doi: 10.1016/s0962-8924(98)01226-4. [DOI] [PubMed] [Google Scholar]
- ————— Evaluation of human-readable annotation in biomolecular sequence databases with biological rule libraries. Bioinformatics. 1999;15:528–535. doi: 10.1093/bioinformatics/15.7.528. [DOI] [PubMed] [Google Scholar]
- Floyd RW. Algorithm 97: Shortest path. Comm ACM. 1962;5:345. [Google Scholar]
- Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci. 2000;97:12115–12120. doi: 10.1073/pnas.220399497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menne KM, Hermjakob H, Apweiler R. A comparison of signal sequence prediction methods using a test set of signal peptides. Bioinformatics. 2000;16:741–742. doi: 10.1093/bioinformatics/16.8.741. [DOI] [PubMed] [Google Scholar]
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- Nakai K. Protein sorting signals and prediction of subcellular localization. Adv Protein Chem. 2000;54:277–344. doi: 10.1016/s0065-3233(00)54009-1. [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Newell WR, Motts R, Beck S, Lehrach H. Construction of genetic maps using distance geometry. Genomics. 1995;30:59–70. doi: 10.1006/geno.1995.0009. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Birney E. Identification of domains from protein sequences. Methods Mol Biol. 2000;143:53–69. doi: 10.1385/1-59259-368-2:53. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Schultz J, Copley RR, Andrade MA, Bork P. Evolution of domain families. Adv Protein Chem. 2000;54:185–244. doi: 10.1016/s0065-3233(00)54007-8. [DOI] [PubMed] [Google Scholar]
- Reinhardt A, Hubbard T. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 1998;26:2230–2236. doi: 10.1093/nar/26.9.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Nurse P. Identification of fission yeast nuclear markers using random polypeptide fusions with green fluorescent protein. Proc Natl Acad Sci. 1996;93:15146–15151. doi: 10.1073/pnas.93.26.15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland HG, Mumford GK, Newton K, Ford LV, Farrall R, Dellaire G, Caceres JF, Bickmore WA. Large-scale identification of mammalian proteins localized to nuclear sub-compartments. Hum Mol Genet. 2001;10:1995–2011. doi: 10.1093/hmg/10.18.1995. [DOI] [PubMed] [Google Scholar]
- Tenenbaum JB, de Silva V, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–2323. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- Torgeson W. Theory and methods of scaling. New York: John Wiley & Sons; 1958. [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]