

Abstract
Exposure to high levels of inspired oxygen leads to respiratory failure and death in many animal models. Endothelial cell death is an early finding, before the onset of respiratory failure. Vascular endothelial growth factor (VEGF) is highly expressed in the lungs of adult animals. In the present study, adult Sprague-Dawley rats were exposed to >95% FiO2 for 24 or 48 hours. Northern blot analysis revealed a marked reduction in VEGF mRNA abundance by 24 hours, which decreased to less than 50% of control by 48 hours. In situ hybridization revealed that VEGF was highly expressed in distal airway epithelial cells in controls but disappeared in the oxygen-exposed animals. Immunohistochemistry and Western blot analyses demonstrated that VEGF protein was decreased at 48 hours. TUNEL staining demonstrated the presence of apoptotic cells coincident with the decline in VEGF. Abundance of VEGF receptor mRNAs (Flt-1 and KDR/Flk) decreased in the late time points of the study (48 hours), possibly secondary to the loss of endothelial cells. We speculate that VEGF functions as a survival factor in the normal adult rat lung, and its loss during hyperoxia contributes to the pathophysiology of oxygen-induced lung damage.
Supplemental oxygen therapy has marked beneficial effects in the treatment of numerous cardiorespiratory diseases, but oxygen use is limited by direct toxicity. Severe pulmonary damage from exposure to high levels of inspired oxygen has been well documented in animal models. For example, rats exposed to >95% FiO2 are asymptomatic for 48 hours, but the animals then develop pulmonary edema and respiratory failure that progresses to death within 72 to 96 hours. 1,2 Endothelial cells have been identified as early targets of hyperoxia-induced lung damage. 3 Although there has been significant focus on the role of free radicals as direct mediators of oxygen toxicity, 4 the exact mechanisms of early endothelial cell death have not been defined.
Cell death can occur as a result injury or as part of normal tissue changes. For example, apoptosis is part of normal development and is important in tissue remodeling. 5 Recently, apoptosis has been implicated in some pathological processes, including acute lung injury. 6 The regulation of apoptosis is complex. In vitro, withdrawal of a growth factor is a well recognized trigger for apoptosis in many cells, including endothelial cells, 7 and these growth factors are frequently referred to as survival factors. The potential consequence of the loss of normal levels of growth factors in vivo has not been extensively addressed.
Vascular endothelial growth factor (VEGF) is an endothelial-cell-specific mitogen and potent angiogenic factor that is highly abundant in the lung. 8 In developing lung, the pattern of expression of the VEGF receptors suggests a role in the formation of the pulmonary circulation, 9 but the function of the large reservoir of VEGF present in the normal adult lung is not known. There are five different molecular weight human isoforms of VEGF of 206, 189, 165, 145, and 121 amino acids, which are generated by alternative splicing. VEGF206 and VEGF189 are highly basic and bind heparin with great affinity, whereas VEGF121 is non-heparin-binding, acidic, and freely diffusible. The heparin-binding capabilities of VEGF165 and VEGF145 are intermediate. 10,11 In rats, only three subtypes have been identified so far having 188, 164, and 120 amino acids, respectively. The abundance of the splice variants varies in different organs, but the biological significance of this finding is not known. 12
VEGF binds to two tyrosine kinase receptors, Flt-1 and KDR (murine Flk), which are found predominantly on endothelial cells. 10 KDR/Flk mediates the endothelial mitogenic response; however, the role of Flt-1 is not clear. In the presence of KDR/Flk, Flt-1 can augment the mitogenic response to VEGF, and it has been implicated in the chemotactic responses of monocytes. 13 The receptors appear to be coordinately up-regulated with VEGF; for example, in tumors hypoxia/ischemia leads to increased expression. 14 The effects of hyperoxia on the expression of Flt-1 and KDR/Flk has not been examined.
Hypoxia has been well established as a powerful stimulus for increasing the abundance of VEGF mRNA in many tissues and cells through both transcriptional and stability pathways. 15-17 We reported that hypoxia is a potent inducer of VEGF mRNA in ovine pulmonary artery smooth muscle cells in vitro. 18 Tuder et al demonstrated up-regulation of VEGF and the VEGF receptors in rats exposed to acute and chronic hypoxia. 19 Although hypoxia is clearly an important stimulus for VEGF, hyperoxia has not been as well studied, but recent work suggests that it leads to decreased levels of VEGF. For example, exposure of newborn rats to hyperoxia resulted in decreased levels of VEGF in the retina with subsequent endothelial cell death. 20 We have previously examined rats exposed to >95% FiO2 and found that there was a significant decrease in lung VEGF mRNA as early as 24 hours, before the onset of clinical symptoms. 21
In the present study, we hypothesized that exposure to hyperoxia would decrease the expression of VEGF in the lung and, furthermore, that the loss of VEGF during hyperoxia may be an important part of the pathophysiology of hyperoxia-induced respiratory failure. We studied the effects of exposure to hyperoxia on the expression of VEGF in rat lungs, including examination of the localization of both mRNA and protein, expression of Flt-1 and KDR/Flk, and appearance of apoptosis. We found that VEGF mRNA and protein decreased during early exposure to hyperoxia. Additionally, the abundance of mRNA for Flt-1 and KDR/Flk also decreases, although not as early in the time course. Although the animals were clinically asymptomatic, apoptotic cells appeared in the lung tissue by 24 hours.
Materials and Methods
Animal Exposure to Hyperoxia
Adult male Sprague-Dawley rats (250 g) were kept in standard plastic chambers with continuous access to food and water. For oxygen exposure, chambers were placed inside an incubator with oxygen or room air flowing at 10 L/minute. Temperature and humidity were monitored and adjusted as needed. Groups of five to six rats were exposed to room air or >95% FiO2 for 24 or 48 hours. Animals were examined every 12 hours and at the end of the experimental period killed with inhalation of CO2.
Tissue Harvesting and Processing
Immediately after death, the chest was opened, the left atrium was cut, and 30 ml of normal saline was injected into the right ventricle to perfuse the pulmonary circulation free of blood. The trachea was cannulated, the lungs were inflated, and a portion of the lung was clamped, excised, and fixed in cold 4% paraformaldehyde in phosphate-buffered saline (PBS). The remaining lung was frozen in liquid nitrogen for RNA isolation. Tissues were fixed in paraformaldehyde overnight at 4°C and then dehydrated through serial ethanols of 70%, 90%, 95%, and 100% for 1 hour each. Tissues were then processed through xylene and embedded in paraffin. Five- to six-micron-thick sections were cut and placed on positively charged slides (Super Frost Plus, Fisher Scientific, Pittsburgh, PA).
Cloning of cDNA for VEGF, KDR, and Flt-1
A rat 448-bp VEGF cDNA fragment was generated by reverse transcription polymerase chain reaction (RT-PCR) using rat lung RNA as a template and previously reported primers and conditions 18,22 (upper primer, 5′-GAAGTGGTGAAGTTCATGGA-3′; lower primer, 5′TTGTCACATCTGCAAGTACG-3′).
Briefly, PCR was performed at 94°C for 4 minutes and then 45 cycles of 94°C, 52°C, and 72°C, followed by 72°C for 10 minutes. A 660-bp KDR cDNA fragment was generated using RT-PCR with primers constructed from the known sequence for human KDR. (GenBank accession number U93306) (antisense primer, 5′-TCCATTGGCCCGCTTAACGGT-3′; sense primer, 5′-CAGATCTACGTTTGAGAACCTC-3′, with RNA from human liver as a template). A 740-bp Flt-1 cDNA fragment was generated using RT-PCR with primers constructed from the known sequence for rat Flt-1 (GenBank accession number D28498) (antisense primer, 5′-CCACGACCCCCTTCTGGTTGGT-3′; sense primer, 5′-GAATCCTTCATCCTGGATTC-3′, using rat lung RNA as a template). 23 A rat VEGF188 cDNA fragment was cloned using primers identical to the 5′ end, 5′-ATATAAAGCTTGCACCCACGACAGAAGGGGAG-3′, or complementary to the 3′ end (5′-ATATAGAATTCTCACCGCCTTGGCTTGTCACA-3′ with RNA from rat lung as a template). 24 The isolated DNA fragments from the above reactions were cloned into pGEM-T vectors, and orientation of each of the generated cDNAs was confirmed by sequencing.
Probes
For Northern analyses, riboprobes were synthesized to detect VEGF, KDR/Flk, and Flt-1 mRNA expression using the constructs described above as templates and the appropriate RNA polymerase (SP6 or T7). These probes were labeled with [32P]UTP to a specific activity of 2 × 10 6 cpm/μg (UTP-800 Ci/mmol; New England Nuclear, Boston, MA). For in situ hybridization, digoxigenin-labeled sense and antisense VEGF riboprobes were generated using the DIG RNA labeling kit (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer’s specifications. For RNAse protection assays, VEGF188 and murine β-actin riboprobes were labeled with [32P]UTP as described above.
RNA Isolation and Analysis
Total RNA was isolated from frozen lung specimens according to standard techniques using a cesium chloride gradient and ultracentrifugation. 18,22 Ten micrograms of RNA per lane was run overnight into a 1% agarose/2.2 mol/L formaldehyde gel at 20 V. RNA was transferred to nitrocellulose filter (Nitro Plus, Micron Separations, Westboro, MA) in 10X SSC (1X SSC = 150 mmol/L NaCl, 15 mmol/L sodium citrate) and immobilized on the filter with ultraviolet cross-linking at 1200 J (UV Strata Linker, Stratagene, La Jolla, CA). The blots were prehybridized for a minimum of 2 hours at 42°C in 50% formamide, 250 ng/ml sheared salmon sperm DNA, 1X Denhardt’s, 50 ng/ml poly(A), 0.1% sodium dodecyl sulfate (SDS), and 5X SSC. Blots were probed at separate times for both VEGF and the VEGF receptors, KDR/Flk and Flt-1, with labeled riboprobe added to blots at a concentration of 2 × 10 6 cpm per ml of hybridization solution, and hybridized for 18 hours at 63°C. Blots were washed in 0.1X SSC and 0.1% SDS and then exposed to phosphorimager plates for 4 hours. Results were quantified using Molecular Dynamics Phosphorimager (Sunnyvale, CA) with ImageQuant software. To normalize for loading, blots were also probed for cyclophilin expression. 18,25
In Situ Hybridization
In situ hybridizations were done using previously described techniques. 22 Briefly, lung biopsy tissues were dewaxed through xylene and hydrated with decreasing concentrations of ethanol. The tissue was post-fixed in 4% paraformaldehyde in PBS for 20 minutes and treated with proteinase K (20 μg/ml in 50 mmol/L Tris, 5 mmol/L EDTA) for 7.5 minutes at 37°C. The sections were treated again with 4% paraformaldehyde in PBS and acetylated (100 mmol/L triethanolamine, 25 mmol/L acetic anhydride). After dehydration through ethanol, the sections were hybridized at 55°C for 18 hours with digoxigenin-labeled sense and antisense probes at a concentration of 10 ng/ml (∼300 ng per section). After hybridization, the slides were washed in 5X SSC, 10 mmol/L dithiothreitol at 50°C followed by 2X SSC containing 100 mmol/L dithiothreitol, 50% formamide at 65°C for 20 minutes. The sections were then treated for 30 minutes with RNAse A (20 μg/ml; Sigma Chemical Co., St. Louis, MO) in 10 mmol/L Tris, 5 mmol/L EDTA, 0.5 mol/L NaCl at 37°C. After washing in 2X SSC and 0.1X SSC for 30 minutes each at 65°C, the digoxigenin-labeled probe was detected immunohistochemically (nonradioactive detection kit, Boehringer Mannheim), and tissues sections were counterstained with Mayer’s hematoxylin.
RNAse Protection Assay
RNAse protection analyses were performed using the RPA II Kit (Ambion, Austin, TX) according to the manufacturer’s specifications. Briefly, radiolabeled VEGF188 and β-actin riboprobes were synthesized as described above, gel purified on a 5% acrylamide/8 mol/L urea gel, and eluted overnight. Samples of total RNA (10 μg) from all of the experimental animals were co-precipitated with molar excesses of riboprobes and then hybridized to completion (>16 hours) at 45°C in a hybridization buffer provided in the kit. Single-stranded RNA was removed by digestion with RNAse (RNase A+T1 combined) at a 1:100 dilution for 30 minutes at 37°C, followed by precipitation in the manufacturer’s buffer. After the pellets were resuspended in the manufacturer’s gel loading buffer, samples were denatured, and the protected species were separated on a 5% acrylamide/8 mol/L urea gel in 1X Tris-buffered ethanolamine for 3 hours at 200 V. The gels were then adhered to filter paper, exposed to the Molecular Dynamics phosphorimager, and analyzed using ImageQuant software. Molar ratios were calculated to adjust for differences in signal intensity as a result of size of the band.
RT-PCR
RT-PCR was performed to identify splice variants of VEGF using primers and conditions described above with RNA from rat lung biopsy tissue specimens of animals exposed to room air or 24 or 48 hours of >95% FiO2. The primers flank the regions containing all exons and thus should identify all splice variants that are present. In addition, a pair of L19 ribosomal protein-specific oligonucleotides primers were used as an internal standard. 20 Upper primer was 5′-GGAGAGATGAGCTTCCTGCAG-3′, and lower primer was 5′-TCACCGCCTTGGCTTGTCACA-3′.
Immunohistochemistry
Tissue sections were dewaxed through xylene, placed in 100% ethanol, and then incubated in methanol/30% hydrogen peroxide to quench endogenous peroxidase. The slides were then hydrated through graded ethanol, followed by three washes in PBS. Normal goat serum (Biogenex, San Ramon, CA) was then applied to sections for 30 minutes to block nonspecific binding. Rabbit polyclonal VEGF antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:50 was applied to tissues for 2 hours at room temperature, and normal rabbit serum was used on additional sections at the same concentration as a negative control. Tissues were washed in PBS and then treated with biotinylated anti-rabbit immunoglobulins (Biogenex) as a secondary linking antibody and labeled with peroxidase-conjugated streptavidin (Biogenex). Diaminobenzidine chromogen was used to visualize the antibody, and tissues were counterstained with Mayer’s hematoxylin for 2 minutes and then dehydrated through serial ethanols.
TUNEL Staining
Apoptosis was detected in rat lung tissues using TUNEL staining. Briefly, apoptotic fragments were detected by adding digoxigenin-labeled nucleotides with terminal deoxynucleotidyl transferase using the ApopTag kit (Oncor, Gaithersburg, MD) according to the manufacturer’s instructions. Tissues were counterstained with methyl green. Sections from rat testis were used as a positive control.
Western Blot Analysis
Frozen lung tissue was homogenized in lysis buffer (150 mmol/L NaCl, 10 mmol/L HEPES, 1 mmol/L EDTA, 0.5 mmol/L phenylmethylsulfonyl fluoride, 0.6% Nonidet P-40). Samples were sonicated for 15 seconds and then centrifuged at 3000 rpm at 4°C for 15 minutes. Supernatants were transferred to clean tubes, and total protein was determined with BCA protein assay (Pierce, Rockford, IL). Forty micrograms of protein was separated on a 12% SDS gel run at 200 V for 50 minutes, 50 mA initially, ending with 20 mA. Proteins were transferred to ECL Hybond membrane (Amersham, Aylesbury, UK) for 1 hour at 100 V. Nonspecific binding was blocked by soaking overnight at 4°C in Tris-buffered saline plus 5% nonfat dry milk, and membranes were washed twice in Tris-buffered saline containing 0.5% Tween 20. The membranes were then incubated with anti-VEGF antibody 1:200 (Santa Cruz) for 1 hour at room temperature, followed by three washes. After incubation with secondary antibody (anti-rabbit IgG horseradish peroxidase conjugate, 1:5000), immunoreactive proteins were detected using the ECL Western blotting detection system (Amersham). Three to four samples from each group were analyzed.
Data Analyses
Results for each of the Northern blots were corrected for loading and normalized to control for each experiment. Mean and SEM were calculated for each experimental group. Data from experimental groups were compared using nonparametric statistics (Mann-Whitney) with a P value of less than 0.05 considered significant.
Results
Clinical Observations of Animals
All animals were observed every 12 hours during the exposure period to either room air or supplemental oxygen. There were no signs of respiratory distress in any of the animals during these interim observation periods, and all continued to be asymptomatic at the completion of the time course.
Expression of VEGF mRNA in Rat Lungs
Northern Analysis
Total RNA from rat lung biopsy specimens was assessed by Northern analysis using a 32P-labeled VEGF riboprobe (Figure 1) ▶ . VEGF mRNA was constitutively expressed in the control rat lung with a predominant band at approximately 3.7 kb. After 24 hours of exposure to >95% FiO2, VEGF mRNA abundance was significantly decreased, and after 48 hours of exposure, mRNA abundance was less than 50% of control (Table 1 ▶ ; n = 6).
Figure 1.

Northern blot analysis of VEGF mRNA from rat lung tissue. VEGF was expressed in control tissue and significantly decreased after exposure to hyperoxia (10 μg of total mRNA per lane). C, control; 24, 24 hours >95% FiO2; 48, 48 hours >95% FiO2.
Table 1.
Abundance of VEGF and VEGF Receptor mRNA after Exposure to Hyperoxia
| Baseline | Exposure to >95% FiO2 | ||
|---|---|---|---|
| 24 hours | 48 hours | ||
| VEGF | 1 | 0.68 ± 0.07* | 0.48 ± 0.08* |
| Flt-1 | 1 | 0.84 ± 0.13 | 0.46 ± 0.06* |
| KDR/Flk | 1 | 0.85 ± 0.14 | 0.58 ± 0.07* |
RNA abundance was corrected for loading and normalized to baseline.
*P < 0.05 (n = 6).
In Situ Hybridization
To localize the expression of VEGF mRNA abundance in lung tissue after hyperoxic exposure, digoxigenin-labeled sense and antisense VEGF probes were used, and similar to the results of the Northern analyses, in situ hybridization revealed a decrease in VEGF mRNA abundance. In tissue biopsy specimens from control animals, VEGF mRNA was abundant and localized to distal epithelial cells (possibly Type II cells; Figure 2A ▶ ). In the rats exposed to 24 hours of >95%FiO2, there was a noticeable decrease in expression of VEGF mRNA (Figure 2B) ▶ , and after 48 hours of exposure to >95%FiO2, VEGF mRNA was almost undetectable (Figure 2C) ▶ . No hybridization was seen with sense probes (data not shown). Tissue was examined from four to five animals in each group.
Figure 2.
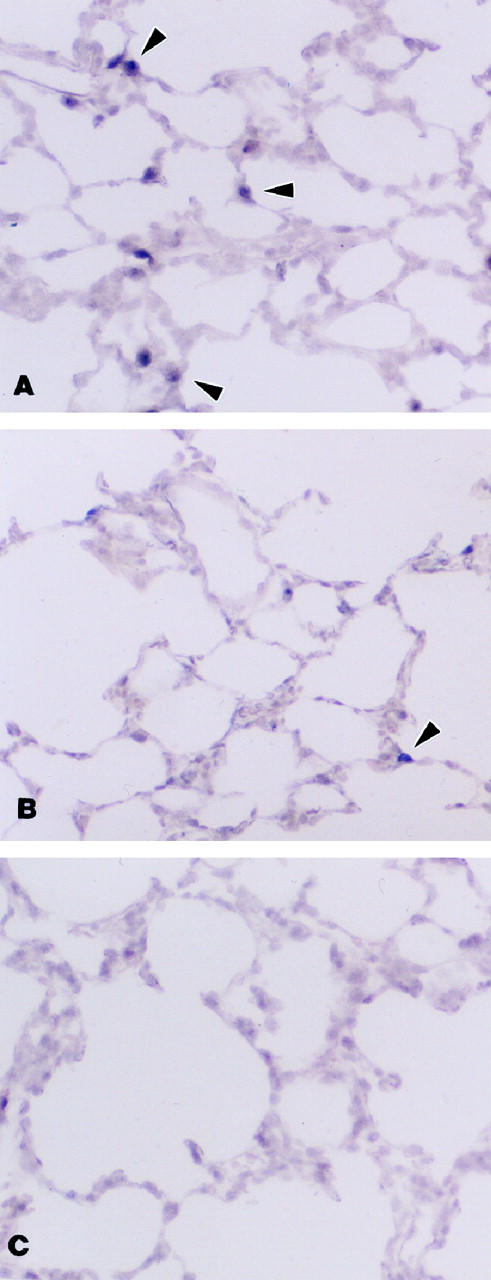
In situ hybridization for VEGF mRNA using digoxigenin-labeled probes. A: Control. Multiple positively stained cells (arrowheads) are seen in the distal airway epithelium. B: Exposure for 24 hours of >95% FiO2. Only a few positive cells are seen compared with control tissue. C: Exposure for 48 hours of >95% FiO2. VEGF mRNA expression can no longer be identified. Magnification, ×200.
RT-PCR
To examine for the presence of the splice variants, RT-PCR was performed. The results suggested that VEGF164 and VEGF188 were the predominant mRNAs expressed in the rat lungs, both control and those exposed to hyperoxia (data not shown). To better assess the abundance of the splice variants, RNAse protection assays were done.
RNAse Protection Assay
RNAse protection assays were done using a VEGF188 riboprobe. This riboprobe would generate a single protected fragment of 564 bp when hybridized to VEGF188 RNA, bands of 341 and 151 bp when hybridized to VEGF164, and bands of 341 and 19 bp when hybridized to VEGF120. All RNAse protection assays demonstrated the anticipated fragments of 564, 341, and 151 bp, confirming the presence of VEGF188 and VEGF164 in all samples (Figure 3) ▶ . The 19-bp band is likely to be too small to be detected, so it is not possible to quantify the abundance of VEGF120, although the 341-bp band presumably contains both of the smaller isoforms of VEGF. Correcting for the size of the fragments, we found no differences in the ratios of VEGF188 to VEGF164 during exposure to hyperoxia, although the total amount of RNA in each sample appeared to decline after exposure to hyperoxia, as was confirmed by Northern analysis. In our samples, VEGF188 constituted 28.0% of the total RNA in control animals, 28.7% in animals exposed to >95%FiO2 for 24 hours, and 28.6% after 48 hours of >95%FiO2.
Figure 3.

RNAse protection assay using VEGF188 riboprobe showed a similar distribution of protected bands of 564, 341, and 151 bp in RNA from lung tissue of both control animals and animals exposed to 24 or 48 hours of >95% FiO2. These bands are consistent with the presence of VEGF188, VEGF164, and possibly VEGF120.
VEGF Protein in Rat Lungs
In tissue from control animals, VEGF immunoreactivity was present not only in the distal airway epithelium but also in proximal epithelial cells (Figure 4, A and B) ▶ . Lung biopsy tissue from animals exposed to >95% FiO2 for 24 hours still demonstrated VEGF immunoreactivity in the bronchiole and peripheral epithelium, perhaps at a slightly decreased intensity (data not shown). However, in tissue specimens from rats exposed to >95%FiO2 for 48 hours (Figure 4, C and D) ▶ there was a marked reduction of VEGF protein both in the large and small airways. There was no staining present with control serum (data not shown).
Figure 4.
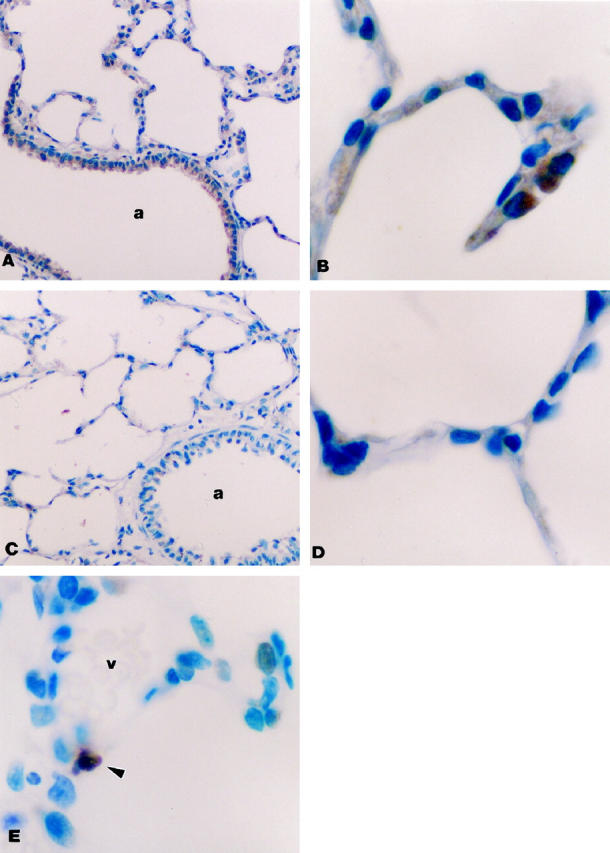
Immunohistochemistry for VEGF protein. A and B: Control. VEGF immunoreactivity was seen in bronchial and distal epithelial cells. Magnification, ×200 (A) and ×1000 (B). C and D: Exposure for 48 hours to >95% FiO2. No immunoreactivity in bronchial or distal epithelium. Magnification, ×200 (C) and ×1000 (D). E: TUNEL stain for apoptosis. Positive apoptotic staining is indicated by arrow. Magnification, ×1000. a, airway; v, vessel.
Western blot analyses identified two proteins bands consistent with VEGF188 and VEGF164, with VEGF164 being most abundant. After 24 hours of exposure to hyperoxia, some but not all tissues showed a decrease in VEGF proteins. However, in tissue from animals exposed to 48 hours of hyperoxia, VEGF188 was decreased to 47% of control levels and VEGF164 decreased to 75% of control.
Apoptosis in Rat Lung
To determine whether cell death by apoptosis was occurring during exposure to hyperoxia, TUNEL staining was performed on the same tissues. There were no positively stained apoptotic cells noted in any of the lung biopsy tissue specimens from control animals (data not shown). Apoptotic cells were easily identified in cells of the peripheral lung from all of the rats exposed to 24 or 48 hours of >95%FiO2 (Figure 4E) ▶ . Some of the apoptotic cells appeared to be endothelial cells according to their location, but there were also apoptotic epithelial cells. The presence of apoptotic cells was confirmed by electron microscopy (data not shown).
Expression of the VEGF Receptors Flt-1 and KDR/Flk mRNA in Rat Lungs
After confirming down-regulation of the VEGF ligand, we explored the possibility of similar regulation of the receptor. All Northern blots were reprobed separately using 32P-labeled riboprobes for the VEGF receptors Flt-1 and KDR/Flk. Abundance of Flt-1 mRNA was not significantly decreased after 24 hours of exposure to >95% FiO2 but was reduced to half of baseline after 48 hours of exposure to >95% FiO2 (Figure 5) ▶ . Similarly, the abundance of KDR/Flk receptor mRNA was significantly reduced only after 48 hours of exposure to >95% FiO2 (Figure 6 ▶ ; Table 1 ▶ ).
Figure 5.

Summary of Northern blot analyses showing the relative abundance of Flt-1 mRNA in lung tissue from animals exposed to >95% FiO2 for 24 or 48 hours (24h and 48h, respectively) compared with mRNA from lung tissue of control animals (C). Results were corrected for loading and normalized to control. There was a greater than 50% reduction in Flt-1 mRNA after 48 hours of exposure to hyperoxia. n = 6; *P < 0.05.
Figure 6.

Summary of Northern blot analyses showing the relative abundance of KDR/Flk mRNA in lung tissue from animals exposed to >95% FiO2 for 24 or 48 hours (24h and 48h, respectively) compared with mRNA from lung of control animals (C). Results were corrected for loading and normalized to control. After 48 hours of exposure to hyperoxia, KDR/Flk mRNA was reduced to 58% of control value. n = 6; *P < 0.05.
Discussion
Lung injury caused by exposure to 100% oxygen is characterized by loss of microvascular integrity resulting in accumulation of edema fluid in the alveolar spaces. In rat studies, the animals are asymptomatic during the initial 48 hours, but microscopic and tracer studies have revealed the onset of vascular leak before 48 hours, before the influx of inflammatory cells. 26 This early increase in permeability is accompanied by endothelial cell death, which may be the critical initiating factor in the subsequent massive respiratory failure and death.
There is evidence in human lung disease that apoptosis accompanies the development of lung injury. Guinee et al found that apoptotic cells were present in the majority of 20 lung biopsy specimens from patients with diffuse alveolar damage from various causes. 6 There is precedent for endothelial cell death as an early trigger in the pathogenesis of lung injury. Radiation injury to the lung causes a pneumonitis (after a brief asymptomatic period) that is characterized by atelectasis, hemorrhage, capillary leak, and extensive interstitial edema, not dissimilar from the clinical and histological appearance of late hyperoxic lung injury. Infusion of basic fibroblast growth factor prevented the development of radiation-induced pneumonitis in mice exposed to lethal doses of whole-lung irradiation, by blocking endothelial cell apoptosis. 27 Although this study demonstrated that the exogenous delivery of an endothelial growth factor could be protective, they did not examine whether there was loss of VEGF in the lung.
In the present study, we found that lung VEGF mRNA, protein and VEGF receptor mRNAs are markedly down-regulated by exposure to >95% inspired oxygen during the initial 48 hours of exposure, before the onset of clinical symptoms. In addition, we detected apoptosis in the lung tissues of the hyperoxic animals, coincident with the timing of the loss of VEGF. The large reservoir of VEGF present in the normal adult lung may function as a survival factor maintaining the endothelial cells. There is precedent for the loss of VEGF leading to cell death. Hyperoxia-induced down-regulation of VEGF has been reported in the rat retina and was associated with death of endothelial cells by apoptosis. 20 Furthermore, the endothelial cell death could be prevented by intraocular injections of VEGF. Additional evidence for VEGF having an anti-apoptotic function was established in a recent study that demonstrated that VEGF could prevent tumor necrosis factor (TNF)-α-induced endothelial cell apoptosis in vitro. 28 The idea that airway epithelial cells could produce a factor that protects endothelial cells was shown in the work by Wendt et al. 29 They found that conditioned media from type II cells was found to prevent TNF-α-induced death of endothelial cells. We speculate that VEGF is one of the factors made by the type II cells in their study that protected the endothelial cells.
Endothelial cells are considered to be the primary targets of the actions of VEGF; however, there are reports of effects on other cell types. For example, VEGF treatment prevented radiation-induced apoptosis in both a human leukemia cell line and normal hematopoietic stem cells. 23 Recent reports have shown that VEGF stimulates epithelial proliferation in human lung explants, suggesting that in the lung VEGF may be influencing cells other than endothelial cells. 30 We demonstrated the presence of VEGF protein in all epithelial cells lining the airways of control animals and its reduction after hyperoxic exposure. Whether the loss of VEGF in hyperoxia alters the survival of epithelial cells as well as endothelial cells needs further study.
Maniscalco et al have examined neonatal rabbits and shown that exposure to hyperoxia decreased VEGF after 9 days; VEGF decreased after 4 days in adult animals. 31 The differences in timing of the loss of VEGF in their study compared with our data may be explained by a combination of age of animals and species differences. It is interesting to speculate whether the increased tolerance of neonatal rabbits to hyperoxia is related to their ability to maintain VEGF levels. Changes in the abundance of VEGF receptor mRNAs in the rabbit model were not examined.
If VEGF is critical to the maintenance of the pulmonary vascular endothelium, then loss of VEGF receptors could also contribute to pathophysiology. We found a significant decrease in the mRNA abundance of both of the VEGF receptors, KDR/Flk and Flt-1, although the decrease occurred later than the loss of VEGF. The predominant expression of VEGF receptors is on endothelial cells, and we speculate that the decreased abundance of receptor mRNA is related in part to the death of endothelial cells. Some studies have suggested a coordinate up-regulation of VEGF ligand and receptors during hypoxia, 19 so it is possible that hyperoxia also alters receptor transcription. Immunohistochemistry studies did not show any decrease in either KDR/Flk or Flt-1 in tissue from animals exposed to hyperoxia (data not shown).
Hypoxia has been shown to be an important potent up-regulator of VEGF abundance through both transcription and stability. 15-17 We speculate that hyperoxia decreases the abundance of VEGF at least partially by eliminating any stimulation that is present at lower oxygen tensions. It is also possible that the decline in VEGF mRNA abundance represents decreased production of VEGF secondary to cell death or injury. However, the changes in mRNA occur very early in the time course when there is no evidence of significant alteration in the lung architecture histologically. As VEGF is produced by numerous alveolar epithelial cells, it seems unlikely that in the setting of a histologically normal appearing lung, a significant proportion of alveolar epithelial cells could be severely injured or dead.
An alternative explanation for the change in VEGF abundance would be a shift in the prevalence among the different isoforms to a species that was not recognized by our methods. However, the probes used in both Northern analysis and in situ hybridization should have hybridized with all species of VEGF that have been identified in the rat. We did explore the possibility of a change in the relative ratio of the major splice variants and did not find any evidence for a change in the pattern of isoform expression during exposure to hyperoxia. This is in contrast to a recent report by Watkins et al, 32 which demonstrated a decline in the proportion of VEGF189 after hyperoxic exposure in neonatal and adult rabbit lungs. In two previous studies of the effects of hypoxia on the relative abundance of the VEGF isoforms, no change in the relative proportions was demonstrated after exposure to low oxygen tension. 15,33
In summary, we have demonstrated down-regulation of VEGF mRNA and protein in rat lung tissues in response to 24 to 48 hours of hyperoxia. Importantly, the timing of these findings, although the animals remained clinically asymptomatic, suggests that there is an opportunity to provide a therapeutic intervention before development of irreversible lung damage. Additional studies will be required to assess the efficacy of providing exogenous VEGF in the setting of high oxygen exposure to prevent or attenuate the injury pattern.
Footnotes
Address reprint requests to Dr. Elizabeth A. Perkett, S-0119 MCN, Pediatric Pulmonary Medicine, Nashville, TN 37232-2586. E-mail: eperkett@mcmail.vanderbilt.edu.
Supported by Turner Scholars Program.
References
- 1.Crapo JD, Barry BE, Foscue HA, Shelburne J: Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis 1980, 122:123-143 [DOI] [PubMed] [Google Scholar]
- 2.Royston BD, Webster NR, Nunn JF: Time course of changes in lung permeability and edema in the rat exposed to 100% oxygen. J Appl Physiol 1990, 69:1532-1537 [DOI] [PubMed] [Google Scholar]
- 3.Kistler GS, Caldwell PRB, Weibel ER: Development of fine structural damages to alveolar and capillary cells in oxygen-poisoned rat lungs. J Cell Biol 1967, 32:605-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryan CL, Jenkinson SG: Oxygen toxicity. Clin Chest Med 1988, 9:141-152 [PubMed] [Google Scholar]
- 5.Majno G, Joris I: Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol 1995, 146:3-15 [PMC free article] [PubMed] [Google Scholar]
- 6.Guinee D, Fleming M, Hayashi T, Woodward M, Zhang J, Walls J, Koss M, Ferrans V, Travis W: Association of p53 and WAF1 expression with apoptosis in diffuse alveolar damage. Am J Pathol 1996, 149:531-538 [PMC free article] [PubMed] [Google Scholar]
- 7.Hase M, Araki S, Kaji K, Hayashi K: Classification of signals for blocking apoptosis in vascular endothelial cells. J Biochem 1994, 116:905-909 [DOI] [PubMed] [Google Scholar]
- 8.Monacci WT, Merrill MJ, Oldfield EH: Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol 1993, 264:C995-C1002 [DOI] [PubMed] [Google Scholar]
- 9.Millauer B, Wizigmann-Voos S, Schrurch H, Martinez R, Mleer NPH, Risau W, Ullrich A: High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72:835-846 [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 11.Park JE, Keller G-A, Ferrara N: The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell 1993, 4:1317-1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacic M, Edwards NA, Merril MJ: Differential expression of vascular endothelial growth factor (vascular permeability factor) forms in rat tissues. Growth Factors 1995, 12:11-15 [DOI] [PubMed] [Google Scholar]
- 13.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D: Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87:3336-3343 [PubMed] [Google Scholar]
- 14.Plate KH, Breier G, Weich HA, Mennel HD, Risau W: Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer 1994, 59:520-529 [DOI] [PubMed] [Google Scholar]
- 15.Minchenko A, Bauer T, Salceda S, Caro J: Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab Invest 1994, 71:374-379 [PubMed] [Google Scholar]
- 16.Namiki A, Brogi E, Kearney M, Kim EA, Wu T, Couffinhal T, Varticorski L, Isner JM: Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem 1995, 270:31189-31195 [DOI] [PubMed] [Google Scholar]
- 17.Brogi E, Wu T, Namiki A, Isner JM: Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cell, whereas hypoxia upregulates VEGF expression only. Circulation 1994, 90:649-652 [DOI] [PubMed] [Google Scholar]
- 18.Klekamp JG, Jarzecka K, Hoover RL, Summar ML, Redmond N, Perkett EA: Vascular endothelial growth factor is expressed in ovine pulmonary vascular smooth muscle cell in vitro and regulated by hypoxia and dexamethasone. Pediatr Res 1997, 42:744-749 [DOI] [PubMed] [Google Scholar]
- 19.Tuder RM, Flook BE, Voelkel NF: Increased gene expression for VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. J Clin Invest 1995, 95:1798-1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Kesher E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nature Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 21.Perkett EA, Klekamp JG: Vascular endothelial growth factor expression is decreased in rat lung following exposure to 24 or 48 hours of hyperoxia. Chest 1998, 114:52S-53S [DOI] [PubMed] [Google Scholar]
- 22.Perkett EA, Pelton RW, Meyrick B, Gold LI, Miller DA: Expression of transforming growth factor β mRNAs and proteins in vascular remodeling in the sheep air embolization model of pulmonary hypertension. Am J Respir Cell Mol Biol 1994, 11:16-24 [DOI] [PubMed] [Google Scholar]
- 23.Katoh O, Tauchi H, Kawaishi K, Kimura A, Satow Y: Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res 1995, 55:5687-5692 [PubMed] [Google Scholar]
- 24.Berkman RA, Merrill MJ, Reinhold WC, Monacci WT, Saxena A, Clark WC, Robertson JT, Ali IU, Oldfield EH: Expression of the vascular permeability factor/vascular endothelial growth factor gene in central nervous system neoplasms. J Clin Invest 1993, 91:153-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danielson PE, Forss-Petters S, Brow MA, Calavetta L, Douglass J, Milner RS, Sutcliffe JG: p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA 1988, 7:261-267 [DOI] [PubMed] [Google Scholar]
- 26.Fox RB, Hoidal JR, Brown DM, Repine JE: Pulmonary inflammation due to oxygen toxicity: involvement of chemotactic factors and polymorphonuclear leukocytes. Am Rev Respir Dis 1981, 123:521-523 [DOI] [PubMed] [Google Scholar]
- 27.Fuks Z, Persuad RS, Alfieri A, McLoughlin M, Ehleiter D, Schwartz JL, Seddon AP, Cordon-Cardo C, Haimovitz-Friedman A: Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res 1994, 54:2582-2590 [PubMed] [Google Scholar]
- 28.Spyridopoulos I, Brogi E, Kearney M, Sullivan AB, Cetrulo C, Isner JM, Losordo DW: Vascular endothelial growth factor inhibits endothelial cell apoptosis induced by tumor necrosis factor-α: balance between growth and death signals. J Mol Cell Cardiol 1997, 29:1321-1330 [DOI] [PubMed] [Google Scholar]
- 29.Wendt CH, Polunovsky VA, Peterson MS, Bitterman PB, Ingbar DH: Alveolar epithelial cells regulate the induction of endothelial cell apoptosis. Am J Physiol 1994, 267:C893-C900 [DOI] [PubMed] [Google Scholar]
- 30.Acarregui MJ, Ramirez K, Brown KR, Mallampalli RK: Vascular endothelial growth factor (VEGF) induces airway epithelial cell proliferation and surfactant protein gene expression in human fetal lung in vitro. Pediatr Res 1998, 43:44A [Google Scholar]
- 31.Maniscalco WM, Watkins RH, D’Angio CT, Ryan RM: Hyperoxic injury decreases alveolar epithelial cell expression of vascular endothelial growth factor (VEGF) in neonatal rabbit lung. Am J Respir Cell Mol Biol 1997, 16:557-567 [DOI] [PubMed] [Google Scholar]
- 32.Watkins RH, D’Angio CT, Ryan RM, Maniscalco WM: Vascular endothelial growth factor (VEGF) mRNA splice variant expression in acute hyperoxic lung injury. Am J Respir Crit Care Med 1998, 153:A579 [Google Scholar]
- 33.Sandner P, Gess B, Wolf K, Kurtz A: Divergent regulation of vascular endothelial growth factor and of erythropoietin gene expression in vivo. Eur J Physiol 1996, 431:905-912 [DOI] [PubMed] [Google Scholar]