

Abstract
Tumor necrosis factor (TNF)-α and transforming growth factor (TGF)-β mRNA and protein expression and the degree of fibroproliferative response to inhaled asbestos fibers are clearly reduced in the 129 inbred mouse strain as compared with typical fibrogenesis observed in the C57BL/6 inbred strain. The C57BL/6 mice showed prominent lesions at bronchiolar-alveolar duct (BAD) junctions where asbestos fibers deposit and responding macrophages accumulate. The 129 mice, however, were generally indistinguishable from controls even though the numbers of asbestos fibers deposited in the lungs of all exposed animals were the same. Quantitative morphometry of H&E-stained lung sections comparing the C57BL/6 and 129 mice showed significantly less mean cross-sectional area of the BAD junctions in the 129 animals, apparent at both 48 hours and 4 weeks after exposure. In addition, fewer macrophages had accumulated at these sites in the 129 mice. Nuclear bromodeoxyuridine immunostaining demonstrated that the number of proliferating cells at first alveolar duct bifurcations and in adjacent terminal bronchioles was significantly reduced in the 129 strain compared with C57BL/6 mice at 48 hours after exposure (P < 0.01). TNF-α and TGF-β1 gene expression, as measured by in situ hybridization, was reduced in the 129 mice at 48 hours after exposure, and expression of TNF-α and TGF-β1 protein, as measured by immunohistochemistry, was similarly reduced or absent in the 129 animals. We postulate that the protection afforded the 129 mice is related to reduction of growth factor expression by the bronchiolar-alveolar epithelium and lung macrophages.
Inhalation of asbestos fibers causes interstitial pulmonary fibrosis in both man and experimental animals. 1 Common features of this disease are epithelial and interstitial cell hyperplasia and extracellular matrix deposition. 2,3 These features are associated with varying degrees of acute and chronic inflammation. The increased connective tissue is known to cause a stiff lung and shortness of breath in millions of individuals worldwide. 2,3 Despite a fairly complete understanding of the clinical and pathological features of the disease process, there are no effective therapeutic approaches because it is not clear how this fibroproliferative process is mediated at the biochemical and molecular levels.
Experimental evidence is mounting to support the view that peptide growth factors elaborated at sites of lung injury mediate the cell proliferation and matrix production that are hallmarks of pulmonary fibrosis. 4 For example, instillation of platelet-derived growth factor (PDGF) causes airway fibrosis in rats; 5 both isoforms of PDGF are up-regulated rapidly in response to a brief asbestos exposure; 6 transforming growth factor (TGF)-β is up-regulated at sites of fiber deposition in rats, 7 and antibodies against tumor necrosis factor (TNF)-α block the development of silicosis. 8 These growth factors are among a large array of cytokines that have been demonstrated as products of lung cells under a variety of conditions. 9 A major problem facing investigators today is to establish which of these many factors is playing a role in disease development.
We use well characterized murine models of asbestos-induced lung fibrosis to test the hypothesis that growth factors such as PDGF, TGF-α, TGF-β1, and TNF-α are involved in the evolution of early fibroproliferative lesions at the sites of asbestos fiber deposition. 10-13 As transgenic technology is increasingly applied to investigations of this nature, 14-19 it becomes important to understand the basis for variations manifested among commonly used inbred mouse strains. In the work presented here, we have compared the fibroproliferative process and growth factor expression consequent to inhaled asbestos fibers in two strains of mice used routinely in making transgenic and knockout model systems. We demonstrate here that compared with C57BL/6 mice, the 129 strain exhibits 1) reduced growth factor expression and cell proliferation at sites of fiber deposition and 2) failure to develop fibrogenic lesions at these same sites. These findings support the postulate that certain peptide growth factors contribute to fibroproliferative lung disease, and investigators using the 129 strain in studies of fibrogenic pulmonary disease should be cautious when interpreting their results.
Materials and Methods
Animals, Exposure, and Tissue Preparation
Specific-pathogen-free 6-week-old male C57BL/6 and 129 mice (Jackson Laboratories, Bar Harbor, ME) weighing 25 to 30 g were exposed to room air for 5 hours in exposure chambers or to an aerosol of chrysotile asbestos (∼10 mg/m 3 respirable mass) in a 39-L inner aluminum chamber containing the exposure atmosphere within a 1.5-m 3 stainless steel Rochester outer chamber. Asbestos aerosol was generated from California chrysotile 20 and passed through a vertical elutriator to allow only particles less than 10 μm aerodynamic equivalent diameter to enter the chamber. Mice were exposed via the nose only. Dust concentrations in the exposure chamber were measured by sampling onto 37-mm PVC membrane filters placed in unused animal ports followed by gravimetric analysis of the samples. Five animals per group were sacrificed at 0 hour, 48 hours, 1 week, 2 weeks, and 4 weeks after the exposure by intraperitoneal injection of tribromoethanol at 200 mg/kg of body weight followed by exsanguination through the renal artery. Eight hours before sacrifice, each animal received an intraperitoneal injection of 5′-bromodeoxyuridine (BrdU) in PBS at 40 mg/kg of body weight. Lungs were perfused through the trachea with 10% neutral buffered formalin (Sigma Chemical Co., St. Louis, MO) at a pressure of 15 to 20 cm H2O for 30 minutes. After perfusion, the trachea was clamped and the lungs were removed from the chest cavity and placed in fresh fixative for 16 hours at 4°C. After fixation, the lungs were embedded in paraffin and 5-μm-thick sections were cut onto positively charged slides (SuperfrostR, CMS, Houston, TX) for immunohistochemistry and in situ hybridization. Small intestine was excised from each animal and similarly fixed as a positive control for BrdU immunostaining.
Morphometry
Hematoxylin and eosin (H&E)-stained sections from exposed and control animals from the 48-hour and 4-week time points were prepared, and the identity of each was masked. The slides were then reordered randomly and renumbered before measurements were made. Bifurcation area was measured using an Olympus microscope with a video camera interfacing with a PC using V150 imaging software. 21 The total area of each of five first alveolar duct bifurcations was measured as previously described. 7 The base of the bifurcation was defined to be delineated by the first lateral alveolar wall to transect the axis of the bifurcation. The resulting measurements from each animal were averaged to give a mean bifurcation size per animal.
Fiber Counts
Lung samples were ashed for 4 hours in an LTA plasma asher. After ashing, samples were washed from the ashing boats into glass vials with 20 ml of filtered bleach. Samples were incubated overnight in filtered bleach before being neutralized with sodium bisulfide and resuspended by sonication. The resulting solution was then filtered onto a 25-mm, 0.2-μm, mixed cellulose ester filter (SKC, Eighty Four, PA), and the filter was then dried in a desiccator for a minimum of 2 days. Sections of the filters were then placed on glass slides and cleared with acetone vapor before being coverslipped. Individual fibers were counted under phase contrast light microscopy according to National Institute of Occupational Safety and Health method 7400. 22
Macrophage Counts at Duct Bifurcations
One H&E-stained histological section randomly selected from each animal was used to count by light microscopy alveolar macrophages that had migrated to first alveolar duct bifurcations. An average of six bifurcations were counted per animal. The mean numbers of cells/bifurcation were then calculated and recorded in StatView 4.02 (Abacus Concepts, Berkeley, CA) and analyzed by analysis of variance.
Immunohistochemistry
Immunohistochemistry for TGF-β1 and TNF-α protein was performed using the immunoperoxidase technique as previously described. 23 For TGF-β1 immunohistochemistry, the Santa Cruz rabbit polyclonal anti-TGF-β1 antibody (catalog item SC-146) was used at a final concentration of 0.1 μg/ml. For TNF-α immunohistochemistry, a protein-A-purified rabbit polyclonal anti-TNF-α antiserum (the generous gift of Dr. Steve Kunkel) was used at a dilution of 1:2000.
BrdU immunohistochemistry using a mouse monoclonal anti-BrdU antibody at a dilution of 1:100 (Beckton Dickinson, San Jose, CA) was as described with the following modifications. Instead of PBS, 0.05% Tween 20 in PBS (PBST) was used in all cases where PBS was called for. After deparaffinization and rehydration, sections were incubated in 2 N HCl for 20 minutes followed by two washes of 5 minutes each in PBST. After washing, sections were incubated for 6 minutes in 0.05 mol/L Tris-Cl, pH 7.8/0.01% Trypsin/0.1% CaCl2 at 37°C, followed by PBST washes. The secondary antibody was biotinylated goat anti-mouse IgG. Negative controls included replacing the primary antibody with an equal concentration of normal mouse IgG and incubation of tissue sections from animals that had not received a BrdU injection before sacrifice.
Quantitative Analysis of Cell Proliferation
One BrdU-stained histological section from each animal was prepared as described above. An average of six fields per animal was analyzed as previously described. 24 The percentage of positive cells/section was then calculated and recorded in StatView 4.02 (Abacus Concepts) and analyzed by analysis of variance.
In Situ Hybridization
Probe Preparation
The in situ hybridization method used to detect TGF-β1 and TNF-α mRNA has been described in detail elsewhere. 23 For TGF-β1, control sense, or antisense cRNA probe was transcribed from a pGEM vector containing an ∼0.9-kb SmaI fragment of TGF-β1 consisting of nucleotides 421 through 1395 of the murine cDNA. This region contains 764 bp of the amino-terminal glycopeptide (precursor) region and 210 bp of the mature region and was a generous gift of Dr. Harold L. Moses (Vanderbilt University School of Medicine, Nashville, TN). 25 For TNF-α, a control sense or antisense cRNA probe was transcribed from a pGEM3 vector containing an ∼1.1-kb PstI-EcoRI fragment of murine TNF-α cDNA (GenBank M13049), a generous gift of Dr. B. Beutler. Linearized plasmids were used for in vitro transcription of digoxigenin-11-UTP-labeled antisense and control sense riboprobes using T7 and SP6 polymerase, respectively (Genius 4 RNA labeling kit, Boehringer Mannheim Corp., Indianapolis, IN).
Hybridization
Hybridization in 4X SSC/50% formamide at 43°C, post-hybridization washes, and RNAse A digestion were carried out as previously described. 23 Positive hybridization was detected using alkaline-phosphatase-conjugated anti-digoxigenin Fab fragments followed by colorimetric development with NBT/BCIP as described by the manufacturer (Boehringer Mannheim).
Results
Histopathology and Morphometry
Mice from two inbred strains (C57BL/6 and SJL) and their F1 hybrid (B6SJLF1) all developed typical fibroproliferative lesions consequent to asbestos exposure (not shown). The 129 strain failed to develop lung lesions as assessed by qualitative histopathology. Here we report that the 129 inbred strain is resistant to the development of asbestos-induced lung lesions. We compared the response of 129 mice with that of C57BL/6 mice, which develop lesions typical of those observed in other animal models (Figure 1) ▶ . 23,24 One of the key features of an early asbestos-induced fibroproliferative process is an enlargement of bronchiolar-alveolar duct (BAD) regions due to selective deposition of asbestos fibers at this anatomical region. 13 Our initial observation was that the 129 mice consistently exhibited reduced lesions compared with those observed in the C57BL/6 mice at BAD junctions as early as 48 hours after exposure (Figure 1, C and D) ▶ . These differences persisted through 4 weeks, the last time point considered in our experiments.
Figure 1.
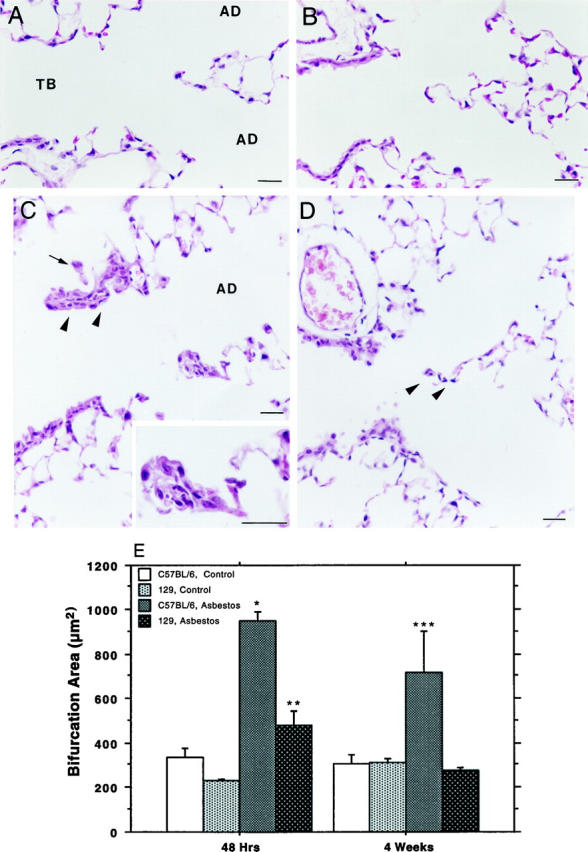
Histology of control and exposed lung from C57BL/6 and 129 inbred mice. A: Normal tissue from unexposed C57BL/6 animals shows no lesions and rare macrophages. TB, terminal bronchiole; AD, alveolar duct. B: Normal tissue from unexposed 129 animals is indistinguishable from unexposed C57BL/6 animals. C: A typical bronchiolar-alveolar duct lesion (arrowheads) and first alveolar duct (AD) bifurcation lesion (enlarged in inset) seen 48 hours after a 5-hour asbestos exposure in a C57BL/6 mouse. Alveolar (arrow) and interstitial macrophages have accumulated around and within these fibrotic lesions. D: Typical normal appearance of first alveolar duct bifurcation region (arrowheads) from a 129 animal 48 hours after exposure. Magnification bars, 20 μm. E: Morphometric analysis comparing exposed and control tissue sections from C57BL/6 and 129 animals at 48 hours and 4 weeks after a single 5-hour exposure. The data are means and standard errors of bifurcation area in square microns. Asterisks indicate degree of statistical significance: *significantly elevated over exposed 129 mice and unexposed controls, P < 0.01; **significantly elevated over unexposed controls, P < 0.01; ***significantly elevated over unexposed controls and exposed 129 mice, P < 0.05.
As shown in Figure 1E ▶ , sections from the 48-hour and 4-week time points after exposure were evaluated by video image analysis for bifurcation area measurement. C57BL/6 mice from both of these time points exhibited a mean bifurcation size significantly increased over controls. In contrast, the mean bifurcation size of the exposed 129 animals was elevated at the 48-hour time point but not at 4 weeks after exposure. Additionally, mean bifurcation area in exposed 129 mice was significantly less than that of exposed C57BL/6 mice at the time points examined.
Cell Proliferation
Because the mean lesion size in the 129 mice was significantly less in comparison with the C57BL/6 animals, we investigated whether cell proliferation at BADs and associated terminal bronchioles correlated with these findings. Cells stained positively for BrdU incorporation were counted at BADs and at terminal bronchioles. 24,26 These results are shown in Figure 2 ▶ . At BAD junctions (Figure 2A) ▶ , the percentage of BrdU-labeled cells at 48 hours in the C57BL/6 mice was increased significantly over the percentage in unexposed mice and in the lungs from 129 mice (P < 0.01 for both). Although the percentage of BrdU-stained cells at BAD junctions in sections from the 129 mice did achieve a significant increase (P < 0.05) over controls, this increase was significantly less (P < 0.05) than that observed for the C57BL/6 over controls. At 1 week after exposure, the percentage of BrdU-labeled cells in the exposed C57BL/6 lung sections remained significantly elevated (P < 0.01) over unexposed controls. By this time point, the percentage of labeled cells at BAD junctions in 129 mice had returned to control levels.
Figure 2.

BrdU labeling in alveolar duct bifurcations and terminal bronchioles. The single, 5-hour exposure to asbestos induced significant increases in the numbers of cells that stain positively for incorporated BrdU in C57BL/6 lung sections at both first alveolar duct bifurcations (A) and at terminal bronchioles (B) at both 48 hours and 1 week after exposure. The data are means and standard errors of positively stained cells counted in five animals from each group. Asterisks indicate degree of statistical significance: *significantly elevated over exposed 129 mice and controls, P < 0.01; **significantly elevated over unexposed controls, P < 0.05; ***significantly elevated over unexposed controls, P < 0.01.
In adjacent terminal bronchioles (Figure 2B) ▶ , we observed similar increased proliferation at 48 hours after exposure in the C57BL/6 animals. This increase was again significantly greater than that observed in the 129 mice (P < 0.01). The increase in the percentage of labeled cells in C57BL/6 mice persisted through 1 week after the exposure and remained significantly elevated over controls (P < 0.05). As with the BAD region, by 1 week after exposure, the percentage of BrdU-labeled cells in the 129 mice had returned to control levels.
Fiber Deposition in the Lung
Critical to the comparisons made above is a demonstration that the numbers of fibers deposited in the lungs of both C57BL/6 and 129 mice were not different. Therefore, the accessory lobe of the right lung of each animal was reserved after sacrifice for fiber counts in two separate experiments. Each lung sample was ashed and the fibers transferred to a filter for counting by phase contrast light microscopy. The numbers of fibers deposited in the lungs of animals of each strain were the same (Figure 3) ▶ . Additionally, over time, both strains exhibited similar rates of fiber clearance (Figure 3) ▶ . We also have shown by scanning electron microscopy that the pattern of initial fiber deposition in the lungs of the 129 mice (Figure 4) ▶ at BAD junctions is essentially the same as has been previously reported in other rodent model systems. 13
Figure 3.

Fiber deposition in the lung. The number of fibers initially deposited in the accessory lobe of the right lung of C57BL/6 and 129 animals is not different. The rate of clearance of fibers from the lungs of the two strains of mice over time is similar.
Figure 4.

Fiber deposition pattern in 129 mouse lung, shown by scanning electron micrograph of asbestos fibers (arrowheads) deposited at a first bronchiolar-alveolar duct bifurcation in a 129 animal. The fibers have attracted a few alveolar macrophages (arrows) as described previously in rats. 10 This pattern of fiber deposition is the same as that observed in other animal models studied. 10-13 Original magnification, 1500×.
Macrophage Counts
Morphometric analysis and cell counting data (Figures 1E and 2 ▶ ▶ , A and B) clearly demonstrate a difference in mean lesion cross-sectional area between the two inbred mouse strains. One known contribution to this increased bifurcation area after a brief asbestos exposure is an overall increase in the number of macrophages present at these sites. 10,13,27 Figures 1 and 4 ▶ ▶ clearly demonstrate that in C57BL/6 and 129 mice, macrophages are attracted to sites of fiber deposition. Thus, we asked whether there is a difference in the number of macrophages attracted to first alveolar duct bifurcations 48 hours after asbestos exposure in the two mouse strains. Alveolar macrophages that could be clearly identified by light microscopy associated with duct bifurcations were counted. Figure 5 ▶ shows that the mean numbers of macrophages attracted to first alveolar duct bifurcations in the asbestos-exposed 129 animals were elevated ∼2.5 times over controls, whereas the mean numbers of macrophages attracted to bifurcations in the asbestos-exposed C57BL/6 animals were ∼5 times that of controls. The increased response of the macrophages in C57BL/6 mice is significantly elevated (P < 0.05) over that of the 129 animals.
Figure 5.

Macrophage numbers at first alveolar duct bifurcations in lung sections from exposed and control C57BL/6 and 129 animals at 48 hours after exposure. The data are means and standard errors of macrophage numbers counted by light microscopy. Asterisks indicate degree of statistical significance: *significantly elevated over exposed 129 mice and unexposed controls, P < 0.01; **significantly elevated over controls, P < 0.01.
TNF-α Gene and Protein Expression
Because TNF-α appears to be a key mediator in pulmonary fibrogenesis, we examined the expression pattern of this pleiotropic cytokine at the sites of initial injury (Figure 6) ▶ . In both the exposed C57BL/6 and 129 mice, expression of TNF-α mRNA appears to be increased over unexposed controls immediately after the exposure (Figure 6, C and D) ▶ . This increase is confined to the epithelium of the terminal bronchioles and apparently precedes the up-regulation of TNF-α protein. By 48 hours after exposure in the C57BL/6 lung sections (Figure 6E) ▶ , TNF-α mRNA expression is clearly abundant at BAD junctions as well as in adjacent terminal bronchioles. In contrast, 129 mice (Figure 6F) ▶ exhibited a pattern of expression similar to that observed immediately after the exposure and confined to the epithelium of the terminal bronchioles. These sections frequently exhibited little positive signal outside of the bronchiolar epithelial compartment, with the exception of an occasional alveolar macrophage. By 1 week after exposure, the expression pattern of TNF-α mRNA returned to background levels (not shown) comparable to unexposed animals (Figure 6A) ▶ .
Figure 6.
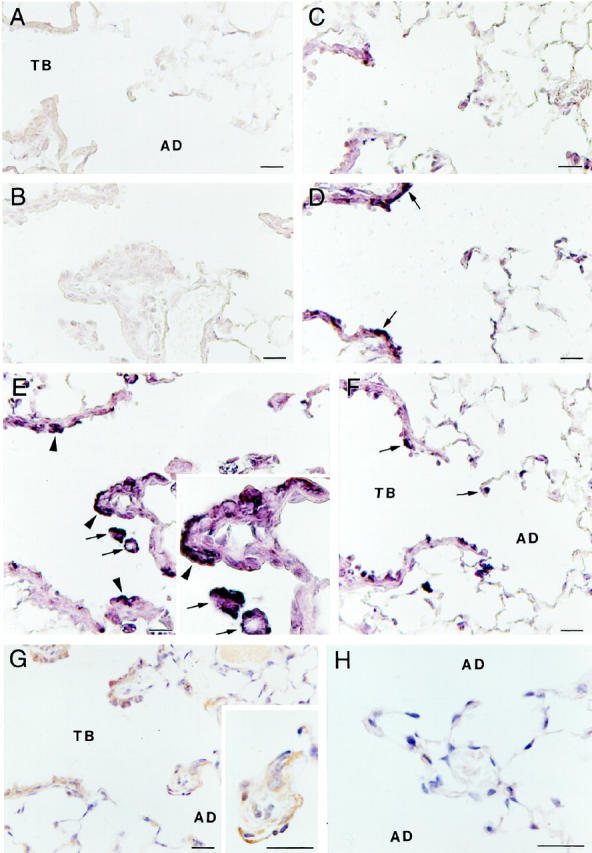
Light micrographs of TNF-α gene expression as detected by non-isotopic in situ hybridization (A to F) and protein expression as detected by immunohistochemistry (G and H). A: Unexposed C57BL/6 shows no positive hybridization signal and histology is normal. B: Exposed C57BL/6 section (serial section, compare with E) hybridized with control sense probe for TNF-α shows no positive hybridization signal. C: 129 animals immediately after the exposure begin to show signal in the epithelium of the terminal bronchioles, but this is reduced compared with C57BL/6 animals (compare with D). D: Immediately after the 5-hour exposure there is some positive signal in the epithelium of the terminal bronchioles (arrows) of C57BL/6 animals. E: At 48 hours after exposure in C57BL/6 animals there is clear hybridization signal for TNF-α evident in responding macrophages (arrows) and bronchiolar epithelial cells (arrowheads). The bronchiolar-alveolar duct lesion is enlarged (inset). F: 129 animals exhibiting a reduced fibroproliferative response 48 hours after an asbestos exposure (see Figure 2 ▶ ) also exhibit reduced TNF-α expression (arrows) at sites of fiber deposition. G: TNF-α protein is expressed 48 hours after the initial exposure in C57BL/6 animals at first alveolar duct bifurcations (enlarged in inset) and in terminal bronchioles similar to gene expression observed in E. H: TNF-α protein expression in 129 animals 48 hours after exposure is largely absent. Magnification bars, 20 μm. TB, terminal bronchiole; AD, alveolar duct.
As stated above, the expression of TNF-α mRNA precedes protein expression. Although there is clear hybridization signal for TNF-α mRNA in the epithelium of the terminal bronchioles at the time point immediately after the exposure, there is not yet any detectable protein expression (not shown). By 48 hours after exposure, however, there is prominent TNF-α protein expressed at BAD junctions in the exposed C57BL/6 mice (Figure 6G) ▶ as identified by immunohistochemistry. There is little apparent up-regulation of TNF-α in the 129 mice (Figure 6H) ▶ at these same sites, and expression generally is absent. As with gene expression, TNF-α protein expression returns to background levels by 1 week after exposure (not shown). The level of protein expression in the 129 mice never exceeded the minimal levels achieved at 48 hours after exposure (not shown).
TGF-β1 Gene and Protein Expression
Based on our previous findings in rats 7 and mice (J.-Y. Liu, unpublished data), after asbestos exposure we expected to see TGF-β1 gene expression up-regulated at sites of fiber deposition in the C57BL/6 mice by in situ hybridization. 25 At 48 hours after exposure, there was a robust signal for TGF-β1 mRNA (Figure 7A) ▶ at BADs and in adjacent terminal bronchioles when compared with control animals. In contrast, the 129 mice (Figure 7B) ▶ showed essentially no increase in TGF-β1 expression at these sites at any time after the exposure. By 1 week after exposure, TGF-β1 mRNA levels in the C57BL/6 animals returned to background levels, similar to unexposed animals (not shown).
Figure 7.

Light micrographs of TGF-β gene expression as detected by nonisotopic in situ hybridization (A and B) and protein expression as detected by immunohistochemistry (C and D). A: At 48 hours after the exposure, TGF-β message is strongly up-regulated at first alveolar duct bifurcations (inset) and in responding macrophages (arrows) in C57BL/6 animals. Sense control hybridization (not shown) completely ablates this signal. B: 129 animals fail to express significant levels of TGF-β mRNA at 48 hours after exposure. C: TGF-β protein expression is up-regulated at first alveolar duct bifurcations (enlarged in inset) 48 hours after exposure in a pattern similar to that seen in A. D: A ×100 magnification of a histologically normal first alveolar duct bifurcation from a 129 animal 48 hours after exposure demonstrates the absence of significant TGF-β protein expression. Magnification bars, 20 μm. TB, terminal bronchiole; AD, alveolar duct.
To confirm that gene expression was translated to protein production, we performed immunohistochemistry for TGF-β1. Figure 7C ▶ shows TGF-β1 protein clearly present at BADs and in adjacent terminal bronchioles in the C57BL/6 animals 48 hours after the initial exposure. In contrast, the 129 mice exhibit essentially no TGF-β1 staining in the bronchiolar regions and little at the duct bifurcations (Figure 7D) ▶ . These results closely follow the mRNA expression pattern demonstrated by in situ hybridization. As with the gene expression, TGF-β1 protein expression returned to background levels in the C57BL/6 mice by 1 week after exposure, and expression in the 129 mice remained at background levels at all time points after exposure.
Discussion
The results of the present study show that the 129 inbred mouse strain is resistant to the early fibrogenic effects of inhaled asbestos fibers. This is interesting for three prominent reasons. 1) The 129 strain commonly is used as a background for preparing knockout animals, and 2) the 129 strain exhibits reduced cell proliferation and reduced TNF-α and TGF-β1 expression, supporting the view that these potent growth factors play a role in the development of fibroproliferative lung lesions. In addition, 3) the 129 inbred mouse strain represents a model that can be used to study the genes that control interstitial fibrogenesis.
The 129 mouse strain has been used extensively to develop knockout animals. 28,29 To our knowledge, this strain has not been tested previously in regard to its pulmonary responses to inhaled fibrogenic particles. This is an important concept for those interested in the mechanisms through which inhaled toxic particles cause lung injury, and these animals could open opportunities to understand the genetic basis for the apparent protected phenotype observed. Here, we have shown that the 129 strain fails to develop a fibroproliferative response to a dose of asbestos fibers that we know causes fibrogenic lesions in several strains of mice 24,30 and rats. 7,27 Thus, we have begun to test our hypothesis that reduced expression of one or more peptide growth factors is fundamental to the apparent protection from an asbestos-induced fibroproliferative response in the 129 mouse strain.
A major problem confronting investigators interested in growth factor biology is the multitude of highly active cytokines known to be elaborated by lung cells. 4 Consequent to injury, many of these are up-regulated, 6,7,23 suggesting an important role in disease development. Previously, we carried out a series of studies focused on three of the factors thought to participate in the development of fibroproliferative lung disease. These are the isoforms of PDGF, 6 TGF-α, 23 and TGF-β1. 7 These factors are of interest because 1) PDGF is the most potent mesenchymal cell mitogen yet described, 31 2) TGF-α is a powerful mitogen for epithelial cells, 32 and 3) TGF-β1 is known as a clear inducer of extracellular matrix proteins by mesenchymal cells. 33 TGF-β1 generally blocks cell proliferation, but under certain circumstances, TGF-β1 has been demonstrated to be mitogenic for some cell types in vitro. 34,35 We showed in asbestos-exposed rats that these growth factor genes and their concomitant proteins are up-regulated significantly within hours after exposure, 6,7,23 and expression is limited to the anatomical sites of asbestos deposition and lung injury, 6,7,23 ie, the bronchiolar-alveolar duct (BAD) junctions. BAD junctions are sites of initial injury consequent to exposure to a number of inhaled agents, including asbestos 13 and silica, 36 and these anatomical regions are commonly used as focal points of study after exposure to toxic materials. 37
In addition to the factors discussed above, there is compelling evidence that TNF-α plays a critical role in fibroproliferative lung disease. 8,14,38 We have supported this finding by demonstrating here that markedly lower TNF-α expression in the lung correlates with a reduced fibroproliferative response (Figure 6) ▶ . With this information and the knowledge that TNF-α apparently can function as an activator of expression of other growth factors, such as TGF-β, 38,39 we postulate that TNF-α acts as a central mediator in controlling the development of interstitial lung fibrosis (see further discussion of this point below). Also, inasmuch as the numbers of macrophages responding to asbestos exposure are fewer in the 129 mice (Figure 5) ▶ , a proven source of TNF-α would be reduced. Alveolar macrophages have been shown to produce TNF-α in our mouse model, 40 and TGF-β is a known product of lung macrophages. 33,34 The mechanisms controlling reduced macrophage migration and how concomitant reduction in growth factor release might affect lesion development in the 129 mice remain unknown at this time.
If these selected growth factors discussed above have any role in fibrogenic lung disease, it would follow that animals exhibiting a reduced fibroproliferative reaction might have less expression of such factors. However, a significant question in interpretation remains; specifically, is a given factor reduced because it is intrinsically genetically programmed as such, or is it reduced because there is less of a lesion developing for some other reason, with fewer cells available for growth factor expression? The comparison we present between the C57BL/6 and 129 strains could shed some light on this important issue. First, gene expression and proliferative rates in the epithelial cells lining the most distal terminal bronchioles clearly were different between the strains after asbestos exposure (Figures 2, 5, and 6) ▶ ▶ ▶ , yet these well defined regions of lung exhibited little apparent anatomical difference throughout the experiment, suggesting the genetic explanation noted above. Furthermore, our studies on knockout mice lacking both the 55- and 75-kd receptors for TNF-α show that these animals also are protected from the fibrogenic effects of inhaled asbestos. 40 Even though the levels of TNF-α expression are up-regulated after exposure in these knockout mice, there is a clear lack of a typical fibroproliferative response to asbestos, similar to that shown here for the 129 strain. In addition, the protected knockout mice exhibit reduced expression of PDGF, TGF-α, and TGF-β1, 40 suggesting that TNF-α plays a central role in the activation of other growth factors as discussed above. This is consistent with the findings of Piguet et al 8 who showed that TNF-α is necessary for silica- and bleomycin-induced fibrosis to develop. Perhaps the reduced expression of TNF-α exhibited by the 129 mice studied here is key to the protected phenotype. Accordingly, recent experiments with primary mouse lung fibroblasts cultured from the C57BL/6 and 129 inbred strains suggest further that TNF-α may indeed be important in the protected phenotype observed in the 129 inbred strain. Our preliminary data show that TNF-α up-regulates production of pro-α1 (I) collagen mRNA in primary mouse lung fibroblasts from the C57BL/6 inbred strain. 41 TNF-α is much less effective in up-regulating this gene in the primary MLFs from the 129 inbred strain. Additional data from these ongoing experiments indicate that primary MLFs from the 129 strain exhibit reduced proliferative capacity in response to both serum and PDGF. 41 Such studies culturing mesenchymal cells should prove highly valuable in understanding the mechanisms controlling protection of the 129 mouse strain.
Thus, we have shown that the 129 mouse strain fails to develop fibroproliferative lesions consequent to asbestos exposure. These mice exhibit a significantly reduced percentage of dividing cells and fewer macrophages accumulating within the first 2 days after exposure, and this results in essentially normal lungs through the 1-month period studied. These animals have reduced expression of TNF-α and TGF-β1 mRNA, and little TNF-α or TGF-β1 protein is detected at the sites of initial lung injury. The dose and clearance rate of asbestos fibers in these animals were the same as that in the C57BL/6 strain. We suggest that the reduced growth factor expression plays some role in the lack of disease development, and our ongoing experiments are designed to test this postulate.
Acknowledgments
We thank the staff of Tulane Medical Center Anatomic Histopathology Laboratory for technical assistance and Ms. Odette Marquez for preparation of the manuscript.
Footnotes
Address reprint requests to Dr. Arnold R. Brody, Department of Pathology, Tulane University Medical Center, 1430 Tulane Avenue, SL-79, New Orleans, LA 70112-2699. E-mail: abrody@tmc.tulane.edu.
Supported by NIH grants RO1ES60766 and RO1HL60532, Tulane/Xavier Center for Bioenvironmental Research, and The Tulane Cancer Center.
References
- 1.Selikoff IJ, Lee DH: Asbestos and Disease. 1978. Academic Press, New York
- 2.Crystal RG: Interstitial lung disease: current concepts of pathogenesis, staging and therapy. Am J Med 1981, 70:542-565 [DOI] [PubMed] [Google Scholar]
- 3.Crouch E: Pathobiology of pulmonary fibrosis. Am J Physiol 1990, 259:L159-L184 [DOI] [PubMed] [Google Scholar]
- 4.Morris GF, Brody AR: Molecular mechanisms of particle-induced lung disease. Rom WN eds. Environmental and Occupational Medicine. 1998, :pp 305-333 Lippincott-Raven, Philadelphia [Google Scholar]
- 5.Yi ES, Lee H, Yin S, Piguet P, Sarosi I, Kaufmann S, Tarpley J, Wang N-S, Ulich TR: Platelet-derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am J Pathol 1996, 149:539-548 [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J-Y, Morris GF, Lei W-H, Hart CE, Lasky JA, Brody AR: Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am J Respir Cell Mol Biol 1997, 17:129-140 [DOI] [PubMed] [Google Scholar]
- 7.Perdue TD, Brody AR: Distribution of transforming growth factor-β1, fibronectin, and smooth muscle actin in asbestos-induced pulmonary fibrosis in rats. J Histochem Cytochem 1994, 42:1061-1070 [DOI] [PubMed] [Google Scholar]
- 8.Piguet PF, Collart MA, Grau GE, Sappino A-P, Vassili P: Requirement of tumor necrosis factor for development of silica-induced pulmonary fibrosis. Nature 1990, 344:245-247 [DOI] [PubMed] [Google Scholar]
- 9.Kelly J: Cytokines of the lung. Am Rev Respir Dis 1990, 141:765-788 [DOI] [PubMed] [Google Scholar]
- 10.Brody AR, Hill LH, Adkins B, O’Connor R: Chrysotile asbestos inhalation in rats: deposition pattern and reaction of alveolar epithelium and pulmonary macrophages. Am Rev Respir Dis 1981, 123:670-679 [DOI] [PubMed] [Google Scholar]
- 11.Brody AR: Asbestos induced lung disease. Environ Health Perspect 1993, 100:21-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brody AR: Asbestos exposure as a model of inflammation inducing interstitial pulmonary fibrosis. Gallin JI Goldstein IM Snyderman R eds. Inflammation: Basic Principles and Clinical Correlates. 1992, :pp 1033-1049 Raven Press, New York [Google Scholar]
- 13.Brody AR, Roe MW: Deposition pattern of inorganic particles at the alveolar level in the lungs of rats and mice. Am Rev Respir Dis 1983, 128:724-729 [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet P-F, Vassalli P: Expression of a tumor necrosis factor-α transgene in murine lung causes lymphocytic and fibrosing alveolitis. J Clin Invest 1995, 96:250-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray P, Tang W, Wang P, Homer R, Kuhn C, III, Flavell RA, Elias JA: Regulated overexpression of interleukin 11 in the lung: use to dissociate development-dependent and -independent phenotypes. J Clin Invest 1997, 100:2501-2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L, Dey CR, Wert SE, Whitsett JA: Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-β1 chimeric gene. Dev Biol 1996, 175:227-238 [DOI] [PubMed] [Google Scholar]
- 17.Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C: PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 1996, 85:863-873 [DOI] [PubMed] [Google Scholar]
- 18.Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh C-G, Gress RE, Karlsson S: Transforming growth factor-β1 null mice: an animal model for inflammatory disorders. Am J Pathol 1995, 146:264-275 [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson DA, Carrigan PE, Brenneise IE, Horton MA, Haczku A, Gelfand EW, Leikauf GD, Lee NA: Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med 1997, 185:2143-2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinkerton KE, Brody AR, McLaurin DA, Adkins B, O’Connor RW, Pratt PC, Crapo JD: Characterization of three types of chrysotile asbestos after aerosolization. Environ Res 1983, 31:32-53 [DOI] [PubMed] [Google Scholar]
- 21.Fermin CD, Degraw S: Colour thresholding in video imaging. J Anat 1995, 186:469-481 [PMC free article] [PubMed] [Google Scholar]
- 22.Eller P, Cassinelli M: NIOSH Manual of Analytical Methods, DHHS publication 94–113, ed 4. Washington, DC, Department of Health and Human Services, 1994
- 23.Liu J-Y, Morris GF, Lei W-H, Corti M, Brody AR: Up-regulated expression of transforming growth factor-α in the bronchiolar-alveolar duct regions of asbestos-exposed rats. Am J Pathol 1996, 149:205-217 [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner SY, Brody AR: Incorporation of bromodeoxyuridine as a method to quantify cell proliferation in bronchiolar-alveolar duct regions of asbestos-exposed mice. Inhal Toxicol 1995, 7:215-224 [Google Scholar]
- 25.Pelton RW, Johnson MD, Perkett EA, Gold LI, Moses HL: Expression of transforming growth factor-β1, -β2, and -β3 and protein in the murine lung. Am J Respir Cell Mol Biol 1991, 5:522-530 [DOI] [PubMed] [Google Scholar]
- 26.Coin PG, Osornio-Vargas AR, Roggli VL, Brody AR: Pulmonary fibrogenesis after three consecutive inhalation exposures to chrysotile asbestos. Am J Respir Crit Care Med 1996, 154:1511-1519 [DOI] [PubMed] [Google Scholar]
- 27.Chang LY, Overby LH, Brody AR, Crapo JD: Progressive lung cell reactions and extracellular matrix production after a brief exposure to asbestos. Am J Pathol 1988, 131:156-170 [PMC free article] [PubMed] [Google Scholar]
- 28.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R: The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol 1985, 87:27-45 [PubMed] [Google Scholar]
- 29.McMahon AP, Bradley A: The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 1990, 62:1073-1085 [DOI] [PubMed] [Google Scholar]
- 30.Dixon D, Bowser AD, Badgett A, Haseman JK, Brody AR: Incorporation of bromodeoxyuridine (BrdU) in the bronchiolar-alveolar regions of the lungs following two inhalation exposures to chrysotile asbestos in strain A/J mice. J Environ Pathol Toxicol Oncol 1995, 14:205-213 [PubMed] [Google Scholar]
- 31.Raines EW, Ross R: Platelet-derived growth factor. I. High yield purification and evidence for multiple forms. J Biol Chem 1982, 257:5154-5160 [PubMed] [Google Scholar]
- 32.Derynck R: The physiology of transforming growth factor-α. Adv Cancer Res 1992, 58:27-52 [DOI] [PubMed] [Google Scholar]
- 33.Pelton RW, Moses HL: The β-type transforming growth factors, mediators of cell regulation in the lung. Am Rev Respir Dis 1990, 142:31-35 [DOI] [PubMed] [Google Scholar]
- 34.Moses HL, Tucker RF, Leof EB, Coffey RJ, Halper J, Jr, Shipley GD: Type β transforming growth factor is a growth stimulator and a growth inhibitor. Feramisco J Ozanne B Stiles B eds. Growth Factors and Transformation. 1985, :pp 65-71 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, [Google Scholar]
- 35.Centrella M, McCarthy TL, Canalis E: Transforming growth factor β is a bifunctional regulator of replication and collagen synthesis in osteoblast-enriched cell cultures from fetal rat bone. J Biol Chem 1987, 262:2869-2874 [PubMed] [Google Scholar]
- 36.Brody AR, Roe MW, Evans JN, Davis GS: Deposition and translocation of inhaled silica in rats. Lab Invest 1982, 47:533-542 [PubMed] [Google Scholar]
- 37.Pinkerton KE, Dodge DE, Cederdahl-Demmler J, Wong VJ, Peake J, Haselton CJ, Mellick PW, Sing G, Plopper CG: Differentiated bronchiolar epithelium in alveolar ducts of rats exposed to ozone for 20 months. Am J Pathol 1993, 142:946-956 [PMC free article] [PubMed] [Google Scholar]
- 38.Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, Gauldie J: Transfer of tumor necrosis factor-α to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-β1 and myofibroblasts. Am J Pathol 1998, 153:825-832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phan SH, Gharaee-Kermani M, McGarry B, Kunkel SL, Wolber FW: Regulation of rat pulmonary artery endothelial cell transforming growth factor-β production by IL-1β and tumor necrosis factor-α1. J Immunol 1992, 149:103-106 [PubMed] [Google Scholar]
- 40.Liu J-Y, Hoyle GW, Brody AR: Tumor necrosis factor-α receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol 1998, 153:1839-1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brass DM, Hoyle GW, Liu J-Y, Brody AR: Primary lung fibroblasts from mice resistant to the fibrogenic effects of asbestos exhibit reduced proliferative capacity in vitro. Am J Respir Crit Care Med 1998, 157:A246 [Google Scholar]