

Abstract
We used reverse transcriptase-polymerase chain reaction and Western blotting techniques to measure the levels of complement mRNAs and their protein products in Alzheimer’s disease (AD) brain compared with non-AD brain. mRNAs for C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, and C9 were detected in the 11 regions of brain that were investigated. The mRNA levels were markedly up-regulated in affected areas of AD brain. In the entorhinal cortex, hippocampus, and midtemporal gyrus, which had dense accumulations of plaques and tangles, C1q mRNA was increased 11- to 80-fold over control levels, and C9 mRNA 10- to 27-fold. These levels were substantially higher than in the livers of the same cases. Western blot analysis of AD hippocampus established the presence of all of the native complement proteins as well as their activation products C4d, C3d, and the membrane attack complex. These data indicate that high levels of complement are being produced in affected areas of AD brain, that full activation of the classical complement pathway is continuously taking place, and that this activation may be contributing significantly to AD pathology.
The role of the complement system is usually considered to be limited to boosting humoral immune responses. Antibodies react with antigens and then bind C1q, which initiates the classical pathway. Liver is regarded as the principal source of complement proteins and serum the delivery vehicle. However, contemporary research is revealing that this traditional view of the complement system is far too restricted. Many cell types produce complement proteins, and there are numerous molecules other than antigen-bound antibodies that can bind C1q. Thus, the system can be activated both locally and systemically in a wide variety of noninfectious conditions. C1q identifies the target, fragments from C2, C4, and C3 opsonize it, and the terminal components C5b to C9 assemble to form the membrane attack complex. This lipophilic assembly inserts into cell membranes, destroying their integrity and causing cell lysis. The small fragments C3a, C4a, and C5a, cleaved during the activation process, stimulate inflammation. These processes of stimulating inflammation, promoting phagocytosis, and destroying cells are intended to be beneficial but can easily cause damage to the host.
Studies of the pathology of Alzheimer’s disease (AD) have done much to focus attention on the larger role of the complement system in health and disease and particularly to highlight the role of endogenous brain cells, including neurons, in inflammatory and immune responses. It has long been known that activated components of the classical complement pathway are associated with AD lesions. 1-5 Initially, it was believed that such activation was secondary to a classical antigen-antibody reaction in AD brain. However, failure to confirm the presence of antibodies inspired the search for alternative activators of complement. β-Amyloid protein (Aβ), the donor peptide of AD amyloid deposits, was found to bind C1q and activate complement in vitro. 6 Other molecules associated with AD lesions which activate complement in vitro are amyloid P, C-reactive protein, and Hageman factor (reviewed in Ref. 5 ). Thus, the complement cascade could be activated in vivo by one or more of these molecules. The possibility that brain itself acts as the source of complement proteins was also investigated, as a serum source was unlikely due to the blood-brain barrier. Astrocytes, microglia, and neuroblastoma cells were all found to produce complement proteins in vitro.7–11 In vivo immunohistochemical 12,13 and in situ hybridization analyses 14,15 have indicated that neurons are the most abundant source of brain complement proteins, at least in the hippocampus and temporal cortex.
The plaque and tangle lesions of AD are chronic, and the association of complement proteins with them could reflect a low level process of little pathological significance, with most of the deposits reflecting events remote in time. On the other hand, they could reflect an aggressive level of activity which could be an important contributor to AD pathogenesis.
To assess the relative level of complement activity in AD compared with control cases, we used the reverse transcriptase-polymerase chain reaction (RT-PCR) technique to measure relative levels of the mRNAs for all of the components of the classical complement pathway, ie, C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, and C9. We assayed 11 regions of brain, as well as liver, heart, kidney and spleen of many of these cases. All tissues studied contained detectable levels of most of the complement protein mRNAs. The levels in brain were strikingly elevated in AD compared with controls, particularly in those areas with heavy pathological involvement. However, levels in the liver and other organs were not affected. Western blot analyses showed that the complement mRNAs were translated into their protein products, and that, in AD brain, the classical complement pathway was strongly activated, including assembly of the membrane attack complex. These data provide evidence of the generation of complement mRNAs in AD brain, with continuous activation of the classical complement pathway. Such activity points to the probable role of complement as a major force driving pathology in the disease.
Materials and Methods
Cases Studied
Fifteen autopsy cases were used in this study. Brain, heart, liver, spleen, and kidney were investigated, but not all organs were obtained from every case. Details are given in Table 1 ▶ . The brains from four cases of AD and five without neurological disease were studied. In each case of AD, the clinical diagnosis was confirmed by routine pathological analysis for plaques and tangles. All cases met standard criteria for moderate to severe AD. No AD pathology was noted in the control brains. Peripheral organs only were obtained from an additional six cases. The age range of the AD cases was 65 to 78 years (70.5 ± 2.8), with postmortem delays varying from 6 to 16 hours. The ages of the control brain cases ranged from 43 to 82 years (72.2 ± 7.3), with postmortem delays from 9 to 48 hours. Postmortem delay was not considered to be a contributing factor to the results since we have previously established that there is almost no observable degradation for the mRNAs of cyclophilin and the complement proteins in tissue stored for periods of up to 6 days in the cold. 16
Table 1.
Details of Cases
| Case No. | Age | Sex | Postmortem delay (hr) | Cause of death | Tissue used |
|---|---|---|---|---|---|
| 1 | 78 | F | 7.5 | Alzheimer’s disease | Brain |
| 2 | 68 | M | 6 | Alzheimer’s disease | Brain, heart, liver, spleen, kidney |
| 3 | 65 | F | 16 | Alzheimer’s disease | Brain, heart, liver, spleen, kidney |
| 4 | 71 | F | 8 | Alzheimer’s disease | Brain, heart, liver, spleen, kidney |
| 5 | 82 | F | 24 | Ovarian cancer | Brain, heart, liver, spleen, kidney |
| 6 | 78 | M | 48 | Cardiac failure | Brain, liver, spleen, kidney |
| 7 | 43 | F | 9 | Diabetes, drug overdose | Brain, heart, spleen, kidney |
| 8 | 78 | M | 21 | Cardiac failure | Brain, liver, spleen, kidney |
| 9 | 80 | F | 24 | Lung cancer | Brain, liver, heart, spleen, kidney |
| 10 | 25 | F | 30 | Leukemia | Liver, spleen, kidney |
| 11 | 80 | F | 41 | Bronchiogenic cancer | Heart, liver, spleen, kidney |
| 12 | 71 | M | 120 | Acute myocardial infarction | Liver, spleen, kidney |
| 13 | 60 | M | 22 | Glioblastoma | Liver, spleen, kidney |
| 14 | 76 | F | 39 | Parkinson’s disease | Liver, spleen, kidney |
| 15 | 53 | F | 120 | Myocardial infarction | Liver, spleen, kidney |
RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction
The techniques used for RNA extraction and preparation of RT-PCR products have been described in detail elsewhere. 16 Briefly, total RNA was extracted from approximately 500 mg of each tissue sample by the acid guanidinium thiocyanate-phenol-chloroform method. The samples were treated with 10 U of RNase-free DNase (Pharmacia) for 60 minutes at 37°C in 25 μl of 1× reverse transcriptase buffer (50 mmol/L Tris-HCl, 75 mmol/L KCl, 3 mmol/L MgCl2) containing 40 U of RNase inhibitor (Pharmacia) and 1 mmol/L dithiothreitol, followed by an incubation at 85°C for 5 minutes to inactivate the enzyme.
Single-strand cDNA synthesis was performed on 5 μg of total RNA extract. The reaction mixture contained the RNA sample, 25 μl of 1× reverse transcriptase buffer containing 1 μg of random hexamer primers (pDN6, Pharmacia), 1 mmol/L deoxynucleotides (GIBCO BRL), 5 mmol/L dithiothreitol, 40 U of RNase inhibitor (Pharmacia) and 500 U of reverse transcriptase (Superscript TMII RT, GIBCO BRL). Duplicate assays were carried out at 42°C for 90 minutes, followed by heat inactivation at 65°C for 10 minutes.
The appropriate quantity of cDNA was covered with 50 μl of mineral oil and amplified in a 50-μl reaction buffer containing 67 mmol/L Tris-HCl (pH 8.8), 16.6 mmol/L ammonium sulfate, 10 mmol/L 2-mercaptoethanol, 200 mmol/L dNTPs, 2 mmol/L MgCl2, 40 pmol of each specific oligonucleotide primer, and 2.5 U of Taq DNA polymerase (GIBCO BRL). The thermal profile used on a Fisher Scientific (Toronto, Canada) programmable thermal controller consisted of a denaturation step of 94°C for 1 minute, an annealing step of 55°C for 30 seconds, and an extension step of 72°C for 1 minute. The extension step in the first cycle was for 3 minutes at 72°C. All samples were initially denatured for a total of 5 minutes (94°C).
To determine appropriate parameters for amplification of the cDNA products, the method of Nakayama et al 17 was followed. The method involves using graded amounts of cDNA and varying cycles of amplification to determine a range where the logarithm of reaction product intensity is linear with the cycle number. In preliminary experiments, we tested amounts of cDNA from 0.01 μg to 1 μg and cycle numbers from 20 to 40, using the standard conditions described above. We found that for cyclophilin the log of PCR product intensity was linearly proportional to cycle number from 20 to 29 cycles, and, for all complement genes, from 31 to 37 cycles. Plateaus were reached after 29 and 37 cycles, respectively. The product intensity was proportional to the cDNA concentration from 0.1 μg to 1 μg. Accordingly, the standard PCR procedure adopted was 0.5 μg of cDNA (1 μl) and 27 cycles of amplification for cyclophilin and 35 cycles for the complement products.
Each PCR reaction product was electrophoresed through a 6% polyacrylamide gel and the product visualized by incubation for 10 minutes in a solution containing 10 ng/ml of ethidium bromide. Resulting gel bands were imaged using a GDS 6700 image analyzer (Ultra Violet Products, Uplands, CA). The relative intensities of the bands, expressed as optical density units, were quantitatively analyzed using NIH Image software 1.61. Each complement mRNA analysis was made in parallel with a cyclophilin mRNA analysis to provide an internal standard. Cyclophilin has been widely used as a gene product with constant expression in tissue including brain. 18 Polaroid photographs of the gels were taken. Table 2 ▶ lists the GenBank accession number for the gene sequences used and the positions chosen for primer design. Except for C1r, they spanned 1 to 3 introns. The primer sequences were as previously published. 16
Table 2.
Gene Sequences and Endonuclease Digestion Products
| Gene | Position in sequence | GenBank accession No. | Product length (bp) | Restriction enzyme | Digestion fragments (bp) |
|---|---|---|---|---|---|
| C1q (1 intron) | 91–448 | KO 3430 | 358 | Ncol | 46, 71, 241 |
| C1r | 1793–2008 | M 14058 | 216 | HaeIII | 90, 126 |
| C1s (1 intron) | 860–1223 | JO 4080 | 364 | HaeIII | 114, 250 |
| C2 (2 introns) | 1710–1924 | XO 4481 | 215 | BgII | 95, 120 |
| C3 (2 introns) | 509–694 | KO 2765 | 186 | HaeIII | 29, 31, 126 |
| C4 (2 introns) | 3228–3483 | KO 2403 | 256 | HaeIII | 78, 178 |
| C5 (3 introns) | 1810–2124 | M 57719 | 315 | AluI | 53, 114, 148 |
| C6 (1 intron) | 1652–1989 | JO 5064 | 338 | AluI | 32, 306 |
| C7 (1 intron) | 455–702 | JO 3507 | 248 | HincII | 67, 181 |
| C8 (1 intron) | 76–333 | M 17999 | 258 | AluI | 32, 226 |
| C9 (1 intron) | 128–307 | KO 2766 | 180 | Sau3AI | 32, 148 |
| Cyclophilin (3 introns) | 15–220 | YO 0052 | 206 | HaeIII | 53, 153 |
For primer sequences see Ref. 16 .
Each PCR product was verified by restriction digest analysis using the ethanol precipitation procedure. An equal volume of phenol/chloroform was added to the PCR mixture, the mixture was centrifuged for 2 minutes, and the supernatant transferred to a fresh microfuge tube. Two volumes of 100% ethanol and 0.1 volume of sodium acetate (3 mol/L, pH 5.2) were added to this supernatant and the mixture cooled for 15 minutes at −80°C to precipitate the double stranded DNA. The pellet was rinsed twice with 1 ml of 70% ethanol, and the nucleic acids were redissolved in 20 μl of distilled water. The DNA solution (5 μl) was incubated with 10 times the volume of restriction enzyme buffer and 10 U of the desired restriction enzyme. The reaction was carried out for 2 hours at 37°C, and the digested PCR products were analyzed by electrophoresis on a 6% nondenaturing polyacrylamide gel. Table 2 ▶ lists the restriction enzymes used and the products anticipated from the known GenBank sequences. In every case, an initial PCR product of the correct size was obtained, and restriction enzyme products of the correct size were obtained (Table 2) ▶ .
Western blots were performed on extracts of the soluble fraction of homogenates of control and AD hippocampus and compared with normal human serum and and serum activated by aggregated IgG. Brain samples were homogenized in 5× vol/protein extraction buffer (0.02 mol/L Tris-HCl, pH 7.5) containing the protease inhibitors phenylmethylsulfonyl fluoride (1 μg/ml) and aprotinin (1 μg/ml), and 1 mmol/L EDTA. Homogenates were centrifuged at 18,000 × g at 4°C for 30 minutes. The protein content of the supernatants was determined, 19 and the samples were diluted in SDS sample buffer (60 mmol/L Tris, pH 6.8; 2.5% SDS, 5% β-mercaptoethanol) to a final protein content of 1 μg/ml and were boiled for 3 minutes. Samples containing 20 μg of protein were loaded onto 7.5% acrylamide minigels.
Normal human serum taken from a 44-year-old male volunteer was diluted 1:20 in Veronal buffer. A 2-ml aliquot of the diluted serum was mixed with 50 ml of a solution of 2 μg/ml heat-aggregated human IgG (Sigma). The mixture was incubated at 37°C for 1 hour. Aliquots of the normal and IgG activated serum were then diluted in 2 volumes of SDS buffer and boiled for 3 minutes. Samples containing 20 μg of protein were loaded onto 7.5% acrylamide minigels. Life Technologies high range prestained standards were used as molecular weight markers. After 45 minutes of electrophoresis (200 V), the proteins were transferred onto nitrocellulose membranes (Immobilon P, Millipore Corp, MA) at 7 V for 48 minutes using a semidry blotter.
Due to the high molecular weight of the membrane attack complex, modifications of the electrophoresis and protein transfer steps were required. A 3% polyacrylamide gel was used, and separation was carried out for 2.5 hours at 100 V in a cold room with the apparatus surrounded by ice. Transfer to membranes was then carried out at 100 V for 5 hours in the cold.
Membranes were blocked in 5% skim milk for 2 hours. The immunoblots were next treated for 2 hours at room temperature with a primary anti-complement antibody, followed by treatment for 1 hour with an appropriate secondary antibody labeled with horseradish peroxidase. The primary and secondary antibodies are listed in Table 3 ▶ . Immunoreactivity was visualized by incubation with Supersignal CL-HRP chemiluminescent substrate (Pierce Chemical Co., Rockford, IL). After draining, the membranes were covered in clear plastic wrapping and exposed to x-ray film (Hyper Film ECL, Amersham Life Sciences) for 0.3 to 2 minutes, depending on the strength of the signal.
Table 3.
Antibodies, Sources, and Dilutions
| Antibodies | Host | Source | Dilution* |
|---|---|---|---|
| Primary | |||
| C1q | Goat | Quidel | 5k (50k) |
| C1r | Goat | ICN Biochemicals | 2k |
| C1s | Goat | Quidel | 2.5k |
| C2 | Goat | Quidel | 2k |
| C3 | Goat | Calbiochem | 10k |
| C4 | Goat | Chemicon | 10k |
| C5 | Goat | Quidel | 3k |
| C6 | Goat | Calbiochem | 2k |
| C7 | Goat | Quidel | 3k |
| C8 | Goat | Calbiochem | 5k |
| C9 | Goat | Quidel | 5k |
| C3d | Rabbit | Quidel | 5k |
| C4d | Mouse | Quidel | 3k (10k) |
| C5b-9 | Mouse | DAKO | 1k (1k) |
| sC5b-9 neo | Rabbit | Advanced Res. Tech | 2k |
| Secondary | |||
| Anti-goat HRP-conjugated IgG | Sigma | 5–10k | |
| Anti-rabbit HRP-conjugated IgG | Sigma | 5k | |
| Anti-mouse HRP-conjugated IgG | Sigma | 5–6k | |
| Anti-goat biotin-conjugated IgG | Vector | (1k) | |
| Anti-rabbit biotin-conjugated IgG | Vector | (1k) | |
| Anti-mouse biotin-conjugated IgG | Vector | (1k) |
Detection kit used: Vectastain ABC HRP Elite from Vector (2k). HRP, horseradish peroxidase.
* Dilutions without parentheses are those used in Western blots. Dilutions in parentheses are those used in immunohistochemistry.
Immunohistochemistry
Immunohistochemistry was performed as previously reported. 13 Briefly, 30-μm sections were cut on a freezing microtome. Free-floating sections were first treated for 30 minutes with 0.3% H2O2 solution in 0.01 mol/L phosphate-buffered saline, pH 7.4, containing 0.3% Triton X-100 to reduce endogenous peroxidase activity. They were then incubated overnight at 4°C with one of the primary antibodies shown in Table 3 ▶ . The sections were washed and treated with appropriate biotinylated secondary antibodies (Table 3) ▶ for 2 hours at room temperature, followed by incubation in avidin-biotinylated horseradish peroxidase complex (ABC Elite, Vector) for 1 hour at room temperature. Peroxidase labeling was visualized by incubation of the sections in 0.01% 3,3′-diaminobenzidine (Sigma) containing 0.6% nickel ammonium sulfate and 0.00015% H2O2 in 0.05 mol/L Tris-HCl buffer, pH 7.6. When a dark purple color developed, sections were washed, mounted on glass slides, and coverslipped with Entellan. Controls were performed by omitting the primary antibody.
Statistical Analysis
Data are expressed as means ± SE. The data were analyzed by analysis of variance, followed by Student t-tests, for the significance of the difference between AD and control across all complement mRNAs and regions, and for the individual complement mRNA by region. Correlation analyses were used to determine whether there was any significant relationship between the level of each brain mRNA studied and postmortem delay in either the control or AD group. The Holm multiple comparison method 20 was applied to each set of analyses. Significance was accepted at P < 0.05. Data were analyzed as directly obtained and also as values normalized to the cyclophilin internal standard in the same sample. Cyclophilin levels typically fell within a range of 1% (Tables 2 and 4) ▶ ▶ , so the normalization adjustment was minor and did not affect the statistical differences found.
Table 4.
Complement mRNAs in Various Tissues
| mRNA | Hippocampus | Liver | Spleen | Kidney | Heart | AD Hipp/AD Liver | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | Control | AD | Control | AD | Control | AD | Control | AD | Control | ||
| C1q | 79.5 ± 9.0 | 3.45 ± 1.5 | 18.1 ± 1.6 | 15.9 ± 3.2 | 2.9 ± 2.4 | 1.8 ± 0.6 | 4.6 ± 3.5 | 2.1 ± 0.9 | 1.1 ± 1.1 | 0.7 ± 0.7 | 4.39 |
| C1r | 59.7 ± 6.3 | 12.9 ± 3.6 | 28.2 ± 2.7 | 30.8 ± 0.7 | 18.7 ± 6.3 | 18.6 ± 1.8 | 21.1 ± 2.0 | 15.3 ± 2.2 | 11.0 ± 4.0 | 8.4 ± 3.2 | 2.11 |
| C1s | 58.0 ± 3.1 | 19.3 ± 5.3 | 37.5 ± 5.3 | 46.0 ± 3.1 | 19.2 ± 5.1 | 23.5 ± 1.7 | 22.0 ± 0.7 | 19.3 ± 1.4 | 11.2 ± 4.5 | 9.2 ± 4.8 | 1.55 |
| C2 | 37.7 ± 4.1 | 24.1 ± 1.7 | 10.3 ± 6.4 | 16.3 ± 2.3 | 17.6 ± 2.5 | 17.8 ± 2.0 | 20.6 ± 4.8 | 15.5 ± 1.6 | 10.3 ± 6.1 | 8.1 ± 4.2 | 3.74 |
| C3 | 141 ± 14 | 73.1 ± 1.9 | 72.9 ± 8.1 | 74.6 ± 4.1 | 58.0 ± 4.8 | 56.1 ± 3.7 | 53.4 ± 7.6 | 53.3 ± 2.3 | 34.8 ± 6.3 | 29.1 ± 5.7 | 1.93 |
| C4 | 141 ± 3 | 74.9 ± 1.4 | 72.5 ± 5.8 | 76.6 ± 1.5 | 52.1 ± 5.2 | 53.6 ± 3.0 | 52.6 ± 7.8 | 50.3 ± 2.8 | 25.4 ± 5.5 | 30.3 ± 7.0 | 1.94 |
| C5 | 36.7 ± 7.7 | 20.2 ± 0.1 | 17.2 ± 2.8 | 16.0 ± 2.5 | 13.2 ± 3.6 | 12.7 ± 1.1 | 11.1 ± 3.8 | 14.4 ± 2.3 | 8.4 ± 5.0 | 5.1 ± 1.9 | 2.13 |
| C6 | 28.9 ± 1.6 | 15.5 ± 2.4 | 24.9 ± 5.5 | 17.3 ± 1.8 | 7.2 ± 1.6 | 7.9 ± 1.5 | 6.3 ± 1.1 | 3.4 ± 1.0 | 1.7 ± 1.7 | 0.8 ± 0.7 | 1.16 |
| C7 | 39.8 ± 3.6 | 6.3 ± 4.1 | 27.3 ± 1.4 | 35.7 ± 3.4 | 15.0 ± 3.8 | 13.4 ± 2.5 | 14.7 ± 3.9 | 14.5 ± 2.1 | 4.1 ± 0.8 | 4.7 ± 0.6 | 1.46 |
| C8 | 42.6 ± 2.9 | 15.3 ± 8.0 | 28.7 ± 8.2 | 41.0 ± 3.9 | 3.6 ± 2.1 | 1.5 ± 0.7 | 3.3 ± 1.7 | 1.8 ± 0.9 | 6.5 ± 6.5 | 0 ± 0 | 1.48 |
| C9 | 56.4 ± 8.0 | 2.55 ± 0.8 | 23.2 ± 7.6 | 27.6 ± 2.4 | 3.9 ± 2.8 | 1.3 ± 0.7 | 3.2 ± 1.7 | 2.6 ± 0.8 | 3.6 ± 2.6 | 0.5 ± 0.5 | 2.43 |
| Cyc/100 | 85.4 ± 1.1 | 85.5 ± 1.8 | 94.6 ± 0.9 | 94.8 ± 0.8 | 107 ± 0.9 | 108 ± 1 | 94.5 ± 0.5 | 93.8 ± 0.8 | 94.3 ± 1.7 | 94.1 ± 1.6 | 0.90 |
| N | 4 | 5 | 3 | 10 | 3 | 11 | 3 | 11 | 3 | 4 |
Data are means ± S.E., with N for each tissue indicated in the bottom line. In no case was there a significant difference by ANOVA between the AD and control data except in the hippocampus as indicated in Figure 2 ▶ .
Results
RT-PCR products were detected in every AD case and in each of the brain areas studied. Figure 1 ▶ is an example of an ethidium bromide-stained gel showing the individual products for each of the complement mRNAs. Single products were obtained of the predicted size. The cDNA of each product was purified and subjected to selective endonuclease digestion. In each case, the predicted number of products of the expected size was obtained. Details are shown in Table 2 ▶ .
Figure 1.

Representative Polaroid photograph showing an ethidium bromide stained gel of RT-PCR products of C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, and C9 mRNAs. The cDNA had been obtained from a total RNA extract of AD hippocampus. Electrophoretic separation was performed on a 6% polyacrylamide gel. Size markers are in lane M, with sizes shown in base pairs. See under Methods for details.
The quantitative levels of the RT-PCR products varied according to the area of brain investigated, the individual complement mRNA, and the type of case. This was in contrast to the housekeeping gene cyclophilin, which was almost constant from case to case and area to area of any given brain. The overall value for all brain areas in AD cases was 8509 ± 27 (N = 44) which was not significantly different from the value of 8405 ± 142 (N = 55) for all brain areas of normal cases. There was no significant correlation with postmortem delay. There was also no significant correlation with postmortem delay of any of the complement mRNAs in normal or AD brain tissue.
Figure 2 ▶ shows bar graphs in which average normalized complement mRNA levels of AD and control cases are compared for each of the 11 areas of brain studied. Notice in all of the bar graphs how closely comparable the cyclophilin values were for normal and AD cases. As can be seen from the graphs, C3 and C4 mRNAs were the most highly expressed of all of the complement mRNAs in all areas of brain in both AD and normal cases. In control brain, C1q and C9 mRNAs were expressed at the lowest levels, with the other complement mRNAs lying in between. The expression of all complement mRNAs varied little from area to area in control brains although C8 mRNA was not detected in the occipital and motor cortices.
Figure 2.
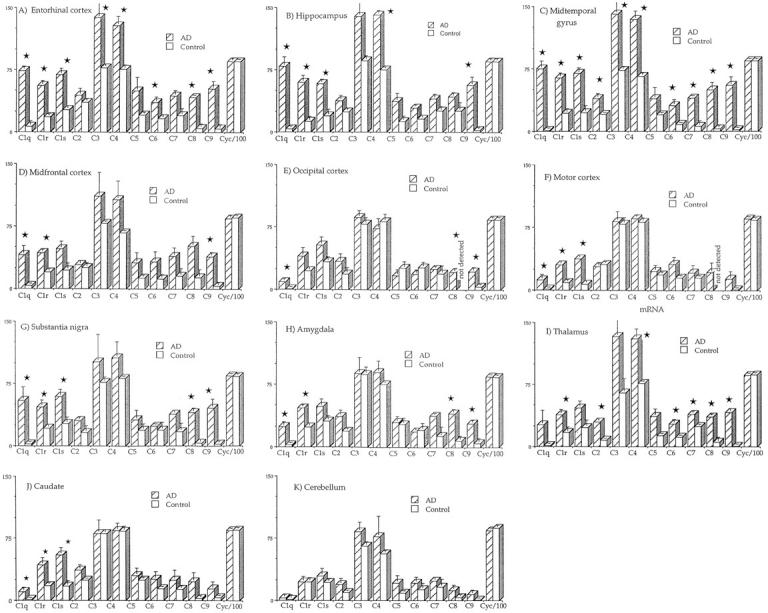
Bar graphs showing relative levels of complement and cyclophilin mRNAs in extracts of AD and non-AD tissue. Optical density units represent readings of ethidium bromide gel band intensities as determined on a GDS6700 image analyzer with quantitative analysis and use of NIH Image software 1.61. Values are averages with bars indicating S.E. Bars marked with a star are those where the AD levels are significantly different from the control levels (P < 0.05) after analysis of variance tests with Holm’s 20 stepdown correction (see under Methods for details).
In AD brains, the situation was quite different. C1q and C9 mRNAs were sharply up-regulated, and this up-regulation was greatest in areas of high pathology such as the entorhinal cortex, hippocampus, and midtemporal gyrus. For example, C1q mRNA increased in the midtemporal gyrus from an average level of 0.93 OD units in controls to 74.9 in AD. The comparable increase in the hippocampus was from 3.45 to 79.5, and in the entorhinal cortex from 6.5 to 73.5. C9 mRNA increased in the midtemporal gyrus from an average level of 1.99 OD units in controls to 55.1 in AD, in the hippocampus from 2.55 to 56.4, and in the entorhinal cortex from 4.07 to 52.1 (see bar graphs in Figure 2 ▶ ). In the cerebellum, which is only mildly affected in AD, C1q mRNA increased from 1.39 in controls to only 3.30 in AD cases. C9 mRNA increased from 0.65 in controls to 6.98 in AD (see graphs in Figure 2 ▶ ). Other complement mRNAs followed the same general pattern of up-regulation in AD, although the up-regulation was not as great as that observed for C1q and C9 mRNAs. The least up-regulation was for C3 and C4 mRNAs, which were expressed at the highest levels. The other areas of brain, which were intermediate in their pathological involvement, were also intermediate in their mRNA up-regulation.
Complement mRNAs were also measured in the liver, heart, spleen, and kidney of AD and control cases. The results are shown in Table 4 ▶ . Inspection of this table shows that the mRNA levels in these peripheral areas were highly comparable between AD and control cases, with no statistically significant differences between them being detected. It is noteworthy, however, that the complement mRNA levels in affected areas of AD brain were substantially higher than those in AD or control liver and other peripheral organs.
Figure 3 ▶ shows the results of Western blot experiments of normal serum (lane 1), of extracts of control hippocampus (lane 2) and AD hippocampus (lane 3), and of the same serum as lane 1 with aggregated IgG added to activate complement (lane 4). Strong bands were detected for all of the complement proteins in the AD hippocampal extract and IgG treated serum, including bands for the activation fragments C3d, C4d, and the membrane attack complex. These same bands were detected to a much lesser extent in the normal serum sample, possibly indicating mild in vivo activation. By contrast, bands were not detected in the control hippocampal extract for C1q, C1r, C1s, C2, C5, C6, C7, C8, C9, and the membrane attack complex. The complement protein fragments represented by these bands have previously been reported for Western blots of heart extracts. 16 These data indicate that the complement mRNAs detected in RT-PCR experiments are being translated into their protein products, and that the classical pathway is being activated in tissue.
Figure 3.
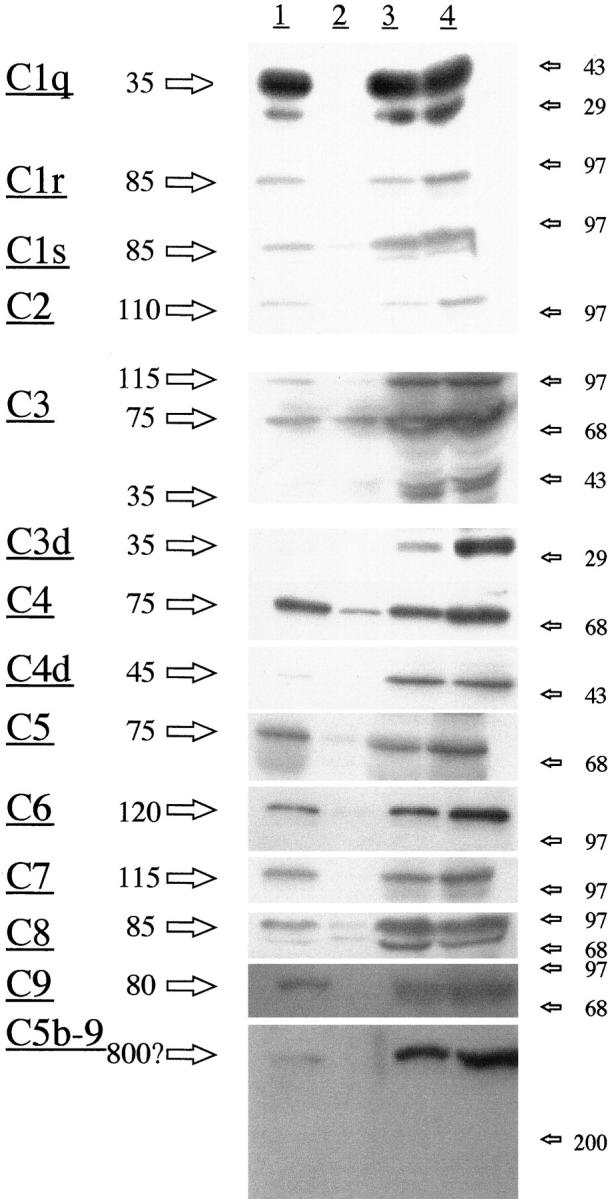
Western blot analysis from protein extracts of AD and control hippocampus compared with normal serum and the same serum activated by aggregated IgG (see under Methods for details). Lane 1, normal serum; lane 2 , normal hippocampal extract; lane 3, AD hippocampal extract; lane 4, serum activated by IgG. Positions of molecular weight markers are shown by the arrows on the right; the estimated molecular weights of the main bands detected are shown by arrows on the left. Note that strong bands are detectable for all complement proteins in the AD extract and the activated serum sample. Detectable bands were not obtained from the control hippocampal extract for C1q, C1r, C1s, C2, C5, C6, C7, C8, C9, and the membrane attack complex. By contrast, very strong bands were obtained for the activated complement fragments C3d and C4d and the membrane attack complex in AD tissue and activated serum.
Figure 4 ▶ illustrates by immunohistochemistry association of some of the complement proteins with the lesions of AD. Antibodies to C1q, the component which identifies the target tissue and initiates the complement cascade, stain senile plaques prominently, as well as tangled pyramidal neurons in the AD transentorhinal cortex (Figure 4A) ▶ . Control transentorhinal cortex, under the same conditions, shows no immunostaining (Figure 4B) ▶ . Very intense staining of senile plaques in the same area in AD is found for C4d, a fragment characteristic of classical pathway activation (Figure 4C) ▶ . Again, no staining is seen in control tissue (Figure 4D) ▶ . The antibody to the membrane attack complex prominently stains dystrophic neurites and intracellular tangles in AD transentorhinal cortex (Figure 4E) ▶ , while no staining is obtained in control tissue (Figure 4F) ▶ . Since the membrane attack complex can destroy host cells through the process of bystander lysis, the evidence of binding to dystrophic neurites in Figure 4E ▶ is suggestive of autodestruction of tissue.
Figure 4.
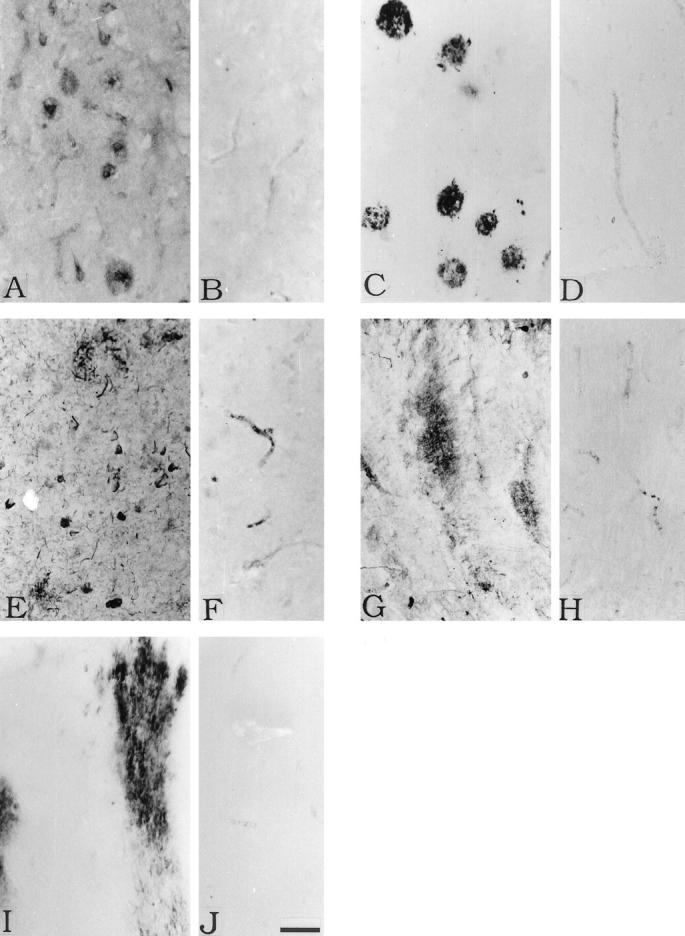
Immunohistochemical staining of AD transentorhinal cortex (A, C, E) and cerebellum (G, I) and control transentorhinal cortex (B, D, F) and cerebellum (H, J). Staining is for C1q (A, B, G, H), C4d (C, D, I, J) and C5b-9 (E, F ). See under Methods for details. In A, the C1q antibody stains senile plaques as well as tangled neurons very weakly. In G, the C1q antibody stains diffuse amyloid deposits. In C, the C4d antibody stains senile plaques, and in I it stains diffuse amyloid deposits. In E, the anti-C5b-9 antibody stains dystrophic neurites and tangled neurons. The control sections B, D, F, H, and J show no specific staining of brain structures. Scale bar in J = 50 μm.
Discussion
The results described here confirm earlier reports that the mRNAs and proteins of all components of the classical complement pathway can be detected in AD brain tissue. 13,14 They expand previous reports which showed up-regulation of the mRNAs for C1q, C3, and C4 15,21 and the protein for C1q 22,23 in AD. They also confirm previous immunohistochemical data showing that the classical complement pathway is activated in AD brain, 1-4 including expression of the membrane attack complex. 4,24
Here we have made semiquantitative comparisons of the mRNA levels of all components of the classical complement pathway, ie, C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, and C9, showing that they are easily detected in all 11 areas of AD brain studied. They are also detectable in normal brain tissue, with the minor exception of C8 in the occipital and motor cortices. C3 and C4 were the most abundantly expressed of the mRNAs in all areas of both normal and AD tissue. They showed little variation from area to area in normal tissue, and showed less than a twofold up-regulation in AD brain tissue, even in the most heavily affected areas. In contrast, C1q, the component which activates the pathway by binding to target tissue, and C9, the component which gives functional capacity to the membrane attack complex, were expressed at the lowest levels in normal tissue but showed dramatic up-regulation in affected AD brain areas.
The mRNA levels for the housekeeping gene cyclophilin were remarkably constant in all areas and for all cases, with values generally falling within 1% of each other and with an outside range of less than 4%. Cyclophilin was also unaffected by postmortem delay, as we have previously reported. 16 The constancy of cyclophilin mRNA levels indicates that the methodology itself is highly reproducible and that the gene product is unaffected by the various pathologies and postmortem delay. The complement mRNAs were similarly unaffected by postmortem delay in normal and AD brains. The high variability in complement mRNA values observed between highly affected and mildly affected areas of AD brain is therefore probably reflective of the physiological state of the tissue and not other extraneous factors.
All of the complement mRNAs were, as expected, strongly detected in liver of AD and non-AD cases. The levels in AD and non-AD liver were highly comparable (Table 4) ▶ . By contrast, the levels in AD hippocampus were substantially higher for all of the complement components than in AD or control liver (Table 4) ▶ . These data are again consistent with the concept of high local production of complement mRNAs in affected areas of AD brain, with continuous activation of the classical pathway.
The mRNAs for all complement components were also detected in spleen, kidney, and heart (Table 4) ▶ . In general, these levels were comparable to those observed in normal brain, with that for C1q being the lowest and those for C3 and C4 the highest. The fact that all organs produce mRNAs for all complement proteins suggests that complement production is far more ubiquitous than is generally recognized and that local production and activation may be associated with a wide spectrum of pathological disorders.
We have previously reported on the expression of complement mRNAs in isolated rabbit hearts and their up-regulation after reperfusion. 25 We have also reported on the presence of complement mRNAs and their protein products in human heart and their up-regulation after myocardial infarcts. 16 Activated complement fragments were detected on damaged myocardium of previously as well as recently infarcted tissue. This indicates that complement-mediated damage can continue to accumulate for years after an initial insult, and, in the case of heart, this insult may be as mild as an anoxic episode.
The previously reported association of activated complement proteins with AD lesions does not indicate when, and to what degree, the complement pathway has been activated. The process could have been proceeding at a low level for years. However, the high up-regulation of mRNAs, and the appearance of strong bands in Western blots for complement activation products, indicates that vigorous activation of the pathway must be continuously taking place. Because the complement system powerfully drives inflammation and tissue destruction in a highly targeted manner, these results imply a substantial contribution of complement to AD pathology.
The process by which the endogenously produced complement proteins are activated is not known with certainty. However, the disease process in AD involves continuous deposition of amyloid deposits. These deposits contain the in vitro activators β-amyloid protein, 6 amyloid P, 26-29 C-reactive protein, 30 and Hageman factor, 31 which should provide a strong stimulus for in vivo activation. Extracellular neurofibrillary tangles, which are also being continuously formed as neurons with intracellular tangles die, have deposited on them the complement activator amyloid P. 32 The high up-regulation of mRNAs in areas where plaques and tangles accumulate may explain why these areas are highly vulnerable to the AD pathological process.
There are now considerable data suggesting that inflammatory processes drive the pathology in AD. Multiple epidemiological studies show that individuals taking anti-inflammatory drugs or suffering from conditions where such drugs are routinely administered, have a substantially reduced prevalence of AD. 33,34 Moreover, a small double-blind, placebo-controlled clinical trial of the anti-inflammatory drug indomethacin demonstrated an apparent arrest of the disease process. 35 Complement may set the pace of neuronal degeneration. If this is true, complement inhibitors might prove to be highly effective therapeutic agents in AD.
Acknowledgments
We thank Dr. Michael Schulzer, Prof. of Medicine and Statistics, University of British Columbia, for invaluable help with the statistical analysis.
Footnotes
Address reprint requests to Dr. Patrick L. McGeer, Kinsmen Laboratory of Neurological Research, University of British Columbia, 2255 Wesbrook Mall, Vancouver, B.C., V6T 1Z3 Canada.
Supported by a grant from the Jack Brown and Family A.D. Research Fund and donations from individual British Columbians.
References
- 1.Eikelenboom P, Stam FC: Immunoglobulins and complement factors in senile plaques: an immunoperoxidase study. Acta Neuropathol 1982, 57:239-242 [DOI] [PubMed] [Google Scholar]
- 2.Eikelboom P, Hack CE, Rozemuller JM, Stam FC: Complement activation in amyloid plaques in Alzheimer’s dementia. Arch Cell Pathol 1989, 56:259-262 [DOI] [PubMed] [Google Scholar]
- 3.Ishii T, Haga S: Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol (Berl) 1984, 63:296-300 [DOI] [PubMed] [Google Scholar]
- 4.McGeer PL, Akiyama H, Itagaki S, McGeer EG: Immune system response in Alzheimer’s disease. Can J Neurol Sci 1989, 16:516-527 [DOI] [PubMed] [Google Scholar]
- 5.McGeer PL, McGeer EG: The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev 1995, 21:195-218 [DOI] [PubMed] [Google Scholar]
- 6.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren S, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I: Complement activation by β-amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1992, 89:10016-10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnum SR: Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med 1995, 6:132-146 [DOI] [PubMed] [Google Scholar]
- 8.Gasque P, Fontane M, Morgan BP: Complement expression in human brain: biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J Immunol 1995, 154:4726-4733 [PubMed] [Google Scholar]
- 9.Shen Y, Sullivan T, Lee CM, Meri S, Shiosaka K, Lin CW: Induced expression of neuronal membrane attack complex and cell death by Alzheimer’s β-amyloid peptide. Brain Res 1998, 796:187-197 [DOI] [PubMed] [Google Scholar]
- 10.Walker DG, McGeer PL: Complement gene expression in neuroblastoma and astrocytoma cell lines of human origin. Neurosci Lett 1993, 157:99-102 [DOI] [PubMed] [Google Scholar]
- 11.Walker DG, Kim SU, McGeer PL: Complement and cytokine gene expression in cultured microglia derived from postmortem human brains. J Neurosci Res 1995, 40:478-493 [DOI] [PubMed] [Google Scholar]
- 12.Singhrao SK, Neal JW, Gasque P, Morgan BP, Newman GR: Role of complement in the aetiology of Pick’s disease. J Neuropathol Exp Neurol 1996, 55:578-593 [DOI] [PubMed] [Google Scholar]
- 13.Terai K, Walker DG, McGeer EG, McGeer PL: Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res 1997, 769:385-390 [DOI] [PubMed] [Google Scholar]
- 14.Shen Y, Li R, McGeer EG, McGeer PL: Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res 1997, 769:391-395 [DOI] [PubMed] [Google Scholar]
- 15.Johnson SA, Lampert-Etchells M, Pasinetti GM, Rozovsky I, Finch CE: Complement mRNA in the mammalian brain: responses to Alzheimer’s disease and experimental brain lesioning. Neurobiol Aging 1992, 13:641-648 [DOI] [PubMed] [Google Scholar]
- 16.Yasojima K, Schwab C, McGeer EG, McGeer PL: Human heart generates complement proteins which are upregulated and activated following myocardial infarction. Circ Res 1998, 83:860-869 [DOI] [PubMed] [Google Scholar]
- 17.Nakayama H, Yokoi H, Fujita J: Quantification of mRNA by non-radioactive RT-PCR and CCD imaging system. Nucleic Acids Res 1992, 20:4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi M, Yamada T, Tooyama I, Moroo I, Kimura H, Yamamoto T, Okada H: Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci Lett 1996, 204:201-204 [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, Randall EJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193:265-275 [PubMed] [Google Scholar]
- 20.Holm S: A simple sequentially repetitive multiple test procedure. Scand J Statist 1979, 6:65-70 [Google Scholar]
- 21.Walker DG, McGeer PL: Complement gene expression in human brain: comparison between normal and Alzheimer disease cases. Mol Brain Res 1992, 14:109-116 [DOI] [PubMed] [Google Scholar]
- 22.Brachova L, Lue L-F, Schultz J, Rashidy TE, Rogers J: Association cortex, cerebellum and serum concentration of C1q and factor B in Alzheimer’s disease. Mol Brain Res 1993, 18:329-334 [DOI] [PubMed] [Google Scholar]
- 23.Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ: Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol 1996, 138:22-32 [DOI] [PubMed] [Google Scholar]
- 24.Webster S, Lue L-F, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J: Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging 1997, 18:415-421 [DOI] [PubMed] [Google Scholar]
- 25.Yasojima K, Kilgore KS, Washington RA, Lucchesi BR, McGeer PL: Complement gene expression by rabbit heart: upregulation by ischemia and reperfusion. Circ Res 1998, 82:1224-1230 [DOI] [PubMed] [Google Scholar]
- 26.Kalaria RN, Galloway PG, Perry G: Widespread amyloid P component immunoreactivity in cortical amyloid deposits and the neurofibrillary pathology of Alzheimer’s disease and other degenerative disorders. Neuropathol Appl Neurobiol 1991, 17:189-201 [DOI] [PubMed] [Google Scholar]
- 27.Coria F, Castano E, Prelli F, Larrondo-Lillo M, Van Duinen S, Shelanski ML, Frangione B: Isolation and characterization of amyloid P component from Alzheimer’s disease and other types of cerebral amyloidosis. Lab Invest 1988, 58:454-458 [PubMed] [Google Scholar]
- 28.Duong T, Pommier EC, Schiebel AB: Immunodetection of the amyloid P component in Alzheimer’s disease. Acta Neuropathol 1989, 78:429-437 [DOI] [PubMed] [Google Scholar]
- 29.Akiyama H, Yamada T, Kawamata T, McGeer PL: Association of amyloid P component with complement proteins in neurologically diseased tissue. Brain Res 1991, 548:349-352 [DOI] [PubMed] [Google Scholar]
- 30.Iwamoto N, Nihiyama E, Ohwada J, Arai H: Demonstration of CRP immunoreactivity in brains of Alzheimer’s disease: immunohistochemical study using formic acid pretreatment of tissue sections. Neurosci Lett 1994, 177:23-26 [DOI] [PubMed] [Google Scholar]
- 31.Yasuhara O, Walker DG, McGeer PL: Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res 1994, 654:234-240 [DOI] [PubMed] [Google Scholar]
- 32.Schwab C, Steele JC, McGeer EG, McGeer PL: Amyloid P immunoreactivity precedes C4d deposition on extracellular neurofibrillary tangles. Acta Neuropathol 1997, 93:87-91 [DOI] [PubMed] [Google Scholar]
- 33.McGeer PL, Schulzer M, McGeer EG: Arthritis and antiinflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiological studies. Neurology 1996, 47:425-432 [DOI] [PubMed] [Google Scholar]
- 34.Stewart WF, Kawas C, Corrada M, Metter EJ: Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48:626-632 [DOI] [PubMed] [Google Scholar]
- 35.Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, Kogan F: Clinical trial of indomethacin in Alzheimer’s disease. Neurology 1993, 43:1609-1611 [DOI] [PubMed] [Google Scholar]