

Abstract
To elucidate the mechanism of tumor extension in human pulmonary adenocarcinoma, we immunohistochemically investigated the expression of cell cycle regulator proteins in 54 small adenocarcinomas less than 3 cm in diameter. The Ki-67-labeling index was significantly higher in the periphery of the tumor nodule than in the center. This proliferative potential correlated well with coexpression of cdk2 and cyclin A. p27, one of the cdk inhibitors, was highly expressed in normal bronchial epithelial cells. Peripherally located tumor cells expressed p27 at an intermediate level, but at a higher frequency and level than those in the center. Expression of p21 was also predominant in the periphery. Furthermore, the expression patterns of p21 and p27 were reciprocal. In vitro kinase assays further demonstrated higher cdk2 kinase activity in the periphery. These results suggest that: (i) within an emerging extension made up of peripherally located tumor cells, their high proliferative potential gradually wanes as their relative topographical position becomes more central in the expanding tumor; (ii) peripherally located tumor cells maintain their proliferative potential by higher cyclin A-cdk2 complex activity; and (iii) intermediate expression of p21/p27 in the peripherally located cells promotes higher cyclin A-cdk2 kinase activity, whereas high p21/p27 expression in nonneoplastic cells inhibits kinase activity.
Adenocarcinoma is the most common histological subtype of lung carcinoma and its incidence is increasing. 1-3 Recently, its developmental basis, and mode of extension, and causative genetic alterations have been fairly well clarified. Following the multistep accumulation of genetic mutations, including oncogenic activation of genes such as K-ras 4,5 and inactivation of tumor suppressor genes such as p53, 6-8 an early stage adenocarcinoma can develop de novo or from its putative preceding lesions. 9,10 One typical histological subtype, well differentiated papillary adenocarcinoma, further pursues sequential morphological changes described as going from “bronchioloalveolar carcinoma replacing alveolar-lining epithelium with thin stroma” to “bronchioloalveolar carcinoma with foci of active fibroblastic proliferation,” finally progressing to an advanced stage of adenocarcinoma characterized by a central scar and remarkable pleural indentation. 11 During this process, cells in the peripheral region of the tumor nodule, particularly around the advancing border, extend outward with higher proliferative activity while those in the central region exhibit attenuated proliferative activity accompanied by surrounding stromal degenerative changes, resulting in the eventual development of fibrotic scar. 12,13 In this sense, a small, well differentiated adenocarcinoma with central fibrosis is a good model in which to examine the mechanism of cell proliferation and tumor extension at the cellular level, including the decrease in proliferative activity as the topographical location of cells changes within the tumor nodule.
Cell proliferation is strictly regulated by a cell cycle control mechanism which depends on the activities of the G1 cyclins and cyclin-dependent kinase (cdk) complexes. 14-21 These complexes are regulated both positively and negatively. Positive regulators include the cyclins and the recently identified cdk-activating kinase (CAK) by which cdks are phosphorylated at specific threonine residues and activated. 22-25 In addition, multiple negative regulators exist, including universal cdk inhibitors p21, p27, and p57 and cdk4/cdk6 inhibitors p16, p15, p18, and p19. 17-19,25-27 Thus, cell proliferation is controlled by complex and redundant mechanisms.
Despite a large body of morphological observations, the mode of tumor extension, the pathological mechanisms of cell proliferation in human pulmonary carcinomas, and, more particularly, the participation of various cell cycle regulators have not been fully analyzed at the cellular level.
To elucidate these mechanisms in lung adenocarcinoma, we examined the expression of cell cycle regulator proteins in the early stage of pulmonary adenocarcinoma by immunohistochemistry, with special emphasis on the G1/S- and S-to-G2 cell cycle transitions.
Materials and Methods
Cases and Histological Classification
This study examined 54 cases of primary well differentiated adenocarcinoma of the lung, each less than 3 cm in maximum diameter and classified into Stage I (T1M0N0) by the TMN classification. 28 These adenocarcinomas were derived from surgically resected materials obtained in the Departments of Pathology, Saiseikai Central Hospital and Kitasato University Hospital between 1987 and 1997. All of these cases were classified as type C (“localized bronchioloalveolar carcinoma with foci of active fibroblastic proliferation”) according to the histopathological classification of early adenocarcinoma of the lung (Figure 1) ▶ . 11
Figure 1.
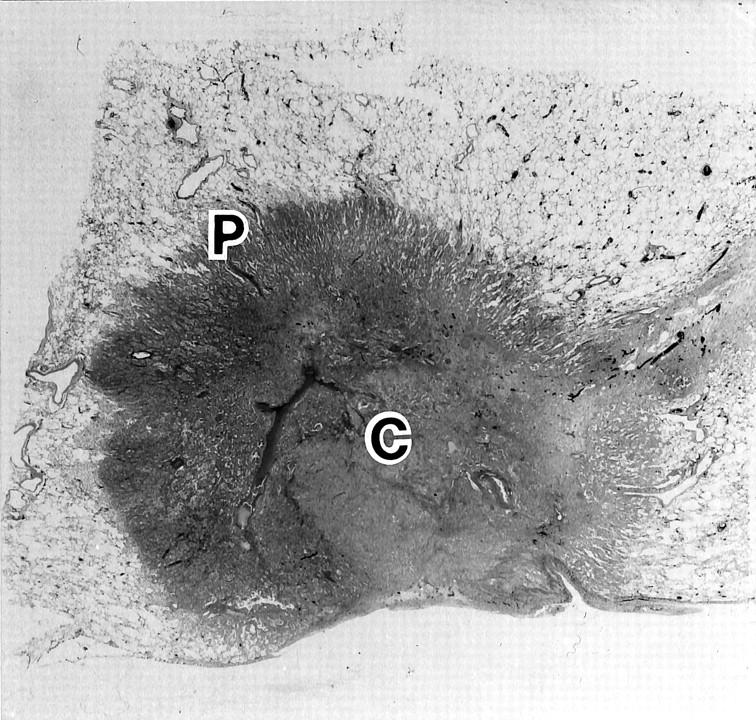
Histological features of typical well differentiated adenocarcinoma, type C, characterized by polarity formation: the peripheral region (P) where tumor cells form an emerging extension on the alveolar surface and the central region (C) with less cellular granulation tissue in a tumor nodule.
Archival Tissue Samples and Immunohistochemistry
All archival tissue samples were routinely fixed in formalin and embedded in paraffin. Deparaffinized sections were autoclaved (120°C, 2 atm., 20 minutes) in 20 mmol/L citrate buffer (pH. 6.0). 29 Immunostaining was performed with primary antibodies at the following dilutions: anti-cyclin A (monoclonal, Novocastra, Newcastle, UK), 1:500 dilution; anti-p21, anti-p27 (monoclonal, Novocastra), 1:100; anti-cdk-activating kinase (anti-CAK, monoclonal, Novocastra), 1:200; anti-p53 (monoclonal, DAKO, Glostrup, Denmark), 1:100; anti-Ki-67 (monoclonal, DAKO), 1:100; anti-cdk2 (polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA), 1:2000; and anti-epithelial keratin (AE-1, monoclonal, ICN, Lisle, IL), 1:200. The specificity of these antibodies was confirmed by immunoblotting (data not shown). The conventional streptavidin-biotinylated horseradish peroxidase complex method (LSAB kit, DAKO, Kyoto, Japan) was used as directed by the manufacturer’s instructions. Colorization was performed by the peroxidase-diaminobenzidine method. In selected cases, serial sections 1 μm in thickness were made and stained alternately for several kinds of proteins known to interact mutually or to form a complex, including cdk2-cyclin A-p21/p27 or p53-p21.
Immunoreactivity Scoring
In each case, positive staining was evaluated in three areas: nonneoplastic lung tissue adjacent to the tumor, the peripheral region around the advancing border of the tumor nodule (designated the “periphery”) where the tumor cells proliferate in emerging extension on the alveolar surface, and in the center of the tumor, often accompanied by granulation or loose fibrosis. The percentage of positive cells was estimated by counting 500 tumor cells in 10 high-power fields in each area and evaluating semiquantatively. In the present study, cases showing >5% positive tumor cells were defined as positive.
Statistical Analysis
Differences in labeling indices (LIs) between the tumor cells from the periphery and those from the center were calculated from the staining results, compared, and analyzed by paired comparison t-test. Degree of correlation between LIs obtained for two marker proteins was calculated by the Spearman’s rank correlation coefficient test.
Paired Tumor/Normal Lung Tissue Collection
Fresh fragments of tissue from the peripheral and the central regions of tumor nodules of well differentiated adenocarcinoma and paired adjacent nonneoplastic lung tissues were obtained from surgically resected specimens and were used for immunoblotting analysis and in vitro kinase reaction assay. These consisted of five cases; their immunohistochemical profiles are listed in Table 1 ▶ .
Table 1.
Histological and Immunohistochemical Profiles of Cases Utilized for in Vitro Kinase Reaction
| Case | % Positive cells on IHC | ||||||
|---|---|---|---|---|---|---|---|
| Ki-67 | cyc A | cdk2 | CAK | p21 | p27 | p53 | |
| 1 | 35 | 9 | 21 | 72 | 2 | 14 | 89 |
| 2 | 27 | 27 | 49 | 71 | 3 | 31 | 79 |
| 3 | 56 | 23 | 58 | 88 | 8 | 30 | 54 |
| 4 | 41 | 2 | 34 | 75 | 8 | 11 | 7 |
| 5 | 22 | 7 | 9 | 81 | 4 | 13 | 6 |
IHC, immunohistochemistry; cyc A, cyclin A; CAK, cdk-activating kinase.
Immunoblotting Analysis
For protein extraction, fresh tissues were homogenized in high-salt lysis buffer (50 mmol/L Tris-HCl (pH 8.0), 0.25 mol/L NaCl, 0.5% Nonidet P-40, 0.1% sodium dodecyl sulfate, 5 mmol/L EDTA, 50 mmol/L NaF, 0.5 mmol/L phenylmethylsulfonyl fluoride, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin) 30 on ice and the resulting lysates were sonicated on ice four times for 10 seconds each time. 31 Lysates were clarified by centrifugation at 10,000 × g for 5 minutes.
Initially, 50-μg aliquots of protein were used for AE-1 blotting. The same specific antibody used in the immunohistochemical staining was used in 1:200 dilution. Protein was detected by the sequential application of a primary antibody followed by an alkaline phosphatase-conjugated secondary antibody (Promega, Madison, WI, 1:6000 dilution). Colorization was performed with nitroblue tetrazolium and 5-bromo-4-chloro-3-indol-phosphate (Bio Rad, Gaithersburg, MD) in 100 mmol/L Tris buffer (pH. 9.6).
In Vitro Kinase Reaction
For immunocomplex kinase reactions to detect the activity of cdk2 or p27-associated cdks, tissues were lysed in NP40 lysis buffer (50 mmol/L Tris-HCl (pH 7.4), 0.5% Nonidet P-40, 0.15 mol/L NaCl, 50 mmol/L NaF, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin) with addition of 1 mmol/L Na3VO4. 30 To standardize the amount of extracts obtained from tumor cells or nonneoplastic epithelial cells from the three areas, the protein amounts from epithelial cells used for in vitro kinase assay were determined by anti-AE1 (keratin) blotting. In brief, lysate protein concentrations were quantitated by protein assay kit (Bio Rad, Hercules, CA), 50 μg of extract were then subjected to anti-AE-1 immunoblotting analysis, and the results were quantified using a GT6500ARTS scanner (Epson Co., Tokyo). All scans were kept as 16-bit PICT files and incorporated into NIH Image software (version 1.56) for densitometric analysis. 32 From the results of densitometric analysis, aliquots of lysates containing equivalent amounts of AE-1 were taken and incubated with anti-cdk2 antibody (diluted 1:150) for 1 hour followed by an additional 2-hour incubation with protein A-Sepharose beads at 4°C. For p27-associated kinase reactions, lysates were incubated with anti-p27 (diluted 1:100) for 1 hour followed by an additional 2-hour incubation with protein G-Sepharose beads at 4°C. A bacterially expressed fragment of the retinoblastoma protein (pRB, amino acids 385–928) fused to glutathione S-transferase was used as a substrate (0.5 μg protein) in 50 μl of kinase reaction buffer (50 mmol/L Tris-HCl (pH 7.2), 10 mmol/L MgCl2, 1 mmol/L dithiothreitol, 20 mmol/L [γ-32P]ATP(5 μCi;1 μCi = 37 kBq) (ICN, Irvine, CA). 30 After incubation for 10 minutes at room temperature, the sample was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by autoradiography.
Results
Immunohistochemical Stains and LI Values
Positive immunohistochemical staining in this study was confined almost exclusively to the nuclei (Figure 2) ▶ . The overall results of the LIs from immunohistochemical analysis are shown in Figures 2 and 3 ▶ ▶ and described below.
Figure 2.
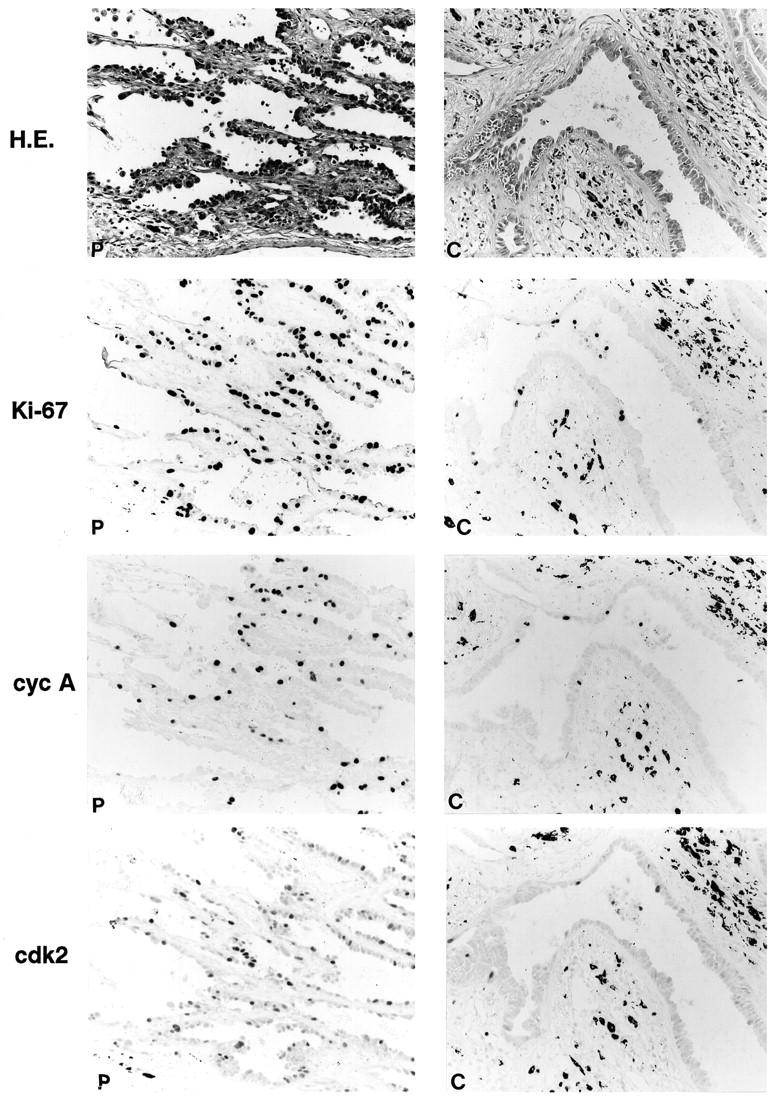
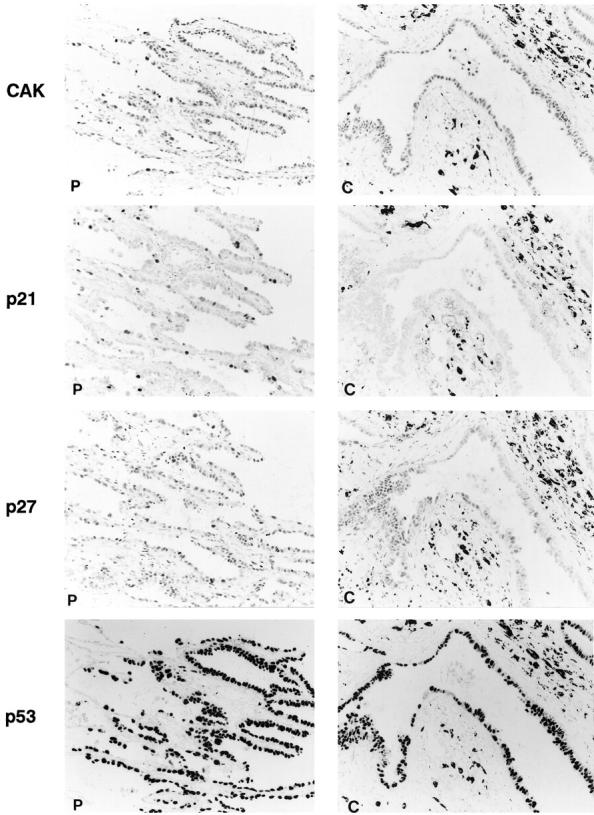
Histological features of tumor nodule of well differentiated adenocarcinoma and immunohistochemical staining for Ki-67 and cell-cycle regulator proteins. Staining results of peripheral (P) and central (C) regions of tumor nodule are shown. Streptavidin-biotin peroxidase method with hematoxylin counterstain; magnification, ×350. In the right panels (“C”), positive staining is found in some nuclei of the epithelium of a centrally located gland. There are some black pigments due to anthracosis in the stroma adjacent to the gland.
Figure 3.

Labeling index obtained from immunohistochemical staining for Ki-67 and cell-cycle regulator proteins. Labeling indices in the peripheral (P) and central (C) regions of tumor nodules were estimated and both indices in each case are connected by a line. Each oblique line represents three cases.
Ki-67
Proliferative activity was evaluated by determining the Ki-67 LI. The Ki-67 LI was 10 to 20 times higher than the mitotic index, which demonstrated the staining reliability of the antibody. 33 Nuclear positivity in the tumor cells was identified in 90.7% (49/54) of the cases. In these 49 cases, the LI of the tumor cells in the periphery ranged from 8 to 65%, whereas that of cells in the center ranged from 1 to 45%. This difference in LIs was statistically significant (P < 0.0001 by t-test, Figure 3 ▶ ). There were three cases in which the center exhibited a higher LI than the periphery. Those cases turned out to contain small foci of tumor nests in solid proliferation, corresponding to moderately or poorly differentiated adenocarcinoma in those particular areas. Most of the cells in the nonneoplastic lung tissue stained negatively except for occasional fibroblasts.
Cyclin A
The distribution of cells staining positive for cyclin A appeared to parallel Ki-67 staining except in nonneoplastic lung tissue, where positivity was identified only in the germinal center of lymphoid follicles. Nuclear positivity in the tumor cells was identified in 72.2% (39/54) of the cases. The cyclin A LIs of tumor cells in the periphery ranged from 7 to 68% and were higher to a statistically significant degree (P < 0.001) than those in the center, for which LIs ranged from 1.5 to 33% (Figures 2 and 3) ▶ ▶ .
Cdk2
Cdk2 positivity was restricted to tumor cells (Figure 2) ▶ . Nuclear positivity in tumor cells was identified in 83.3% (45/54) of the cases. In general, the cdk2 LI of the tumor cells was higher in the periphery (9–69%) compared with the center (2–50%) to a statistically significant degree (P < 0.05).
CAK
Nuclear positivity for CAK was ubiquitously identified in the tumor cells and in the nonneoplastic cells, including bronchial epithelial cells and alveolar macrophages. In the tumor cells, CAK-positive staining was identified in 94.4% (51/54) of the cases and the LI values among the tumor cells were quite similar at the periphery and the center, ranging from 11 to 93%. Staining intensity was almost identical in all areas and among various kinds of cells, except for a few instances in which the LIs and staining intensity were slightly higher in the periphery (Figure 3) ▶ . Statistically, no obvious difference in the LI between the periphery and the center of the tumors was found (P > 0.1).
p21
Cells in the nonneoplastic lung tissue generally showed no or very rare and faint staining. The exceptions were occasional intense positivity in the infiltrating lymphocytes and macrophages. Nuclear positivity in the tumor cells was identified in 72.2% (39/54) of the cases. In the positive cases, the LI was approximately, 8 to 67% in the periphery and 2 to 42% in the center (Figures 2 and 3) ▶ ▶ . However, the difference between p21 LIs for the two areas was not statistically significant (P > 0.05).
p27
In nonneoplastic lung tissue, the bronchial and alveolar epithelial cells and lymphoid cells showed intense nuclear staining for p27 protein. In tumor, positive staining was demonstrated in 94.4% (51/54) of the cases. Cells located in the periphery showed higher LI (range, 12–88%) than those in the center (range, 2–68%) (Figures 2 and 3) ▶ ▶ . The difference in p27 LIs between the two areas was statistically significant (P < 0.0001). In addition, staining in the periphery was more intense, although not as intense as in nonneoplastic cells.
p53
63.0% (34/54) of the cases exhibited positive staining for the p53 protein. No significant difference in LI was found between cells in the periphery and those in the center of the tumors (P > 0.1) (Figures 2 and 3) ▶ ▶ .
Specific Correlations in Staining Patterns
The signal cascade in which many of these cell cycle regulators are involved, and their interactions with one another have been extensively studied in the past few years. Based on the known relationship among some of these molecules, we statistically evaluated the immunohistochemical results (LI) obtained in the periphery of the tumor nodule as follows (Table 2) ▶ .
Table 2.
Correlation Coefficients (ρ) between Expression Frequencies of the Related Proteins in Tumor Cells
| Cyclin A | Cdk2 | CAK | P21 | p27 | p53 | |
|---|---|---|---|---|---|---|
| Cyclin A | — | 0.74** | 0.12 | 0.078 | 0.312 | N.D. |
| Cdk2 | 0.74** | — | 0.11 | 0.089 | 0.53* | N.D. |
| CAK | 0.12 | 0.11 | — | N.D. | N.D. | N.D. |
| P21 | 0.078 | 0.089 | N.D. | — | −0.573** | −0.171 |
| p27 | 0.312 | 0.53* | N.D. | −0.573** | — | N.D. |
| p53 | N.D. | N.D. | N.D. | −0.171 | N.D. | — |
*P < 0.01
**P < 0.001.
CAK, cdk-activating kinase; N.D., not determined.
Positive ratios of the tumor cells on immunohistochemical staining against respective proteins counted in the peripheral region of the tumor nodule in each case were analyzed by Spearman’s rank test.
p53 and p21
No specific relationship between p53 and p21 staining (ie, coexpression or reciprocal staining) was identified in either individual tumor cells or individual cases. The correlation coefficient as determined by Spearman’s test between these two LIs was −0.171 (P > 0.1).
p21 and p27
Overall, there was a weak but significant inverse correlation between these two LIs with a correlation coefficient of −0.573 (P < 0.001). Those cases which revealed high p21 LI showed no or weak p27 staining in occasional tumor cells at the periphery and vice versa. Furthermore, in serial sections, expression of p21 and that of p27 were observed to be reciprocal at the cellular level, not only in the tumor cells, but also in the bronchial or alveolar epithelial cells and lymphocytes (Figure 4) ▶ .
Figure 4.
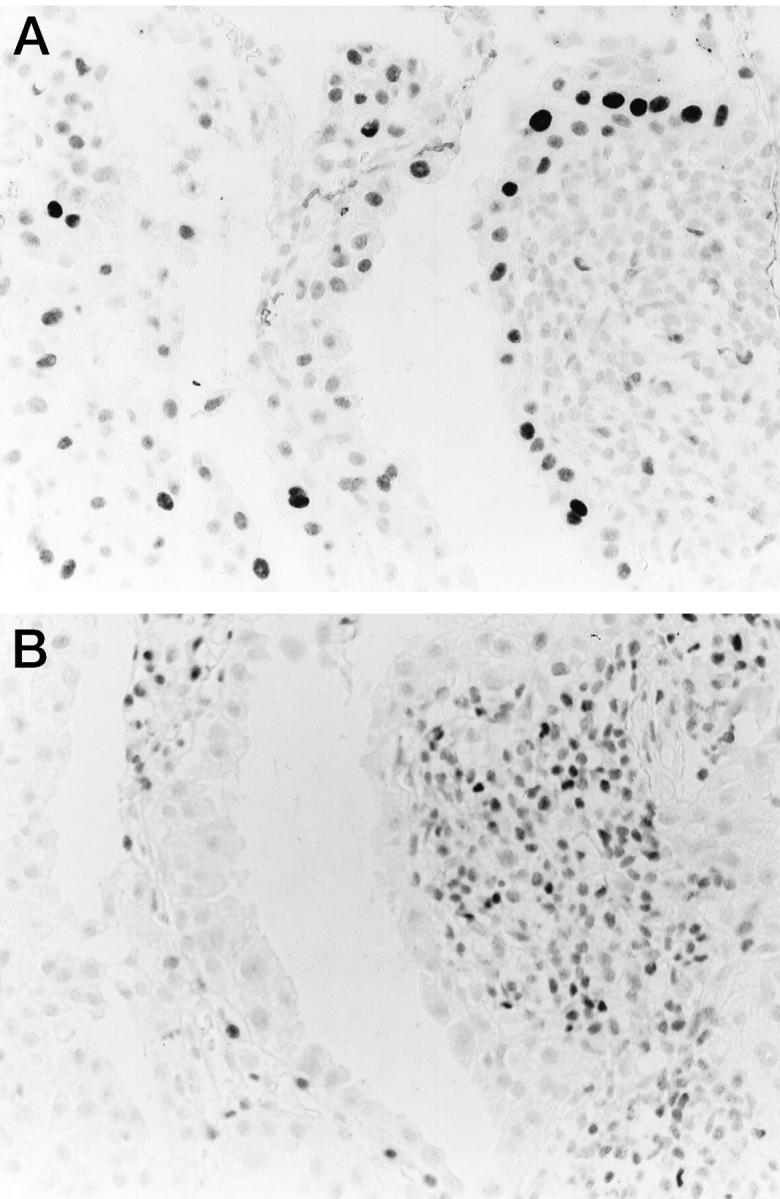
Immunohistochemical staining for p21 and p27 proteins in serial sections. Reciprocal expression of p21 (A) and p27 (B) in the cells around the advancing border of the tumor nodule as well as infiltrating lymphocytes. Streptavidin-biotin peroxidase method with hematoxylin counterstain; magnification, ×350.
Cyclin A and cdk2
The correlation between cyclin A and cdk2 LIs was statistically significant with a coefficient of 0.74 (P < 0.001). Analysis of serial sections demonstrated that cells positive for cyclin A staining also exhibited positive cdk2 staining at a high frequency (Figure 5) ▶ .
Figure 5.
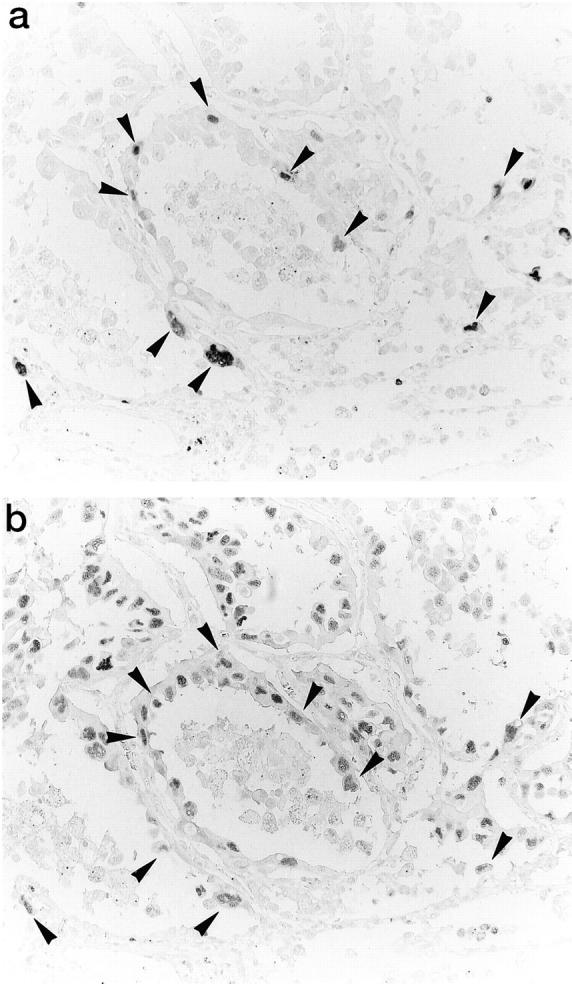
Immunohistochemical staining for cyclin A and cdk2 proteins on serial sections. Coexpression of cyclin A (a) and cdk2 (b) in the cells around the advancing border of the tumor nodule. Streptavidin-biotin peroxidase method with hematoxylin counterstain; magnification, ×350.
CAK and Cyclin A-cdk2
Although cyclin A-cdk2 positivity was observed predominantly at the periphery, positive CAK staining was observed ubiquitously, ie, in both the periphery and the center (Figures 2 and 3) ▶ ▶ . The correlation coefficient between LIs of CAK and cyclin A was 0.12 and between CAK and cdk2 it was 0.11. Neither was statistically significant.
Cyclin A-cdk2 and p21 or p27
p21 and p27 are universal inhibitors of cyclin-cdk complexes, including cyclin A-cdk2, which was examined in this study. There was occasional overlap of the staining patterns for cyclin A-cdk2 and p21 or p27 in the tumor cells. However, the correlations between the cyclin A and the p21 LIs (ρ = 0.078) or the cyclin A and the p27 LIs (ρ = 0.312) were not statistically significant.
Cdk2- or p27-Associated Kinase Activity
Cdk2- or p27-associated kinase activity was examined in fresh tissue samples from five selected cases which showed immunohistochemical positivity for cyclin A, cdk2, and p27 (Table 1) ▶ . Protein amounts used for the assay were standardized by anti-AE1 blotting (Figure 6 ▶ , lower panel). This standardization gave us equal amounts of protein from epithelial tumor cells and normal epithelial cells, because almost all of the tumor cells as well as nonneoplastic bronchial and alveolar epithelial cells stained positive by AE-1 on immunohistochemistry (data not shown). We found that the periphery exhibited significantly higher levels of cdk2 kinase activity compared with the center. As a control, matched nonneoplastic lung tissues from the same case revealed no detectable cdk2 kinase activity (Figure 6 ▶ , upper panel). Furthermore, the periphery exhibited higher levels of p27-associated kinase activity than the center. Adjacent nonneoplastic lung tissues from the same case revealed no detectable p27-associated kinase activity (Figure 6 ▶ , middle panel). We obtained similar results of kinase assay in all five cases examined. Because the p21 LI was lower than the p27 LI in all samples tested, we were unable to detect p21-associated kinase activity in any area of the five samples (data not shown).
Figure 6.
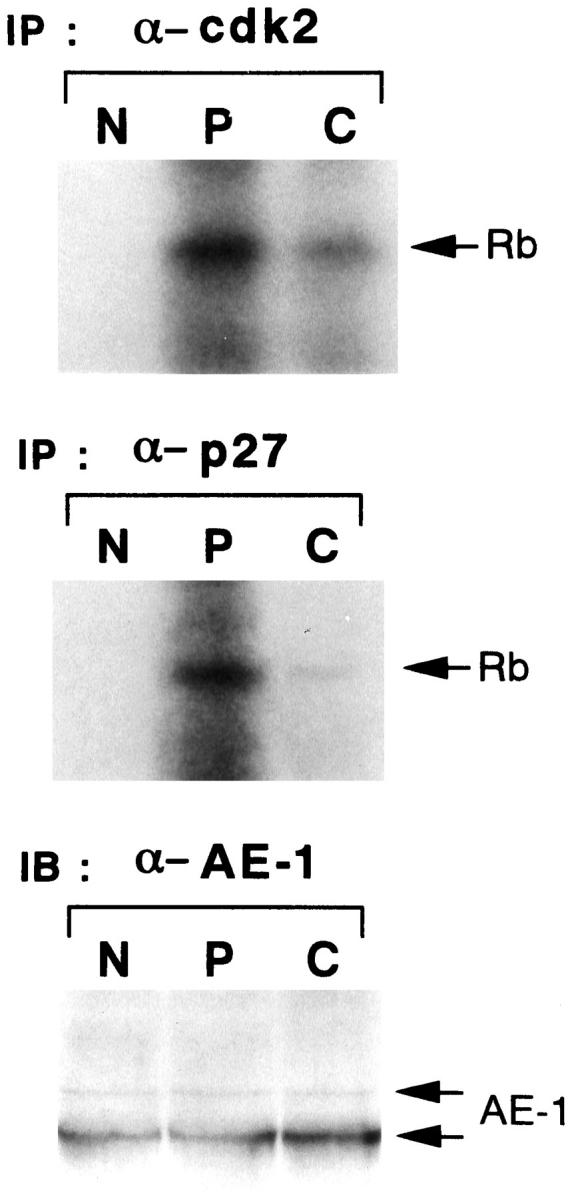
Kinase activities associated with cdk2 or p27. Cdk2-associated (upper) or p27-associated (middle) kinase activity are shown in normal lung tissue (N) and in peripheral (P) and central (C) regions of the tumor nodules of surgically resected lung tissue. Lysates (200–400 μg) were immunoprecipitated with anti-cdk2 or anti-p27 antibodies and the immunocomplex was assayed for kinase activity using glutathione S-transferase-RB fusion protein as a substrate. The result of immunoblotting analysis with anti-AE1 antibody (lower) showing standardization of the amount of protein from epithelial cells in three areas described above. IP, immunoprecipitation; IB, immunoblotting.
Discussion
Tumor extension in well differentiated pulmonary adenocarcinoma has been accompanied by development of a centrally located scar as a desmoplastic host reaction. 13,34-37 It could be hypothesized that the tumor cells in the periphery proliferate actively at the advancing border and thus extend outward while other cells, rather static in their proliferative activity, end up in a relatively central location as the tumor nodule enlarges. 12 Subsequently, attenuation of proliferation in the center brings about development of polarity, ie, a peripheral proliferating zone and a central scar. However, the mechanisms by which this process occurs have been unclear at cellular level.
To elucidate this mechanism, we selected 54 cases of small well differentiated adenocarcinoma. This was a very useful model for our experimental purpose because all cases showed tumor polarity without significant secondary changes such as necrosis or dense hyalinization. The clinicopathological implications could not be evaluated in this study because all of the cases were classified into Stage I and were expected to show a favorable prognosis. However, these studies were able to clarify several essential mechanisms in this process, particularly the involvement of cell cycle regulators.
First of all, Ki-67 staining revealed heterogeneous proliferative activity in the tumor nodules characterized by a significantly higher LI in the periphery.
Second, these actively proliferating, peripherally located cells stained positively for cyclin A and cdk2, as we previously demonstrated in squamous cell carcinoma of the lung. 31 Presumably, their coexpression and consequent high kinase activity should serve to promote active proliferation. 31 Indeed, in vitro cdk2 kinase assay revealed higher kinase activity in the periphery.
Third, this up-regulation of cdk2 activity is not induced through a CAK-dependent pathway. This is in contrast to a previous report in which the proliferative activity in human breast and urinary bladder carcinomas was attributed to up-regulated expression of CAK protein. 38
Assuming that the cyclin A-cdk2 complex is not activated by the positive regulator CAK, one alternative interpretation is that its activity is up-regulated by the suppression of kinase inhibitors. However, our results show that p21 and p27 exhibit higher LIs in the periphery. To test the idea that these molecules do not function as kinase inhibitors in these cases, we examined p27-associated cdk2 kinase activity in fresh tissue samples using a cdk2-specific protein extraction procedure. 30,39 Our results showed that p27 probably forms complexes with cyclins-cdk2 and that these complexes exhibit higher kinase activity in the periphery despite higher p27 expression. As illustrated in Figure 7 ▶ , these results can be interpreted in two ways.
Figure 7.
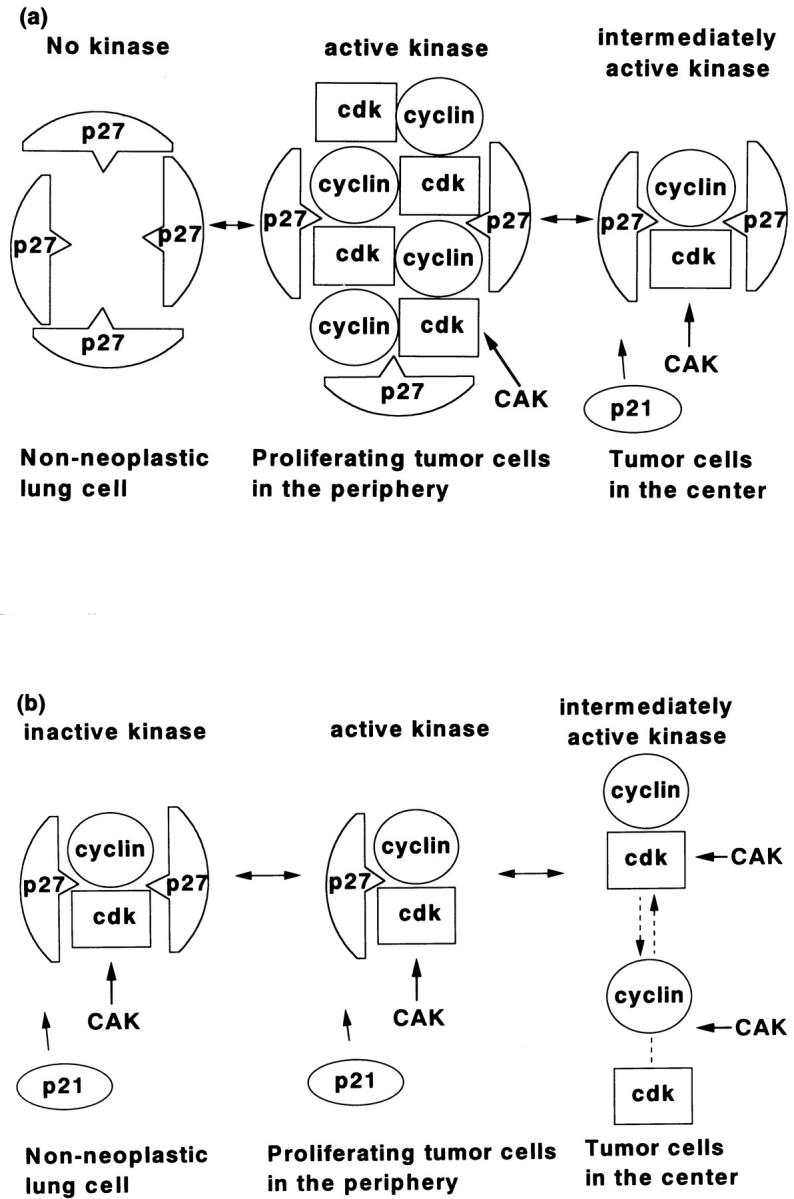
Diagrams showing possible interactions of cyclin A, cdk2, and p27. a: Inhibition of kinase activity by the elevated expression of p27 and/or p21 suppresses the proliferative activity of nonneoplastic lung cells, revealing no detectable kinase activity in addition to the very low level expression of cyclin A-cdk2 (left). Inhibition of kinase activity by moderately elevated expression of p27/p21 was overcome by highly elevated expression of cyclin A-cdk2 complexes in the periphery, resulting in active kinase (middle). The expression of both cyclin A-cdk2 and p27/p21 are lower in the tumor cells of the center resulting in moderate cyclin A-cdk2 complex activity (right) and consequent lower proliferation, relative to that in the periphery. b: p27 and/or p21 have dual roles depending on expression level. In nonneoplastic tissue, p27/p21 expression is elevated and functions as a kinase inhibitor, rendering cyclin A-cdk2 inactive as a kinase complex (left). In the periphery of the tumor, p27/p21 expression is moderately up-regulated and they function here as assembly factors promoting cyclin A-cdk2 assembly and kinase activity (middle). In tumor cells in the center, p27/p21 expression is suppressed and thus cannot promote the assembly of stable cyclin-cdk complexes, resulting in low kinase activity compared with in the periphery (right).
The first (Figure 7a) ▶ is that the inhibitory effects of up-regulated p21 or p27 on kinase activity in the periphery are overcome by the greater up-regulation of cyclin A-cdk2 activity (Figure 7a ▶ , middle). In the central region of the tumor nodules, expression of p21/p27 is lower, but cyclin A-cdk2 expression is also lower, resulting in low kinase activity (Figure 7a ▶ , right). In nonneoplastic lung tissue, cyclin A-cdk2 is not significantly detectable; thus, p21/p27 inhibition is more effective (Figure 7a ▶ , left). This may be a reasonable interpretation of the results of immunohistochemistry.
The second interpretation (Figure 7b) ▶ is that the functions of p21, p27, or both change depending on the stoichiometry of their interactions with cyclin A-cdk2. 40 In the nonneoplastic tissue, p21 and p27 are up-regulated and exert their function as kinase inhibitors, inactivating the cyclin A-cdk2 kinase complex (Figure 7b ▶ , left). However, in the proliferating cells of the periphery, expression of p21/p27 is intermediate. Here, they function as positive regulators by, for example, serving as assembly factors for the cyclin-cdk complex (Figure 7b ▶ , middle). In contrast, low expression of p21/p27 in the central region of the tumor results in low cyclin A-cdk2 complex formation due to insufficient assembly factor activity (Figure 7b ▶ , right).
We support the latter hypothesis for several reasons. First, although p21 has been characterized as a negative regulator of the cell cycle, we have successfully established cell lines constitutively expressing p21 derived from rat pheochromocytoma PC12 cells. 30 Furthermore, the one among those cell lines which exhibited the highest expression of p21 protein also showed a slightly enhanced proliferation rate compared with parental cells. These results are consistent with the idea that at some expression level, p21 can function as an assembly factor and activator of cdk rather than a kinase inhibitor. This is also supported by Zhang et al, who demonstrated that addition of recombinant p21 protein to cyclin A-cdk2 complex in vitro resulted in a dose-dependent, biphasic change in cdk2-kinase activity; activity initially increased at lower p21 concentrations and then rapidly disappeared as p21 concentration was increased. 40 From this, we imagine that p21 and p27 could function as assembly factors of cdk and cyclin to form stable ternary or quaternary complexes, similar to the way p37/MAT1 functions to promote assembly of cyclin H and cdk7 into the CAK complex. 41,42
Our analysis of serial sections further revealed that expression of p21 and p27 are inversely correlated in a statistically significant manner, suggesting that they function as reciprocal and redundant cdk regulators.
Finally, the expression of p21 seemed to be regulated by a p53-independent pathway in adenocarcinoma of the lung, similar to what has been described in several other kinds of human tumors including pancreatic or non-small cell lung carcinomas. 43-45
From a pathological viewpoint it appears that despite their lower proliferative activity, centrally located tumor cells still retain their malignant potential as ascertained by such criteria as vascular invasiveness. 13,46 Although one explanation for this biological aggressiveness may be the abundance of capillaries and lymph vessels in the center, the discrepancy between proliferative capability and high invasiveness should be elucidated by future study.
In conclusion, we propose the following hypothesis for the mechanism of extension of pulmonary adenocarcinoma at the cellular level:
Peripherally located tumor cells maintain their proliferative potential and extend outward on the alveolar surface. During this process, high kinase activity manifested by cyclin A-cdk2 complexes plays a crucial role. 31 Cyclin A and cdk2 are associated and form a stable complex 31 and p21/p27 expression is mildly suppressed below the level at which p21 and p27 can exert an inhibitory effect on cdk.
As the advancing border of the tumor extends, centrally located tumor cells adapt themselves to the fibrotic process, probably by host response. During that process, p21/p27 expression is down-regulated, possibly by up-regulation of some type of suppressor of p21/p27. Concomitantly, cyclin and cdk are also down-regulated, thus resulting in an overall decrease in kinase activity.
Acknowledgments
We thank the technical staffs of the Departments of Pathology, Kitasato University School of Medicine and Saiseikai Central Hospital, for their excellent technical support.
Footnotes
Address reprint requests to Toru Kameya, M.D., Department of Pathology, Kitasato University School of Medicine, 1–15-1, Sagamihara, Kanagawa 228-8555, Japan. E-mail: ydobashi@med.kitasato-u.ac.jp.
Supported by grant-in-aid for Scientific Research (no. 09770130) from the Ministry of Education, Science and Culture in Japan and by a Mitsui Life Social Welfare Foundation Research Grant.
References
- 1.Barsky SH, Cameron R, Osann KE, Tomita D, Holmes EC: Rising incidence of bronchioloalveolar carcinoma and its unique clinicopathologic features. Cancer 1994, 73:1163-1170 [DOI] [PubMed] [Google Scholar]
- 2.Kerr KM, Carey FA, King G, Lamb D: Atypical alveolar hyperplasia: relationship with pulmonary adenocarcinoma, p53, and c-erbB-2 expression. J Pathol 1994, 174:249-256 [DOI] [PubMed] [Google Scholar]
- 3.Kitamura H, Kodama Y, Nakamura N, Nakatani Y, Inayama Y, Iida M, Noda K, Ogawa N, Shibagaki T, Kanisawa M: Proliferative potential and p53 overexpression in precursor and early stage lesions of bronchioalveolar lung carcinoma. Am J Pathol 1995, 146:876-887 [PMC free article] [PubMed] [Google Scholar]
- 4.Ohshima S, Shimizu Y, Takahama M: Detection of c-Ki-ras gene mutation in paraffin sections of adenocarcinoma and atypical bronchioloalveolar cell hyperplasia of human lung. Virchows Arch 1994, 424:129-134 [DOI] [PubMed] [Google Scholar]
- 5.Westra WH, Bass IO, Hurban RH, Askin FB, Wilson K, Offerhaus GJA, Slebos RJC: K-ras oncogene activation in atypical alveolar hyperplasia of the human lung. Cancer Res 1996, 56:2224-2228 [PubMed] [Google Scholar]
- 6.Iggo R, Gatter K, Bartek J, Lane D, Harris AL: Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335:675-679 [DOI] [PubMed] [Google Scholar]
- 7.Li ZH, Zheng J, Weiss LM, Shibata D: c-K-ras and p53 mutation occur very early in adenocarcinoma of the lung. Am J Pathol 1994, 144:303-309 [PMC free article] [PubMed] [Google Scholar]
- 8.Kishimoto Y, Murakami Y, Shiraishi M, Hayashi K, Sekiya T: Aberrations of the p53 tumor suppresser gene in human non-small cell carcinomas of the lung. Cancer Res 1992, 52:4799-4804 [PubMed] [Google Scholar]
- 9.Kitamura H, Kameda Y, Nakamura N, Inayama Y, Nakakita Y, Shibagaki T, Ito T, Hayashi H, Kimura H, Kanisawa M: Atypical adenomatous hyperplasia and bronchoalveolar lung carcinoma.: Analysis by morphometry and the expression of p53 and carcinoembryonic antigen. Am J Surg Pathol 1996, 20:553-562 [DOI] [PubMed] [Google Scholar]
- 10.Nakayama H, Noguchi M, Tsuchiya R, Kodama T, Shimosato Y: Clonal growth of atypical adenomatous hyperplasia of the lung: cytofluorometric analysis of nuclear DNA content. Mod Pathol 1990, 3:314-320 [PubMed] [Google Scholar]
- 11.Noguchi M, Morikawa A, Kawasaki M, Matsuno Y, Yamada T, Hirohashi S, Kondo H: Small adenocarcinoma of the lung: histologic characteristic and prognosis. Cancer 1995, 75:2844-2852 [DOI] [PubMed] [Google Scholar]
- 12.Yoshida K, Morinaga S, Shimosato Y, Hayata Y: A cell kinetic study of pulmonary adenocarcinoma by an immunoperoxidase procedure after bromodeoxyuridine labeling. Cancer 1989, 64:2284-2291 [DOI] [PubMed] [Google Scholar]
- 13.Shimosato Y, Suzuki A, Hashimoto T, Nishizaki Y, Kodama T, Yoneyama T, Kameya T: Prognotic implications of fibrotic focus (scar) in small peripheral lung cancers. Am J Surg Pathol 1980, 4:365-373 [DOI] [PubMed] [Google Scholar]
- 14.Pardee AB: G1 events and regulation of cell proliferation. Science 1989, 246:603-608 [DOI] [PubMed] [Google Scholar]
- 15.Lees E, Faha B, Dulic V, Reed SI, Harlow E: Cyclin E/cdk2, and cyclin A/cdk2 kinase associated with p107, and E2F in a temporary distinct manner. Genes Dev 1992, 6:1874-1885 [DOI] [PubMed] [Google Scholar]
- 16.Xiong Y, Connolly T, Futcher B, Beach D: Human D-type cyclin. Cell 1991, 65:691-699 [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ: G1 phase progression: cycling on cue. Cell 1994, 79:551-555 [DOI] [PubMed] [Google Scholar]
- 18.Hunter T, Pines J: Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994, 79:573-582 [DOI] [PubMed] [Google Scholar]
- 19.Sherr CJ: Cancer cell cycle. Science 1996, 274:1672-1678 [DOI] [PubMed] [Google Scholar]
- 20.Dulic V, Lees E, Reed SI: Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992, 257:1958-1961 [DOI] [PubMed] [Google Scholar]
- 21.Koff A, Giordanao A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM: Formation and activation of cyclin E-cdk2 complex during the G1-phase of the human cell cycle. Science 1992, 257:1689-1694 [DOI] [PubMed] [Google Scholar]
- 22.Fisher RP, Morgan DO: A novel cyclin associated with MO15/CDK7 to form the CDK-activating kinase. Cell 1994, 78:713-724 [DOI] [PubMed] [Google Scholar]
- 23.Makela TP, Tassan JP, Nigg EA, Frutiger S, Hughes GJ, Weinberg RA: A cyclin associated with the CDK-activating kinase MO15. Nature 1994, 371:254-257 [DOI] [PubMed] [Google Scholar]
- 24.Tassan JP, Schultz SJ, Bartek J, Nigg EA: Cell cycle analysis of the activity, subcellular localization, and subunit composition of human CAK (CDK-activating kinase). J Cell Biol 1994, 127:467-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan DO: Principles of CDK regulation. Nature 1995, 374:131-134 [DOI] [PubMed] [Google Scholar]
- 26.El-Deiry W, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M: WAF1/CIP1 is induced in p53-mediated G1 arrest, and apoptosis. Cancer Res 1994, 54:1169-1174 [PubMed] [Google Scholar]
- 27.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D: p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366:701-707 [DOI] [PubMed] [Google Scholar]
- 28.: International Union Against Cancer: TNM Classification of Malignant Tumours. Sobin LH Wittekind CH eds. Fifth, Fully Revised Edition. 1997, :pp 91-97 Wiley-Liss, New York [Google Scholar]
- 29.Jiang SX, Kameya T, Sato Y, Yanase N, Yoshimura H, Kodama T: Bcl-2 protein expression in lung cancer and close correlation with neuroendocrine differentiation. Am J Pathol 1996, 148:837-846 [PMC free article] [PubMed] [Google Scholar]
- 30.Dobashi Y, Kudoh T, Matsumine A, Toyoshima K, Akiyama T: Constitutive overexpression of cdk2 inhibits neuronal differentiation of rat pheochromocytoma PC12 cells. J Biol Chem 1995, 270:23031-23037 [DOI] [PubMed] [Google Scholar]
- 31.Dobashi Y, Shoji M, Jiang SX, Kobayashi M, Kawakubo Y, Kameya T: Active cyclin A-cdk2 complex, a possible critical factor for cell proliferation in human primary lung carcinomas. Am J Pathol 1998, 153:963-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobashi Y, Shoji M, Wakata Y, Kameya T: Expression of HuD protein is essential for initial phase of neuronal differentiation in rat pheochromocytoma PC12 cells. Biochem Biophys Res Comm 1998, 244:226-229 [DOI] [PubMed] [Google Scholar]
- 33.Weidner N, Moore DH, Vartanian R: Correlation of Ki-67 antigen expression with mitotic figure index and tumor grade in breast carcinoma using the novel “paraffin”-reactive MIB-1 antibody. Hum Pathol 1994, 25:337-342 [DOI] [PubMed] [Google Scholar]
- 34.Friedrich G: Periphere lungenkrebse auf dem boden pleuranaher narben. Virchows Arch Pathol Anat 1939, 304:230-247 [Google Scholar]
- 35.Madri JA, Carter D: Scar cancer of the lung. Hum Pathol 1984, 15:625-631 [DOI] [PubMed] [Google Scholar]
- 36.Meyer EC, Liebow AA: Relationship of interstitial pneumonia, honeycombing and atypical epithelial proliferation to cancer of the lung. Cancer 1965, 18:322-351 [DOI] [PubMed] [Google Scholar]
- 37.Raeburn C, Spencer H: A study of the origin and development of lung scar cancer. Thorax 1953, 8:1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartokova J, Zemonava M, Bartek J: Expression of cdk7/CAK in normal and tumour cells of diverse histogenesis, cell-cycle position and differentiation. Int J Cancer 1996, 66:732-737 [DOI] [PubMed] [Google Scholar]
- 39.Dobashi Y, Kudoh T, Toyoshima K, Akiyama T: Persistent activation of cdk4 during neuronal differentiation of rat pheochromocytoma pheochromocytoma PC12 cells. Biochem Biophys Res Comm 1996, 221:351-355 [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Hannon GJ, Beach D: p21-containing cyclin kinases exist in both active and inactive states. Genes Dev 1994, 8:1750-1758 [DOI] [PubMed] [Google Scholar]
- 41.Devault A, Martinez AM, Fesquet D, Labbe JC, Morin N, Tassan JP, Nigg EA, Cavadore JC, Doree M: MAT1 (‘menage a trois’) a new Ring finger protein subunit stabilizing cyclin H-cdk 7 complexes in starfish, and Xenopus CAK. EMBO J 1995, 14:5027-5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fisher RP, Jin P, Chamberin HM, Morgan DO: Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell 1995, 83:47-57 [DOI] [PubMed] [Google Scholar]
- 43.DiGiuseppe JA, Redston MS, Yeo CJ, Kern SE, Hruban RH: p53-independent expression of the cyclin-dependent kinase inhibitor p21 in pancreatic carcinoma. Am J Pathol 1995, 147:884-888 [PMC free article] [PubMed] [Google Scholar]
- 44.Marchetti A, Doglioni C, Barbareschi M, Buttitta F, Pellegrini S, Dalla Palma P, Bevilacqua G: p21/WAF1 expression in lung carcinomas: association with differentiation. Oncogene 1996, 12:1319-1324 [PubMed] [Google Scholar]
- 45.Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Giro ID: Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res 1994, 54:3391-3395 [PubMed] [Google Scholar]
- 46.Yokoo H, Suckow EE: Peripheral lung cancers arising in scars. Cancer 1961, 14:1205-1215 [DOI] [PubMed] [Google Scholar]